International Journal of Biological Sciences
ISSN: 1449-2288
10
Impact Factor
ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2010; 6(2):172-186. doi:10.7150/ijbs.6.172 This issue Cite
Research Paper
The complete mitochondrial genome of the fall webworm, Hyphantria cunea (Lepidoptera: Arctiidae)
Fang Liao1,2, Lin Wang3, Song Wu4, Yu-Ping Li4, Lei Zhao1, Guo-Ming Huang2, Chun-Jing Niu2, Yan-Qun Liu4, ![]() , Ming-Gang Li1,
, Ming-Gang Li1, ![]()
1. College of Life Sciences, Nankai University, Tianjin 300071, China
2. Tianjin Entry-Exit Inspection and Quarantine Bureau, Tianjin 300457, China
3. Beijing Entry-Exit Inspection and Quarantine Bureau, Beijing 101113, China
4. College of Bioscience and Biotechnology, Shenyang Agricultural University, Liaoning, Shenyang 110866, China
Received 2010-2-3; Accepted 2010-3-26; Published 2010-3-29
Citation:
Liao F, Wang L, Wu S, Li YP, Zhao L, Huang GM, Niu CJ, Liu YQ, Li MG. The complete mitochondrial genome of the fall webworm, Hyphantria cunea (Lepidoptera: Arctiidae). Int J Biol Sci 2010; 6(2):172-186. doi:10.7150/ijbs.6.172. https://www.ijbs.com/v06p0172.htm
Other stylesAbstract
The complete mitochondrial genome (mitogenome) of the fall webworm, Hyphantria cunea (Lepidoptera: Arctiidae) was determined. The genome is a circular molecule 15 481 bp long. It presents a typical gene organization and order for completely sequenced lepidopteran mitogenomes, but differs from the insect ancestral type for the placement of tRNAMet. The nucleotide composition of the genome is also highly A + T biased, accounting for 80.38%, with a slightly positive AT skewness (0.010), indicating the occurrence of more As than Ts, as found in the Noctuoidea species. All protein-coding genes (PCGs) are initiated by ATN codons, except for COI, which is tentatively designated by the CGA codon as observed in other lepidopterans. Four of 13 PCGs harbor the incomplete termination codon, T or TA. All tRNAs have a typical clover-leaf structure of mitochondrial tRNAs, except for tRNASer(AGN), the DHU arm of which could not form a stable stem-loop structure. The intergenic spacer sequence between tRNASer(AGN) and ND1 also contains the ATACTAA motif, which is conserved across the Lepidoptera order. The H. cunea A+T-rich region of 357 bp is comprised of non-repetitive sequences, but harbors several features common to the Lepidoptera insects, including the motif ATAGA followed by an 18 bp poly-T stretch, a microsatellite-like (AT)8 element preceded by the ATTTA motif, an 11 bp poly-A present immediately upstream tRNAMet. The phylogenetic analyses support the view that the H. cunea is closerly related to the Lymantria dispar than Ochrogaster lunifer, and support the hypothesis that Noctuoidea (H. cunea, L. dispar, and O. lunifer) and Geometroidea (Phthonandria atrilineata) are monophyletic. However, in the phylogenetic trees based on mitogenome sequences among the lepidopteran superfamilies, Papillonoidea (Artogeia melete, Acraea issoria, and Coreana raphaelis) joined basally within the monophyly of Lepidoptera, which is different to the traditional classification.
Keywords: Fall webworm, Hyphantria cunea, Mitochondrial genome, Lepidoptera, Arctiidae, Phylogeny
1. Introduction
The fall webworm, Hyphantria cunea Drury (Lepidoptera: Arctiidae), is a severe invasive and quarantine pest which has a wide range of habitats. It is a polyphagus pest that feeds on about 160 species of broad leaf trees. The preferred host plants include mulberry, oak, hickory, pecan, walnut, elm, alder, willow, sweetgum, and poplar. This insect has caused serious damage to forests throughout its range and appears to be continuing to spread. It also damages the roadside and garden trees around urban areas. The species was introduced from North America to Central Europe and Eastern Asia in the early 1940s [1, 2]. In China, this species was first found in Dandong (124°N/40°E) of Liaoning Province in 1979, and now has spread southwards to Shanghai (129°N/31°E) and westwards to Xianyang (108°N/34°E) of Shanxi Province. The southern populations in China may complete three generations in one year, while in the north the fall webworm completes only one life cycle. Many studies have been done on aspects of adaptability, sex pheromones, host preference and natural enemies of the fall webworm [3]. As H. cunea is a devastating invasive species, the mitochondrial genome (mitogenome) information of the species may provide fundamental information for future phylogenetic analyses and evolutionary biology.
Insect mitochondrial DNA (mtDNA) is a circular DNA molecule with 14-20 kb in size and has a remarkably conserved set of 37 genes, including 13 protein-coding genes (PCGs; subunits 6 and 8 of the F0 ATPase [ATP6 and ATP8]; cytochrome oxidase subunits 1-3 [COI-III]; cytochrome b [Cytb]; NADH dehydrogenase subunits 1-6 and 4L [ND1-6 and ND4L]), two ribosomal RNA genes (large and small ribosomal RNAs [lrRNA and srRNA]), and 22 tRNA genes [4, 5]. It additionally contains a control region of variable length, known as the adenine (A) + thymine (T)-rich region in insect mtDNA, which is involved in the regulation and initiation of mtDNA replication and transcription [6]. The mitochondrial genes and genomes have been widely used as an informative molecular marker for diverse evolutionary studies of animals, including phylogenetics and population genetics [7-9], with the development of long range PCR for amplification of partial sequence of mtDNA genes and whole mitochondrial genome [10].
At present, the complete or nearly complete mitogenome sequences from more than 100 species of insects have been determined. However, only 19 complete or nearly complete mitogenomes are currently available in the GenBank for lepidopteran species (Table 1). The Ostrinia sequences each lack the sequence information of the A+T-rich region, partial tRNAMet and srRNA sequence. The L. chinensis and P. xuthus sequences each lack the sequence information of the partial srRNA, A+T-rich region, tRNAMet-Ile-Gln, and partial ND2. Within the insects, the Lepidoptera order accounts for more than 160 000 species. Despite this huge taxonomic diversity the existing information on lepidopteran mtDNA is very limited and limited to six superfamiles among the 45-48 known and to 13 families of the recognized 120. Newly added lepidopteran mitogenomes can provide further insights into our understanding of diversity of lepidopteran mitogenomes and evolution.
Table 1
List of the complete mitogenome of Lepidoptera
| Superfamily / Family | Species | Acc. number | Reference |
|---|---|---|---|
| Noctuoidea | |||
| Arctiidae | Hyphantria cunea | GU592049 | This study |
| Lymantriidae | Lymantria dispar | FJ617240 | Zhu et al. unpublished |
| Notodontidae | Ochrogaster lunifer | AM946601 | [11] |
| Geometroidea | |||
| Geometridae | Phthonandria atrilineata | EU569764 | [12] |
| Bombycoidea | |||
| Saturniidae | Antheraea pernyi | AY242996 | [13] |
| Saturniidae | Antheraea yamamai | EU726630 | [14] |
| Saturniidae | Caligula boisduvalii | EF622227 | [15] |
| Saturniidae | Eriogyna pyretorum | FJ685653 | [16] |
| Bombycidae | Bombyx mori | AB070264 | [17] |
| Bombycidae | Chinese Bombyx mandarina | AY301620 | [18] |
| Bombycidae | Japanese Bombyx mandarina | NC_003395 | [17] |
| Sphingidae | Manduca sexta | EU286785 | [19] |
| Pyraloidea | |||
| Crambidae | Diatraea saccharalis | FJ240227 | Li and Yue, unpublished |
| Crambidae | Ostrinia furnicalis | NC_003368 | [20] |
| Crambidae | Ostrinia nubilalis | NC_003367 | [20] |
| Tortricoidea | |||
| Tortricidae | Adoxophyes honmai | DQ073916 | [21] |
| Papilionoidea | |||
| Nymphalidae | Acraea issoria | NC_013604 | [22] |
| Pieridae | Artogeia melete | EU597124 | [23] |
| Lycaenidae | Coreana raphaelis | DQ102703 | [24] |
| Papilionidae | Luehdorfia chinensis | EU622524 | Liu et al. unpublished |
| Papilionidae | Papilio xuthus | EF621724 | Feng et al. unpublished |
The H. cunea mitochondrial COI, COIII and Cytb have been utilized for biological identification as DNA barcode and phylogenetic studies [25, 26], but the genetic information on the complete mtDNA of the species remains largely unknown. In this study, we describe the complete mitogenome sequence of the fall webworm, H. cunea, and compare its sequence with other available lepidopteran mitogenomes. Furthermore, the mitogenome sequence of H. cunea was used to provide further insight into the phylogenetic relationships among lepidopteran superfamilies.
