Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Liver stem cells in human liver...
Human Liver Progenitor Cells and...
Liver cancer stem cell and...
Molecular signaling of Liver...
Therapeutic target of Molecular...
Future Directions
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2011; 7(5):517-535. doi:10.7150/ijbs.7.517 This issue Cite
Review
Novel therapeutic Strategies for Targeting Liver Cancer Stem Cells
Naoki Oishi, Xin Wei Wang ![]()
Liver Carcinogenesis Section, Laboratory of Human Carcinogenesis, Center for Cancer Research, National Cancer Institute, Bethesda, MD, USA
Received 2011-3-28; Accepted 2011-4-14; Published 2011-4-26
Abstract
The cancer stem cell (CSC) hypothesis was first proposed over 40 years ago. Advances in CSC isolation were first achieved in hematological malignancies, with the first CSC demonstrated in acute myeloid leukemia. However, using similar strategies and technologies, and taking advantage of available surface markers, CSCs have been more recently demonstrated in a growing range of epithelial and other solid organ malignancies, suggesting that the majority of malignancies are dependent on such a compartment.
Primary liver cancer consists predominantly of hepatocellular carcinoma (HCC) and intrahepatic cholangiocarcinoma (ICC). It is believed that hepatic progenitor cells (HPCs) could be the origin of some HCCs and ICCs. Furthermore, stem cell activators such as Wnt/β-catenin, TGF-β, Notch and Hedgehog signaling pathways also expedite tumorigenesis, and these pathways could serve as molecular targets to assist in designing cancer prevention strategies. Recent studies indicate that additional factors such as EpCAM, Lin28 or miR-181 may also contribute to HCC progression by targeting HCC CSCs. Various therapeutic drugs that directly modulate CSCs have been examined in vivo and in vitro. However, CSCs clearly have a complex pathogenesis, with a considerable crosstalk and redundancy in signaling pathways, and hence targeting single molecules or pathways may have a limited benefit for treatment. Many of the key signaling molecules are shared by both CSCs and normal stem cells, which add further challenges for designing molecularly targeted strategies specific to CSCs but sparing normal stem cells to avoid side effects. In addition to the direct control of CSCs, many other factors that are needed for the maintenance of CSCs, such as angiogenesis, vasculogenesis, invasion and migration, hypoxia, immune evasion, multiple drug resistance, and radioresistance, should be taken into consideration when designing therapeutic strategies for HCC.
Here we provide a brief review of molecular signaling in liver CSCs and present insights into new therapeutic strategies for targeting liver CSCs.
Keywords: Liver cancer stem cell, Hepatocellular carcinoma, Cholangiocellular carcinoma, Wnt/β-catenin signaling, TGF-β signaling, Notch signaling, Hedgehog signaling, BMI-1 signaling, EpCAM, Lin-28, miR-181, Self-renewal, Self-protection
Introduction
Hepatocellular carcinoma (HCC) is the fifth most common cancer worldwide and the third leading cause of cancer death [1]. Despite some progress in the treatment of cancers, existing therapies are limited in their ability to cure malignancies and to prevent metastases and relapses. Surgery, radiofrequency ablation therapy and chemotherapy are all directed at reducing the bulk of the tumor mass. However, on completion of therapy there is ultimately regrowth of tumor and relapse of diseases in the majority of cases [2-5]. Although the idea of tumor stem cells has been proposed for a number of decades, demonstration of their existence has only occurred within the last ten years. Recently, HCC progression has been thought to be driven by cancer stem cells (CSC) through their capacity for self-renewal, production of heterogeneous progeny, resistance to chemotherapy and to limitlessly divide. Advances were first achieved in hematological malignancies, with the first CSC demonstrated in acute myeloid leukemia. However, using similar strategies and technologies, and taking advantage of available surface markers, CSCs have been more recently demonstrated in a growing range of epithelial and other solid organ malignancies, suggesting that the majority of malignancies are dependent on such a compartment. Furthermore, many potentially and biologically significant surface markers and pathways that can modulate these stem/progenitor cells in cancer tissue have been successfully identified based on their dual role both in embryogenic stem cell development and tumor activation or suppression. In this review, we demonstrate a brief and up-to date review of molecular signaling in liver CSC and present insights into new therapeutic strategies for liver CSCs.
Liver stem cells in human liver regeneration
The liver is both an exocrine and an endocrine gland, which performs complex functions and has the phenomenal ability to regenerate. This process enables the recovery of the lost mass without endangering the viability of the entire organism [6, 7]. Many studies suggest that the existence of two basic types of liver regeneration [8, 9]. After acute liver injury, hepatic stem cells take part in normal tissue repair and homeostasis quickly [10]. In contrast, liver regeneration after loss of hepatic tissue does not depend on these kinds of cells, but on the proliferation of the existing mature hepatocytes, the parenchymal cells of the organ. In addition, other cells such as endothelial cells, Kuppfer cells, and Ito cells may also contribute to regeneration of the lost hepatic tissue [6].