2. Materials and Methods
Insect and total DNA extraction
The H. cunea larvae were collected on the mulberry trees on the campus of the Shenyang Agricultural University, Shenyang, China. The larvae were then fed on the leaves of mulberry trees in room until pupation. The fresh pupae were directly frozen and kept in the laboratory at - 80 °C. A single pupa was used to extract the total DNA using the TIANamp Genomic DNA Kit (TIANGEN, Beijing, China) according to the manufacturer's instruction.
PCR amplification and sequencing
The full mitogenome of H. cunea was amplified in four overlapping fragments by PCR amplification using universal primers and specific primers designed for this study (Fig. 1; Table 2). All PCR reactions were performed in a 50 μl volume with 1 U of LA Taq (TaKaRa Co., Dalian, China), 1 μl (about 20 ng) of DNA, 5 μl 10 × LA Taq buffer (Mg2+ plus), 200 μM dNTPs, and 10 pmol each primer. Initially, the H. cunea COI gene of ~600 bp was amplified using the primer set LYQ1/LYQ2 as previously reported [25], and the Cytb gene of ~400 bp was amplified using the primer set LYQ5/LYQ6 as previously reported [27]. The PCR amplification was performed under the following procedure: 2 min at 94 °C, followed by 35 cycles of 1 min at 94 °C, 30 sec at 50 °C, and 1 min at 72 °C, with a subsequent 10 min final extension at 72 °C. After purification with TIANgel Midi Purification Kit (TIANGEN, Beijing, China), the PCR fragments were directly sequenced with the PCR primers.
Fig 1
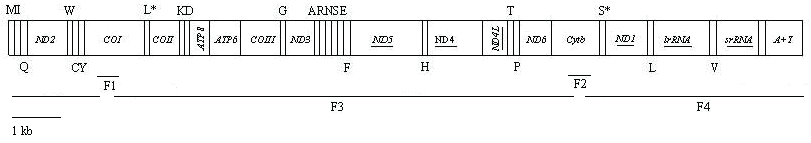
Linear map of the mitogenome of Hyphantria cunea. The tRNAs are labeled according to the IUPAC-IUB single letter amino acid codes above the bar indicating coding sequence on major strand or below the bar showing on minor strand. One-letter symbol L, L*, S and S* denote codon tRNALeu(CUN), tRNALeu(UUR), tRNASer(AGN), and tRNASer(UCN), respectively. Underlined PCGs or rRNA genes are located on minor strand and PCGs that are not underlined are located on major strand. Overlapping lines (F1-F4) under the map denote four overlapping PCR fragments amplified for sequencing. The line at the lower left represents the map scale.
Table 2
Primers used to amplify the Hyphantria cunea mitogenome
| Primer | Sequence (5'- 3') | Fragment | Reference |
|---|---|---|---|
| LYQ1 | GGTCAACAAATCATAAAGATATTGG | F1 | [25] |
| LYQ2 | TAAACTTCAGGGTGACCAAAAAATCA | ||
| LYQ5 | TATGTACTACCATGAGGACAAATATC | F2 | [27] |
| LYQ6 | ATTACACCTCCTAATTTATTAGGAAT | ||
| LYQ29 | CTTTTCTATTACTTCTTTCTCTTCCTGTTTTA | F3 | This study |
| LYQ32 | TAAAATAATAAATGGTAATAAAAAATGAAATG | ||
| LYQ30 | TAAAACAGGAAGAGAAAGAAGTAATAGAAAAG | F4 | This study |
| LYQ31 | CATTTCATTTTTTATTACCATTTATTATTTTA |
On the basis of the information from the determined fragments, two new primer pairs LYQ29/LYQ32 and LYQ30/LYQ31 were designed to amplify the remaining longer fragments of the mitogenome of H. cunea (F3 and F4 in Fig. 1 and Table 1). The two fragments were amplified with denaturation at 94 °C for 2 min, followed by 35 cycles of 1 min at 94 °C, 10 min at 65 °C, with a subsequent 10 min final extension at 72 °C. These PCR products were then utilized to construct a shotgun sequencing library. In brief, DNAs were sheared into 1-3 kb fragments using DNase I, and the DNA fractions were collected with a Chromaspin TE 1000 column. The DNA fractions were then cloned into the pGEM-T easy vector (Promega, USA), and each of the resultant plasmid DNAs was isolated with a Wizard Plus SV Minipreps DNA Purification System (Promega, USA). DNA sequencing was conducted using the ABI PRISM BigDyeTerminator v3.1 Cycle Sequencing Kit and the ABI PRISMTM 3100 Genetic Analyzer (PE Applied Biosystems, USA). All fragments were sequenced from both strands. The number of clones sequenced was sufficient to fulfill the six times coverage of the mitogenome.
Sequence analysis
The sequence alignment was carried out using Clustal X [28]. The PCGs and rRNA genes were determined by BLAST on NCBI Entrez Database and by comparing them with homologous regions in other lepidopteran mitogenome sequences. The PCG nucleotide sequences were translated on the basis of the Invertebrate Mitochondrial Genetic Code. The tRNA genes and its secondary structure were predicted using the tRNAscan-SE Search [29]. The two tRNASer secondary structure not found by tRNAscan-SE Search was developed using the constraints proposed by Steinberg and Cedergren [30]. Composition skew analysis was carried out to describe the base composition of nucleotide sequences, which measures the relative number of As to Ts (AT skew=[A-T]/[A+T]) and Gs to Cs (GC skew=[G-C]/[G+C]) [31]. Codon usage was calculated using the Countcodon program version 4 (http://www.kazusa.or.jp/codon/countcodon.html). The entire A+T-rich region was subjected to a search for the tandem repeats using Tandem Repeats Finder program [32]. The sequence data has been deposited in GenBank under accession No. GU592049.
Phylogenetic analysis
To illustrate the phylogenetic relationship of Lepidoptera, the other complete mitogenomes were obtained from GenBank. The L. chinensis and P. xuthus sequences lacking more sequence information were excluded. The mitogenomes of Drosophila yakuba (NC_001322) [33] and Anopheles gambiae (NC_002084) [34] were used as outgroups. The alignment of the amino acid sequences of each 13 mitochondrial PCGs was aligned with Clustal X [28] using default settings and concatenated. As for the ND4 genes, the insertion of A nucleotide in O. furnacalis (position 8211 bp) and O. nubilalis (position 8206 bp) resulted in transcript frameshifts [13], the amino acid sequences of which were therefore revised for further phylogenetic analyses. The concatenated set of amino acids sequences from the 13 PCGs was used in phylogenetic analyses, which was performed using maximum parsimony (MP) and Nerghbor-joining (NJ) methods by using MEGA ver 4.0 [35].
3. Results and Discussion
Genome organization
The H. cunea mitogenome presents the typical gene content observed in metazoan mitogenomes (Table 3, Fig. 1): 13 PCGs, 22 tRNA genes, two rRNA subunits, and a major non-coding region known as the A+T-rich region in insects [5]. The complete mitogenome of H. cunea consists of 15 481 bp, which is well within the range observed in the completely sequenced lepidopteran insects, with size ranging from 15 140 in A. melete to 15 928 in Japanese B. mandarina (Table 4). The gene order and orientation of the H. cunea mitogenome are identical to the completely sequenced lepidopteran mitogenomes. By the translocation of tRNAMet to a position 5' upstream of tRNAIle, the lepidopteran arrangement differs from that of D. yakuba, the hypothesized ancestral gene order of insects [36]. This suggests that the mitochondrial gene arrangement in lepidopteran insects evolved independently after splitting from its stem lineage [24].
Genome composition and skewness
The genome composition of the major strand of the H. cunea mitogenome is heavily biased toward As and Ts, accounting for 80.38%: A 40.58%, G 7.55%, T 39.80% and C 12.07%, as is the case with other insect sequences (Table 4). The bias value is similar to the completely sequenced lepidopteran insects, with the range from 77.84% in O. lunifer to 82.66% in C. raphaelis. The A+T content in the sequence of the A+T-rich region is 94.96%, also within the range observed in the completely sequenced lepidopteran insects, with the value from 89.17% in A. melete to 98.25% in P. atrilineata.
Table 3
Annotation and gene organization of the Hyphantria cunea mitogenome
| Gene | Strand | Nucleotide no. | Size(bp) | Anticodon | Non | OL | Start codon | Stop codon |
|---|---|---|---|---|---|---|---|---|
| tRNAMet | J | 1-67 | 67 | CAT | ||||
| tRNAIle | J | 68-136 | 69 | GAT | 11 | |||
| tRNAGln | N | 148-216 | 69 | TTG | 50 | |||
| ND2 | J | 267-1277 | 1011 | 18 | ATT | TAA | ||
| tRNATrp | J | 1 296-1 364 | 69 | TCA | 8 | |||
| tRNACys | N | 1 357-1 419 | 63 | GCA | 4 | |||
| tRNATyr | N | 1 424-1 489 | 66 | GTA | 11 | |||
| COI | J | 1 501-3 034 | 1534 | CGA | T-tRNA | |||
| tRNALeu(UUR) | J | 3 035-3 100 | 66 | TAA | ||||
| COII | J | 3 101-3 782 | 682 | ATG | T-tRNA | |||
| tRNALys | J | 3 783-3 853 | 71 | CTT | 1 | |||
| tRNAAsp | J | 3 853-3 918 | 66 | GTC | ||||
| ATP8 | J | 3 919-4 080 | 162 | 7 | ATA | TAA | ||
| ATP6 | J | 4 074-4 750 | 677 | 5 | ATG | TA-COIII | ||
| COIII | J | 4 756-5 547 | 792 | 9 | ATG | TAA | ||
| tRNAGly | J | 5 557-5 621 | 65 | TCC | ||||
| ND3 | J | 5 622-5 975 | 354 | ATT | TAA | |||
| tRNAAla | J | 5 976-6 042 | 67 | TGC | 7 | |||
| tRNAArg | J | 6 050-6 116 | 67 | TCG | 5 | |||
| tRNAAsn | J | 6 122-6 188 | 67 | GTT | 9 | |||
| tRNASer(AGN) | J | 6 198-6 263 | 66 | GCT | 21 | |||
| tRNAGlu | J | 6 285-6 352 | 68 | TTC | 2 | |||
| tRNAPhe | N | 6 351-6 418 | 68 | GAA | 3 | |||
| ND5 | N | 6 422-8 167 | 1746 | ATA | TAA | |||
| tRNAHis | N | 8 168-8 235 | 68 | GTG | ||||
| ND4 | N | 8 236-9 574 | 1339 | ATG | T-tRNA | |||
| ND4L | N | 9 575-9 862 | 288 | 5 | ATG | TAA | ||
| tRNAThr | J | 9 868-9 932 | 65 | TGT | ||||
| tRNAPro | N | 9 933-9 997 | 65 | TGG | 7 | |||
| ND6 | J | 10 005-10 535 | 531 | 13 | ATT | TAA | ||
| Cytb | J | 10 549-11 697 | 1149 | 17 | ATA | TAA | ||
| tRNASer(UCN) | J | 11 715-11 782 | 68 | TGA | 34 | |||
| ND1 | N | 11 817-12 755 | 939 | 1 | ATG | TAA | ||
| tRNALeu(CUN) | N | 12 757-12 824 | 68 | TAG | ||||
| lrRNA | N | 12 825-14 250 | 1426 | |||||
| tRNAVal | N | 14 251-14 316 | 66 | TAC | ||||
| srRNA | N | 14 317-15 124 | 808 | |||||
| A+T-rich region | 15 125-15 481 | 357 |
J-strand, majority-coding strand; N-strand, minority-coding strand; Non, non-coding region; OL, overlapping region.
Table 4
Composition and skewness in the major strand of lepidopteran mitogenomes
| Species | size (bp) | A% | G% | T% | C% | A+T % | AT skew | GC skew |
|---|---|---|---|---|---|---|---|---|
| Whole genome | ||||||||
| Hyphantria cunea | 15 481 | 40.58 | 7.55 | 39.80 | 12.07 | 80.38 | 0.010 | -0.230 |
| Lymantria dispar | 15 569 | 40.58 | 7.57 | 39.30 | 12.55 | 79.88 | 0.016 | -0.247 |
| Ochrogaster lunifer | 15 593 | 40.09 | 7.56 | 37.75 | 14.60 | 77.84 | 0.030 | -0.318 |
| Phthonandria atrilineata | 15 499 | 40.78 | 7.67 | 40.24 | 11.31 | 81.02 | 0.007 | -0.192 |
| Antheraea pernyi | 15 566 | 39.22 | 7.77 | 40.94 | 12.06 | 80.16 | -0.021 | -0.216 |
| Antheraea yamamai | 15 338 | 39.26 | 7.69 | 41.04 | 12.02 | 80.29 | -0.022 | -0.219 |
| Caligula boisduvalii | 15 360 | 39.34 | 7.58 | 41.28 | 11.79 | 80.62 | -0.024 | -0.217 |
| Eriogyna pyretorum | 15 327 | 39.17 | 7.63 | 41.65 | 11.55 | 80.82 | -0.030 | -0.204 |
| Bombyx mori | 15 656 | 43.06 | 7.31 | 38.30 | 11.33 | 81.36 | 0.059 | -0.216 |
| Chinese Bombyx mandarina | 15 682 | 43.11 | 7.40 | 38.48 | 11.01 | 81.59 | 0.057 | -0.196 |
| Japanese Bombyx mandarina | 15 928 | 43.08 | 7.21 | 38.60 | 11.11 | 81.68 | 0.055 | -0.213 |
| Manduca sexta | 15 516 | 40.67 | 7.46 | 41.11 | 10.76 | 81.79 | -0.005 | -0.181 |
| Diatraea saccharalis | 15 490 | 40.87 | 7.42 | 39.15 | 12.56 | 80.02 | 0.021 | -0.257 |
| *Ostrinia nubilalis | 14 535 | 41.36 | 8.02 | 38.81 | 11.82 | 80.17 | 0.031 | -0.192 |
| *Ostrinia furnicalis | 14 536 | 41.46 | 7.91 | 38.92 | 11.71 | 80.37 | 0.032 | -0.194 |
| Adoxophyes honmai | 15 680 | 40.15 | 7.88 | 40.24 | 11.73 | 80.39 | -0.001 | -0.178 |
| Acraea issoria | 15 245 | 38.94 | 7.74 | 40.81 | 12.50 | 79.76 | -0.023 | -0.235 |
| Artogeia melete | 15 140 | 40.38 | 7.87 | 39.41 | 12.35 | 79.78 | 0.012 | -0.222 |
| Coreana raphaelis | 15 314 | 39.37 | 7.30 | 43.29 | 10.04 | 82.66 | -0.047 | -0.158 |
| *Luehdorfia chinensis | 13 860 | 40.07 | 7.74 | 40.44 | 11.75 | 80.51 | -0.005 | -0.206 |
| *Papilio xuthus | 13 964 | 39.53 | 7.85 | 40.45 | 12.17 | 79.98 | -0.012 | -0.216 |
| A+T-rich region | ||||||||
| Hyphantria cunea | 357 | 45.66 | 1.12 | 49.30 | 3.92 | 94.96 | -0.038 | -0.556 |
| Lymantria dispar | 435 | 45.29 | 1.61 | 50.50 | 2.30 | 96.09 | -0.054 | -0.176 |
| Ochrogaster lunifer | 319 | 44.51 | 1.57 | 48.90 | 5.02 | 93.42 | -0.047 | -0.524 |
| Phthonandria atrilineata | 457 | 40.70 | 0.66 | 57.55 | 1.09 | 98.25 | -0.171 | -0.246 |
| Antheraea pernyi | 552 | 41.12 | 4.17 | 49.28 | 5.43 | 90.40 | -0.090 | -0.127 |
| Antheraea yamamai | 334 | 41.62 | 3.59 | 47.90 | 6.89 | 89.52 | -0.070 | -0.315 |
| Caligula boisduvalii | 330 | 42.12 | 2.12 | 49.39 | 6.36 | 91.52 | -0.079 | -0.500 |
| Eriogyna pyretorum | 358 | 42.18 | 2.51 | 50.00 | 5.31 | 92.18 | -0.085 | -0.358 |
| Bombyx mori | 494 | 44.94 | 1.62 | 50.61 | 2.83 | 95.55 | -0.059 | -0.272 |
| Chinese Bombyx mandarina | 484 | 46.49 | 2.69 | 47.93 | 2.89 | 94.42 | -0.015 | -0.036 |
| Japanese Bombyx mandarina | 747 | 45.52 | 2.41 | 49.67 | 2.41 | 95.18 | -0.043 | 0 |
| Manduca sexta | 324 | 45.00 | 1.54 | 50.31 | 3.29 | 95.37 | -0.055 | -0.334 |
| Diatraea saccharalis | 335 | 43.28 | 0.60 | 51.64 | 4.48 | 94.93 | -0.088 | -0.764 |
| Adoxophyes honmai | 489 | 48.47 | 2.86 | 45.81 | 2.86 | 94.27 | 0.028 | 0 |
| Acraea issoria | 430 | 45.81 | 1.40 | 50.23 | 2.56 | 96.05 | -0.046 | -0.293 |
| Artogeia melete | 351 | 43.87 | 3.13 | 45.30 | 7.69 | 89.17 | -0.016 | -0.421 |
| Coreana raphaelis | 375 | 44.27 | 1.33 | 49.87 | 4.53 | 94.13 | -0.059 | -0.545 |
* partial mitogenome lacking of the A+T-rich region.
The lepidopteran AT skewness values vary from -0.047 in C. raphaelis to 0.059 in B. mori (Table 4). The AT skewness for the major strand of the H. cunea mitogenome is slightly positive (0.010), indicating the occurrence of more As than Ts. This case is also found in L. dispar (0.016), O. lunifer (0.030), P. atrilineata (0.007), B. mori (0.059), Japanese B. mandarina (0.055), Chinese B. mandarina (0.057), A. melete (0.012), D. saccharalis (0.021), O. nubilalis (0.031), and O. furnicalis (0.032). In contrast, the AT skews are negative in the other lepidopteran mitogenomes. When considering the A+T-rich region, however, the bias toward the use of Ts over As is more obvious in the analyzed lepidopteran mitogenomes with the H. cunea mitogenome exhibiting a slightly value (-0.038). The only one exception is represented in A. honmai where the A+T-rich region exhibits a slightly positive AT skewness (0.028).
In all sequenced lepidopteran mitogenomes, the GC skewness values vary from -0.158 in C. raphaelis to -0.318 in O. lunifer with the H. cunea mitogenome exhibiting a moderate skewness value (-0.230), referring to the occurrence of more Cs than Gs in the lepidopteran mitogenomes.
Protein-coding genes
All of the PCGs in the H. cunea mitogenome are initiated by typical ATN codons (six with ATG, three with ATT, and three with ATA), except for COI (Table 3). The open reading frame of the H. cunea COI gene also starts at a CGA codon for arginine as found in all lepidopteran insects (Fig. 2). The typical ATN initiator for mitochondrial PCGs is also not found at the start site for H. cunea COI or near the tRNATyr. The plausible translation initiator for H. cunea COI is ATA, located within the tRNATyr gene, overlapping 19 bp with the tRNATyr; however, a codon following this triplet has a TAG-stop codon before the CGA codon. This ATA sequence is unlikely to be the start site for H. cunea COI, and there are no other probable start codons for H. cunea COI. Thus, the COI gene must use an atypical start site. In the previous studies, some tetranucleotide (ATAA, TTAA, GTAA and ATTA) and a hexanucleotide (ATTTAA) have been proposed as an initiator of COI for Diptera insects including mosquitoes and Drosophila [34, 37-41]. Among the completely sequenced lepidopteran insects, including A. pernyi, A. yamamai, B. mori, B. mandarina, and C. raphaelis, the TTAG has been designated as an initiator for COI [13, 14, 17, 18], however, Ostrina species were designated as ATTTAG [20], and A. issoria and C. boisduvalii were designated as TTG [15, 22], and six species (A. honmai, A. melete, E. pyretorum, M. sexta, P. atrilineata, and O. lunifer) were designated as CGA [11, 12, 16, 19, 21, 23]. A recent study by analysis of the transcript information from the cDNA sequence of the mtDNA-encoded protein gene revealed that the translation initiation codon for the COI gene is TCG (Serine), rather than those atypical and longer codons in Diptera [41] Therefore, we tentatively designated the CGA as the COI start codon although no mRNA expression data for H. cunea are available until now.
Nine of 13 PCGs in H. cunea harbor the usual termination codon TAA, but the remaining four possess the incomplete termination codons T for COI, COII, ND4, and TA for ATP6 (Table 3). The COI, COII, and ND4 terminate with T exactly adjacent to tRNAs, and ATP6 terminate with TA immediately followed by the ATG translation initiation codon of COIII. These incomplete stop codons are commonly found in metazoan mitochondrial genes [33]. The common interpretation of this phenomenon is that TAA termini are created via posttranscriptional polyadenylation [42].
The Relative Synonymous Codon Usage (RSCU) in PCGs was investigated and the results are summarized in Table 5. In PCGs of the H. cunea mitogenome, the codons CCG, GCG, TGC, CGC, AGC, and AGG are not represented. The genome-wise A+T bias is also reflected in the codon usage of H. cunea mitogenome. The codons TTA (Leu), ATT (Ile), TTT (Phe), and ATA (Met) are the four most frequently used codons in the H. cunea mitogenome, accounting for 39.2%. These codons are all composed of A or T nuleotides, thus indicating the biased usage of A and T nucleotides in the H. cunea PCGs. These four codons were also most frequently used in the sequenced lepidopteran insects. Leucine (14.82%), isoleucine (11.99%), phenylalanine (9%), and serine (8.52%) are the most frequent amino acids in H. cunea mitochondrial proteins, accounting for 44.33%. These amino acids are also the most frequently represented in other insects, averaging 45.08% [43].
Fig 2

Alignment of initiation region for the cytochrome oxidase subunit I (COI) genes of lepidopteran insects. The Diptera insect Anopheles funestus was included due to fact that the translation initiation codon for the COI gene was determined by analysis of the transcript information analysis [41]. The first four or five codons and their amino acids are shown on the right-hand side of the figure. Boxed nucleotides are the presumed translation initiators, which have been postulated as the initiation codon for COI in each species. Underlined nucleotides indicate the adjacent partial sequence of tRNATyr. Arrows indicate the direction of transcription.
Table 5
Codon usage of the protein-coding genes in Hyphantria cunea mitogenome*
| Codon (aa) | n | % | RSCU | Codon (aa) | n | % | RSCU | Codon (aa) | n | % | RSCU |
|---|---|---|---|---|---|---|---|---|---|---|---|
| UUU(F) | 332 | 8.95 | 1.99 | UCU(S) | 113 | 3.05 | 2.86 | UAU(Y) | 180 | 4.85 | 1.82 |
| UUC(F) | 2 | 0.59 | 0.01 | UCC(S) | 10 | 0.27 | 0.25 | UAC(Y) | 18 | 0.49 | 0.18 |
| UUA(L) | 449 | 0.10 | 4.85 | UCA(S) | 81 | 2.18 | 2.05 | UAA(*) | - | - | - |
| UUG(L) | 35 | 0.94 | 0.38 | UCG(S) | 2 | 0.05 | 0.05 | UAG(*) | - | - | - |
| CUU(L) | 43 | 1.16 | 0.47 | AGU(S) | 28 | 0.75 | 0.71 | CAU(H) | 56 | 1.51 | 1.65 |
| CUC(L) | 1 | 0.03 | 0.01 | AGC(S) | 0 | 0 | 0 | CAC(H) | 12 | 0.32 | 0.35 |
| CUA(L) | 20 | 0.54 | 0.22 | AGA(S) | 82 | 2.21 | 2.08 | CAA(Q) | 60 | 1.62 | 1.97 |
| CUG(L) | 2 | 0.05 | 0.02 | AGG(S) | 0 | 0 | 0 | CAG(Q) | 1 | 0.03 | 0.03 |
| AUU(I) | 410 | 11.05 | 1.84 | ACU(U) | 73 | 1.97 | 1.93 | AAU(N) | 215 | 5.79 | 1.69 |
| AUC(I) | 35 | 0.94 | 0.16 | ACC(U) | 14 | 0.38 | 0.37 | AAC(N) | 39 | 1.05 | 0.31 |
| AUA(M) | 265 | 7.14 | 1.91 | ACA(U) | 62 | 1.68 | 1.64 | AAA(K) | 100 | 2.69 | 1.82 |
| AUG(M) | 12 | 0.32 | 0.09 | ACG(U) | 2 | 0.05 | 0.05 | AAG(K) | 10 | 0.27 | 0.18 |
| GUU(V) | 90 | 2.43 | 2.37 | GCU(A) | 84 | 2.26 | 2.63 | GGU(G) | 68 | 1.83 | 1.35 |
| GUC(V) | 5 | 0.13 | 0.13 | GCC(A) | 6 | 0.16 | 0.19 | GGC(G) | 3 | 0.08 | 0.06 |
| GUA(V) | 50 | 1.35 | 1.32 | GCA(A) | 38 | 1.02 | 1.19 | GGA(G) | 108 | 2.91 | 2.14 |
| GUG(V) | 7 | 0.19 | 0.18 | GCG(A) | 0 | 0 | 0 | GGG(G) | 23 | 0.62 | 0.26 |
| UGU(C) | 28 | 0.75 | 2.00 | CGU(R) | 14 | 0.38 | 1.08 | CCU(P) | 75 | 2.02 | 2.36 |
| UGC(C) | 0 | 0 | 0 | CGC(R) | 0 | 0 | 0 | CCC(P) | 11 | 0.30 | 0.35 |
| UGA(W) | 92 | 2.48 | 1.88 | CGA(R) | 36 | 0.97 | 2.77 | CCA(P) | 41 | 1.10 | 1.29 |
| UGG(W) | 6 | 0.16 | 0.12 | CGG(R) | 2 | 0.05 | 0.15 | CCG(P) | 0 | 0 | 0 |
| GAU(D) | 59 | 1.59 | 1.84 | GAA(E) | 66 | 1.78 | 1.74 | ||||
| GAC(D) | 5 | 0.13 | 0.16 | GAG(E) | 10 | 0.27 | 0.26 |
*A total of 3711 codons were analyzed excluding the initiation and termination codons. RSCU, relative sunonymous codon usage.
rRNA genes and tRNA genes
As in all other insect mitogenome sequences, two rRNA genes are present in H. cunea. They are located between tRNALeu(CUN) and tRNAVal, and between tRNAVal and the A+T-rich region, respectively. The length of the H. cunea lrRNA is 1 426 bp, which is the longest among the available completely sequenced lepidopteran insects, with the size range from1 412 bp in D. saccharalis to 1 319 bp in A. melete. The length of the H. cunea srRNA is 808 bp, which is well within the range observed in the available completely sequenced lepidopteran insects, with the size range from 806 bp in O. lunifer to 774 bp in C. boisduvalii.
The H. cunea mitogenome harbors 22 tRNA genes, which are scattered around the molecule (Table 2; Fig. 1). The predicted secondary structure of the H. cunea tRNAs are shown in Fig. 3. Twenty tRNA genes were identified by tRNAscan Search [29]. These 20 tRNA genes vary from 63 bp (tRNACys) to 71 bp (tRNALys) in size, and present a typical clover-leaf secondary structure of previously published mitochondrial tRNA genes. The H. cunea tRNASer(AGN) and tRNASer(UCN) genes not identified by tRNAscan-SE Search were determined to be 66 bp and 68 bp in size, respectively. Their sizes were determined by comparing the conserved relative genome position and sequence similarity with other lepidopteran mitogenome sequences. The tRNASer(UCN) also shows a typical clover-leaf secondary structure. However, the tRNASer(AGN) presents an unusual secondary structure lacking a stable stem-loop structure in the DHU arm, which has been observed in several other metazoan species including insects [4]. The anticodons are identical to those observed in other lepidopteran insects.
A total of 23 unmatched base pairs occurred in the H. cunea tRNA genes. Twelve of 22 tRNA genes, including tRNAGln, tRNATrp, tRNACys, tRNAPhe, tRNAGly, tRNAAla, tRNALeu(CUN), tRNALeu(UUR), tRNAHis, tRNAPro, tRNAThr, and tRNAVal, were found to have 18 G-U mismatches in their secondary structures, which forms a weak bond. Three U-U mismatches were found in the amino acid acceptor stem of tRNAAla, tRNALeu(CUN), and tRNALeu(UUR). The tRNAThr gene was proposed to contain an A-A mismatch in the TψC stem, and the tRNASer(AGN) gene contained an A-A mismatch in the anticodon stem. Moreover, the tRNAHis and tRNALys genes were found to contain an extra nucleotide A in the TψC stem, respectively. Mismatches observed in tRNAs can be corrected through RNA-editing mechanisms that are well known for arthropod mtDNA [44]. The number of mismatches in the H. cunea is similar to those observed in other available lepidopteran insects: 24 in A. pernyi [13], and 24 in E. pyretorum [16]; but lower than in O. lunifer where 35 mismatches were found [11]. No mechanism, however, has been deduced for such high numbers of mismatches in insect mitochondrial tRNAs.
Fig 3
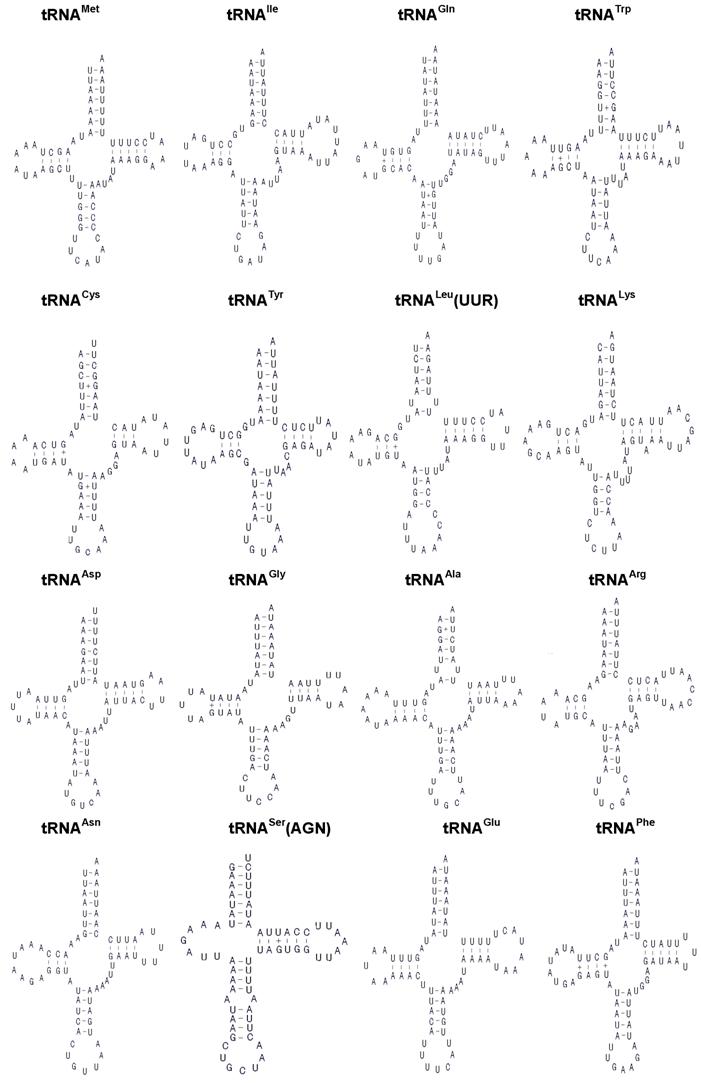
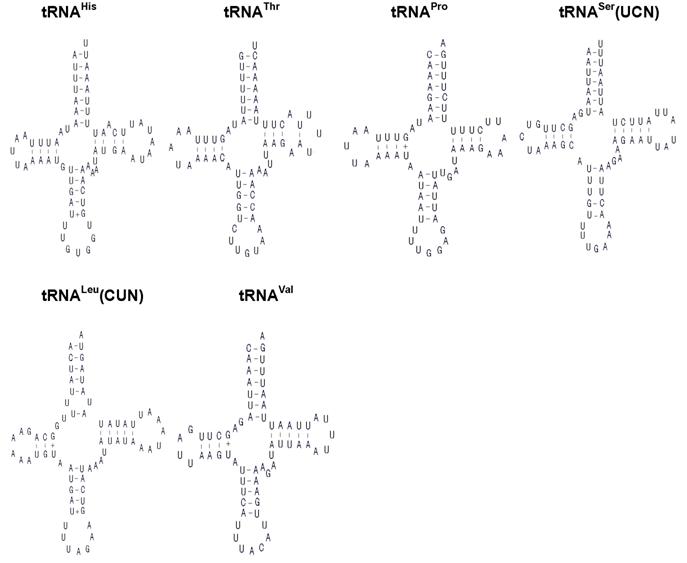
Inferred secondary structure of the 22 tRNAs of the Hyphantria cunea mitogenome. +, GT pairs; -, AT/GC pairs. The tRNAs are labeled with the abbreviations of their corresponding amino acids. Arms of tRNAs (clockwise from top) are the amino acid acceptor arm, the TψC arm, the anticodon arm, and the dihydrouridine arm.
Noncoding and overlapping regions
The H. cunea mitogenome harbors a total of 230 bp intergenic spacer sequences, which are spread over 18 regions ranging in size from 1 to 50 bp (Table 3). The largest intergenic spacer sequence is present between tRNAGln and ND2 gene, with an extreme richness in A and T nucleotides (92%). This spacer is not found in non-lepidopteran insect species [19], but is found to be a feature common to the 21 lepidopteran mitogenomes which have been sequenced to date. By alignment analysis, whilst invariant between each of the congeneric species-pairs which have been examined (O. furnicalis and O. nubilalis; B. mori and B. mandarina; A. pernyi and A. yamamai), this region showed limited sequence conservation between even closely related lepidopteran groups such as within Bombycoidea or between bombycoids and other macrolepidopterans [11, 19], indicating it would imply no functional significance or might not serve as another origin of replication [15].
Additionally, two other intergenic spacer sequences of more than 20 bp are present between tRNASer(AGN) and tRNAGlu (21 bp), and between tRNASer(AGN) and ND1 (34 bp), respectively. The spacer region of 34 bp also contains the ATACTAA motif [19], which is conserved across the Lepidoptera order (Fig. 4). This 7 bp motif is possibly fundamental to site recognition by the transcription termination peptide (mtTERM protein) [45]. This region is present in most insect mtDNAs even if the nucleotide sequence can be quite divergent [19].
Eighteen base pairs were identified as overlapping sequences varying from 1 to 8 bp in four regions (Table 3). The longest overlap is 8 bp between tRNATrp and tRNACys. Similarly sized overlaps are also observed in other sequenced lepidopteran species [14]. The 7 bp overlap with the reading frame involving the ATP8/ATP6 genes was found. This feature is common to other sequenced lepidopteran mitogenomes, and was found in many animal mitogenomes [5]. As for the two remaining overlaps, one is 1 bp between tRNALys and tRNAAsp, the other is 2 bp between tRNAGlu and tRNAPhe.
Fig 4

Alignment of the intergenic spacer region between tRNASer(UCN) and ND1 of lepidopteran insects. The shaded ATACTAA motif [19] was conserved across the Lepidoptera order.
A+T- rich region
The H. cunea A+T-rich region is located between the srRNA gene and tRNAMet (Table 3; Fig. 1), which includes the origin sites for transcription and replication [45]. This region was identified to be 357 bp in length, which is well within the range observed in the completely sequenced lepidopteran insects, with size ranging from 319 bp in O. lunifer to 747 bp in Japanese B. mandarina (Table 4). The A+T-rich region shows the highest A+T content (94.96%) of any region of the H. cunea mitogenome.
The presence of extra tRNA-like structures in the A+T-rich region has been reported in the lepidopteran insects. In the case of Chinese B. mandarina, one tRNASer(TGA)-like sequence is located within the A+T-rich region forming a structure with four stem-loops and one big loop [18]. In A. yamamai A+T-rich region, two tRNA-like structures are present: tRNASer(UCN)-like sequence and tRNAPhe-like sequence, which possess the proper anticodon and form a clover-leaf structure, indicating they may be functional although there are many mismatches in both aminoacyl and anticodon stem regions [14]. However, no tRNA-like structure was detected in the H. cunea A+T-rich region.
The presence of varying copy numbers of tandemly-repeated elements has been reported to be one of the characteristics of the insect A+T-rich region [7]. Some lepidopteran insects have been observed to possess the repeat element in the A+T-rich region. In the case of Antheraea, the A. pernyi A+T-rich region harbors a repeat element of 38 bp tandemly repeated six times [13], whereas the A. roylei A+T-rich region has five repeat elements [9, 13]. In Japanese B. mandarina, the A+T-rich region harbors a tandem triplication of a 126 bp repeat unit, whereas in B. mori and Chinese B. mandarina the A+T-rich region has only one repeat element [17, 18]. It has also been reported that Arethusana arethusa (Nymphalidae: Satyrinae), Leptidea sinapis (Pieridae), and Parnassius apollo (Papilionidae) have a longer A+T-rich region (~500-700 bp), due mainly to an increase in the size and copy number of repeat units [46]. However, the H. cunea A+T-rich region is comprised of non-repetitive sequences, but harbors several features (Fig. 5) common to the Lepidoptera A+T-rich region [11, 19]. In B. mori, the ON (origin of minority or light strand replication) is located 21 bp downstream from srRNA gene, which contains the motif ATAGA followed by an 18 bp poly-T stretch [47]. A very similar pattern occurs in H. cunea where the ATAGA motif is located 20 bp downstream from srRNA gene and is followed by an 18 bp poly-T stretch. A microsatellite-like (AT)8 element preceded by the ATTTA motif is present in the 3' end of the H. cunea A+T-rich region. The presence of a microsatellite preceded by the ATTTA motif is common in the control regions of insect mitogenomes, and has been found in all other lepidopteran species which have been sequenced [19]. Finally, an 11 bp poly-A is present immediately upstream tRNAMet. This poly-A element is still a common feature of the A+T-rich region in Lepidoptera [19, 46]. This sequence has been suggested to be involved in the control of transcription and/or replication initiation in other insects or have some other unknown functional role [7].
Phylogenetic relationships
To place the H. cunea mtDNA sequence in perspective relative to other lepidopteran insect mitogenomes and to probe into the phylogenetic relationships among the lepidopteran superfamilies, a data set containing the concatenated amino acid sequences of 13 PCGs was generated. The sequences of the 13 PCGs were concatenated, rather than analyzed separately, to reconstruct the phylogenetic relationships, which may result in a more complete analysis [48]. The final alignment resulted in 3838 amino acid sites for the 19 ingroup and two outgroup taxa, including gaps. Of these sites, 1606 were conserved, 2170 were variable, and 1555 were informative for parsimony. The MP and NJ analyses generate overall similar topology except for the branching superfamily among A. honmai, and Papillonoidea species (Fig. 6). The phylogenetic analyses support a close relationship between H. cunea and L. dispar with 100% bootstrapping value, which is consistent with the morphological classification. The phylogenetic analyses also support a sister relationship between Noctuoidea (H. cunea, L. dispar, and O. lunifer) and Geometroidea (P. atrilineata).
The M. sexta is sometimes placed in their own superfamily Sphingidea. However, phylogenetic analyses based on the complete mitogenome in this study strongly support the placement within the superfamily Bombycoidea, which is consistent with the previous findings by morphological analysis [49] and by molecular analysis based on some nuclear genes [50].
These 19 sequences represent six superfamilies within the lepidopteran suborder: Bombycoidea, Geometroidea, Noctuoidea, Papillonoidea, Pyraloidea, and Tortricidea. Based on morphological analysis, Bombycoidea, Noctuoidea, Papillonoidea and Geometroidea are designated as the Macrolepidoptera; Pyraloidea together with Macrolepidoptera are designated as Obtectornera; Tortricoidea is the sister to the remaining lepidopteran superfamilies covered in the present study (Fig. 6A) [49]. In the phylogenetic trees constructed (Fig. 6B and C), the butterflies of Papillonoidea (A. melete, A. issoria, and C. raphaelis) are sisters to the remaining lepidopteran superfamilies, also showing a basal position within the monophyly of Lepidoptera [16]. This result is different to the traditional morphological analyses (Fig. 6A). Also, recent phylogenetic analyses of 123 species representing 27 superfamilies of Ditrysia based on five protein-coding nuclear genes (6.7 kb total) provide sufficient information to conclusively demonstrate that several prominent features of the current morphology-based hypothesis, including the position of the butterflies, need revision [51]. These results present in this study suggest that the complete insect mitogenome sequence has a power to resolve the majority of family relationships within superfamilies, however, the deeper nodes among superfamilies need more efforts.
Fig 5

The features present in the A+T-rich region of Hyphantria cunea. The sequence is shown in the N strand.
Fig 6
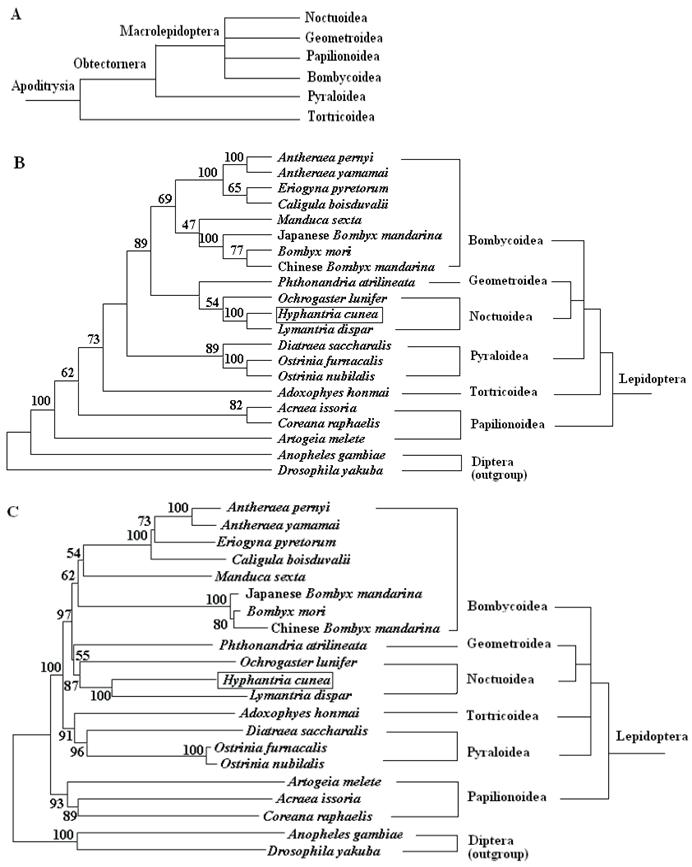
Phylogeny of lepidopteran insects. (A) Current hypothesis of lepidopteran superfamily relationships after Kristensen and Skalski (1999) [50]. Phylogenetic trees inferred from amino acid sequences of 13 PCGs of the mitogenome by using MP analysis (B) and NJ analysis (C). Drosophila yakuba [33] and Anopheles gambiae [34] were used as outgroups. The numbers above branches specify bootstrap percentages (1000 replicates).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant 30800803), the National 863 program of China (grant 2007AA06Z323), the National Key Technology R&D Program in the 11th Five year Plan of china (grant 2006BAK10B06), and the Key Technology R&D Program of Tianjin City (grant 06YFGZNC00700).
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
1. Warren LO, Tadic M. The fall webworm, Hyphantria cunea (Drury). Ark Agric Exp Stn Bull. 1970;759:1-106
2. Umeya K, Itô Y. Invasion and establishment of a new insect pest in Japan. Hidaka T (ed) Adaptation and speciation in the fall webworm. Kodansha, Tokyo. 1977
3. Ji R, Xie BY, Li XH. et al. Research progress on the invasive species, Hyphantria cunea. Entomol Knowledge (China). 2003;40:13-18
4. Wolstenholme DR. Animal mitochondrial DNA: Structure and evolution. Int Rev Cytol. 1992;141:173-216
5. Boore JL. Animal mitochondrial genomes. Nucleic Acids Res. 1999;27:1767-1780
6. Shadel GS, Clayton DA. Mitochondrial transcription initiation: Variation and conservation. J Biol Chem. 1993;268:16083-16086
7. Zhang DX, Szymura JM, Hewitt GM. Evolution and structural conservation of the control region of insect mitochondrial DNA. J Mol Evol. 1995;40:382-391
8. Nardi F, Spinsanti G, Boore JL. et al. Hexapod origins: Monophyletic or paraphyletic? Science. 2003;299:1887-1889
9. Arunkumar KP, Metta M, Nagaraju J. Molecular phylogeny of silkmoths reveals the origin of domesticated silkmoth, Bombyx mori from Chinese Bombyx mandarina and paternal inheritance of Antheraea proylei mitochondrial DNA. Mol Phylogenet Evol. 2006;40:419-427
10. Hwang UW, Park CJ, Yong TS. et al. One-Step PCR Amplification of Complete Arthropod Mitochondrial Genomes. Mol Phylogenet Evol. 2001;19:345-352
11. Salvato P, Simonato M, Battisti A. et al. The complete mitochondrial genome of the bag-shelter moth Ochrogaster lunifer (Lepidoptera, Notodontidae). BMC Genomics. 2008;9:331
12. Yang L, Wei ZJ, Hong GY. et al. The complete nucleotide sequence of the mitochondrial genome of Phthonandria atrilineata (Lepidoptera: Geometridae). Mol Biol Rep. 2009;36:1441-1449
13. Liu Y, Li Y, Pan M. et al. The complete mitochondrial genome of the Chinese oak silkmoth, Antheraea pernyi (Lepidoptera: Saturniidae). Acta Biochim Biophys Sin (Shanghai). 2008;40:693-703
14. Kim SR, Kim M, Hong MY. et al. The complete mitogenome sequence of the Japanese oak silkmoth, Antheraea yamamai (Lepidoptera: Saturniidae). Mol Biol Rep. 2009;36:1871-1880
15. Hong MY, Lee EM, Jo YH. et al. Complete nucleotide sequence and organization of the mitogenome of the silk moth Caligula boisduvalii (Lepidoptera: Saturniidae) and comparison with other lepidopteran insects. Gene. 2008;413:49-57
16. Jiang ST, Hong GY, Yu M. et al. Characterization of the complete mitochondrial genome of the giant silkworm moth, Eriogyna pyretorum (Lepidoptera: Saturniidae). Int J Biol Sci. 2009;5:351-365
17. Yukuhiro K, Sezutsu H, Itoh M. et al. Significant levels of sequence divergence and gene rearrangements have occurred between the mitochondrial genomes of the wild mulberry silkmoth, Bombyx mandarina, and its close relative, the domesticated silkmoth, Bombyx mori. Mol Biol Evol. 2002;19:1385-1389
18. Pan MH, Yu QY, Xia YL. et al. Characterization of mitochondrial genome of Chinese wild mulberry silkworm Bomyx mandarina (Lepidoptera Bombycidae). Sci China Ser C-Life Sci. 2008;51:693-701
19. Cameron SL, Whiting MF. The complete mitochondrial genome of the tobacco hornworm, Manduca sexta (Insecta: Lepidoptera: Sphingidae), and an examination of mitochondrial gene variability within butterflies and moths. Gene. 2008;408:112-113
20. Coates BS, Sumerford DV, Hellmich RL. et al. Partial mitochondrial genome sequences of Ostrinia nubilalis and Ostrinia furnicalis. Int J Biol Sci. 2005;1:13-18
21. Lee ES, Shin KS, Kim MS. et al. The mitochondrial genome of the smaller tea tortix Adoxophyes honmai (Lepidoptera: Tortricidae). Gene. 2006;373:52-57
22. Hu J, Zhang DX, Hao JS. et al. The complete mitochondrial genome of the yellow coaster, Acraea issoria (Lepidoptera: Nymphalidae: Heliconiinae: Acraeini): sequence, gene organization and a unique tRNA translocation event. Mol Biol Rep. 2010 In press
23. Hong G, Jiang S, Yu M. et al. The complete nucleotide sequence of the mitochondrial genome of the cabbage butterfly, Artogeia melete (Lepidoptera: Pieridae). Acta Biochim Biophys Sin (Shanghai). 2009;41:446-455
24. Kim I, Lee EM, Seol KY. et al. The mitochondrial genome of the Korean hairstreak, Coreana raphaelis (Lepidoptera: Lycaenidae). Insect Mol Biol. 2006;15:217-225
25. Hebert PD, Cywinska A, Ball SL. et al. Biological identifications through DNA barcodes. Proc R Soc Lond B Biol Sci. 2003;270:313-321
26. Gomi T, Muraji M, Takeda M. Mitochondrial DNA analysis of the introduced fall webworm, showing its shift in life cycle in Japan. Entomol Sci. 2004;7:183-188
27. Simon C, Frait F, Bechenback A. et al. Evolution, weighting, and phylogenetic utility of mitochondrial gene sequence and a compilation of conserved polymerase chain reaction primers. Ann Entomol Soc Am. 1994;87:651-701
28. Thompson JD, Gibson TJ, Plewniak F. et al. The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876-4882
29. Lowe TM, Eddy SR. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955-964
30. Steinberg S, Cedergren R. Structural compensation in atypical mitochondrial tRNAs. Struct Biol. 1994;1:507-510
31. Perna NT, Kocher TD. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J Mol Evol. 1995;41:353-358
32. Benson G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573-580
33. Clary DO, Wolstenholme DR. The mitochondrial DNA molecular of Drosophila yakuba: Nucleotide sequence, gene organization, and genetic code. J Mol Evol. 1985;22:252-271
34. Beard CB, Mills D, Collins FH. The mitochondrial genome of the mosquito Anopheles gambiae: DNA sequence, genome organization, and comparisons with mitochondrial sequences of other insects. Insect Mol Biol. 1993;2:103-124
35. Tamura K, Dudley J, Nei M. et al. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596-1599
36. Boore JL, Lavrov D, Brown WM. Gene translocation links insects and crustaceans. Nature. 1998;393:667-668
37. Clary DO, Wolstenholme DR. Genes for cytochrome c oxidase subunit I, URF2, and three tRNAs in Drosophila mitochondrial DNA. Nucleic Acids Res. 1983;11:6859-6872
38. de Bruijn MHL. Drosophila melanogaster mitochondrial DNA: a novel organisation and genetic code. Nature. 1983;304:234-241
39. Ballard JWO. Comparative genomics of mitochondrial DNA in members of the Drosophila melanogaster subgroup. J Mol Evol. 2000;51:48-63
40. Nardi F, Carapelli A, Fanciulli PP. et al. The complete mitochondrial DNA sequence of the basal hexapod Tetrodontophora bielanensis: Evidence for heteroplasmy and tRNA translocations. Mol Biol Evol. 2001;18:1293-1304
41. Krzywinski J, Grushko OG, Besansky NJ. Analysis of the complete mitochondrial DNA from Anopheles funestus: An improved dipteran mitochondrial genome annotation and a temporal dimension of mosquito evolution. Mol Phylogenet Evol. 2006;39:417-423
42. Ojala D, Montoya J, Attardi G. tRNA punctuation model of RNA processing in human mitochodria. Natute. 1981;290:470-474
43. Lessinger AC, Junqueira AC, Lemos TA. et al. The mitochondrial genome of the primary screwworm fly Cochliomyia hominivorax (Diptera: Calliphoridae). Insect Mol Biol. 2000;9:521-529
44. Lavrov DV, Brown WM, Boore JL. A novel type of RNA editing occurs in the mitochondrial tRNAs of the centipede Lithobius forficatus. Proc Natl Acad Sci USA. 2000;97:13738-13742
45. Taanman JW. The mitochondrial genome: structure, transcription, translation and replication. Biochim Biophys Acta. 1999;1410:103-123
46. Vila M, Björklund M. The utility of the neglected mitochondrial control region for evolutionary studies in Lepidoptera (Insecta). J Mol Evol. 2004;58:280-290
47. Saito S, Tamura K, Aotsuka T. Replication origin of mitochondrial DNA in insects. Genetics. 2005;171:1695-1705
48. Hassanin A. Phylogeny of Arthropoda inferred from mitochondrial sequences: strategies for limiting the misleading effects of multiple changes in pattern and rates of substitution. Mol Phylogenet Evol. 2006;38:100-116
49. Kristensen NP, Skalski AW. Phylogeny and paleontology. In: (ed.) Kristensen NP. Lepidoptera: Moths and Butterflies, 1 Evolution, Systematics, and Biogeography, Handbook of Zoology Vol IV, Part 35. Berlin and New York: De Gruyter. 1999:7-25
50. Kawahara AY, Mignault AA, Regier JC. et al. Phylogeny and Biogeography of Hawkmoths (Lepidoptera: Sphingidae): Evidence from Five Nuclear Genes. PLoS ONE. 2009;4(5):e5719
51. Regier JC, Zwick A, Cummings MP. et al. Toward reconstructing the evolution of advanced moths and butterflies (Lepidoptera: Ditrysia): an initial molecular study. BMC Evol Biol. 2009;9:280
Author contact
![]() Corresponding author: Y. Q. Liu, College of Bioscience and Biotechnology, Shenyang Agricultural University, Liaoning, Shenyang 110866, China; Tel: 86-24-88487163; E-mail: liuyanqunedu.cn. Or to: M. G. Li, College of Life Sciences, Nankai University, Tianjin 300071, China; Tel: 86-22-23508237; E-mail: mgledu.cn.
Corresponding author: Y. Q. Liu, College of Bioscience and Biotechnology, Shenyang Agricultural University, Liaoning, Shenyang 110866, China; Tel: 86-24-88487163; E-mail: liuyanqunedu.cn. Or to: M. G. Li, College of Life Sciences, Nankai University, Tianjin 300071, China; Tel: 86-22-23508237; E-mail: mgledu.cn.
Citation styles
APA
Liao, F., Wang, L., Wu, S., Li, Y.P., Zhao, L., Huang, G.M., Niu, C.J., Liu, Y.Q., Li, M.G. (2010). The complete mitochondrial genome of the fall webworm, Hyphantria cunea (Lepidoptera: Arctiidae). International Journal of Biological Sciences, 6(2), 172-186. https://doi.org/10.7150/ijbs.6.172.
ACS
Liao, F.; Wang, L.; Wu, S.; Li, Y.P.; Zhao, L.; Huang, G.M.; Niu, C.J.; Liu, Y.Q.; Li, M.G. The complete mitochondrial genome of the fall webworm, Hyphantria cunea (Lepidoptera: Arctiidae). Int. J. Biol. Sci. 2010, 6 (2), 172-186. DOI: 10.7150/ijbs.6.172.
NLM
Liao F, Wang L, Wu S, Li YP, Zhao L, Huang GM, Niu CJ, Liu YQ, Li MG. The complete mitochondrial genome of the fall webworm, Hyphantria cunea (Lepidoptera: Arctiidae). Int J Biol Sci 2010; 6(2):172-186. doi:10.7150/ijbs.6.172. https://www.ijbs.com/v06p0172.htm
CSE
Liao F, Wang L, Wu S, Li YP, Zhao L, Huang GM, Niu CJ, Liu YQ, Li MG. 2010. The complete mitochondrial genome of the fall webworm, Hyphantria cunea (Lepidoptera: Arctiidae). Int J Biol Sci. 6(2):172-186.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.