The normal liver has been estimated to be replaced by normal tissue approximately once a year or more [11]. Therefore replacement rate of the normal adult liver was calculated to be 0.005-0.0025% at any time [12]. However, this slow normal renewal rate differs from the rapid proliferate response to loss of hepatic mass. In rodents, when two-thirds of the liver is resected (partial hepatectomy) the remaining remnant can regrow to the original liver size in approximately 10 days [7, 13]. In response to this stimulus, the normally quiescent hepatocytes leave G0 to enter the cell cycle under the influence of many growth factors. Hepatocyte proliferation begins in the periportal region of the liver and spreads to the centrilobular region. This regenerative response requires each hepatocyte to undergo only 2 rounds of replication to restore normal liver size. Hepatocytes are capable of large-scale clonal expansion within a diseased liver. Following very extensive liver damage or in situations in which hepatocyte regeneration after damage is compromised, a potential stem cell component located within the smallest branches of the intrahepatic biliary tree is activated. Hepatic progenitor cells (HPCs) amplify a biliary population of transit amplifying cells that are bipotential, capable of differentiating into either hepatocytes or cholangiocytes. These cells have been observed after severe hepatocellular necrosis, chronic viral hepatitis, alcoholic liver disease, and nonalcoholic fatty liver disease. It is thought that the activation of a potential stem cell compartment leads to the formation of reactive ductules, anastomosing cords of immature biliary cells with an oval nucleus and small rim of cytoplasm. Differentiation toward the hepatocyte lineage occurs via intermediate hepatocytes, polygonal cells with a size and phenotype intermediate between progenitor cells and hepatocytes. Intermediate hepatocytes become more numerous with time and extend further into the liver lobules. This sequence of changes suggests gradual differentiation of human progenitor cells into intermediate hepatocytes.
The Hepatocyte proliferation rate increases in chronic hepatitis with increased histological appearance of cellular damages until cirrhosis is reached, at which point the proliferation rate falls [14]. This fall probably reflects replicative senescence, although the diversion of blood flow through the liver probably plays a part [15]. The reduction in hepatocyte proliferation indices in chronic hepatitis occurs concurrently with the activation of HPCs [16, 17]. The development of an oval cell reaction in response to hepatocyte replicative senescence has been demonstrated in a transgenic mouse model of fatty liver and DNA damage [18]. In this model, mice developed fatty livers and large number of senescent hepatocytes. A striking oval cell response related to the number of senescent mature hepatocytes. The hepatocytes generated from oval cells in severely-damaged cirrhotic livers may have a high risk for neoplastic transformation.
Stem cells in the liver are proposed to be from two origins: endogenous or intrahepatic and exogenous or extrahepatic. Included in the intrahepatic stem cell compartment are the HPCs which are greater in number but with short-term proliferative capacity. HPCs are thought to be localized within the canals of Hering, interlobular bile ducts [19, 20]. Included in the extrahepatic stem cell compartment are cells derived from bone marrow and peripheral blood cells; these cells are usually few but with long-term proliferation capacity [21-23].
Human Liver Progenitor Cells and Cancer Stem Cells
Human hepatic stem cells most likely give rise to HCC as well as ICC [7, 24-26]. The hypothesis that HCC arises from HPCs is supported by the finding that many tumors contain an admixture of mature cells and cells phenotypically similar to HPCs. Detailed immunophenotyping of HCCs revealed that 28-50% of HCCs express markers of progenitor cells such as CK7 and CK19 [27]. These tumors also consist of cells that have an intermediate phenotype between progenitors and mature hepatocytes. In fact, patients who have HCCs that express hepatocyte and biliary cell markers such as albumin, CK7 and CK10 carry a significantly poorer prognosis and have a higher recurrence rate after surgical resection and liver transplantation [28]. Cells resembling HPCs have also been noted in hepatoblastoma; the most common liver tumors in children which are widely believed to be stem cell derived given there can be both epithelial and mesenchymal tissue components. These tumors can even have structures mimicking intrahepatic bile ducts and form ductal plate-like structures [29] (Fig. 1).
Implication of Stem Cells in Liver Development and Hepatic Tumorigenesis. Normal hepatic stem cells are characterized by their ability to self-renew and differentiate, which leads to formation of a normal liver tissues. Oncogenic mutations in normal stem/progenitor cells or even in differentiated cells enhance or endow the cells with self-renewal capability. Consequently, these cells function as cancer stem cells and contribute to the formation of bulk tumors.
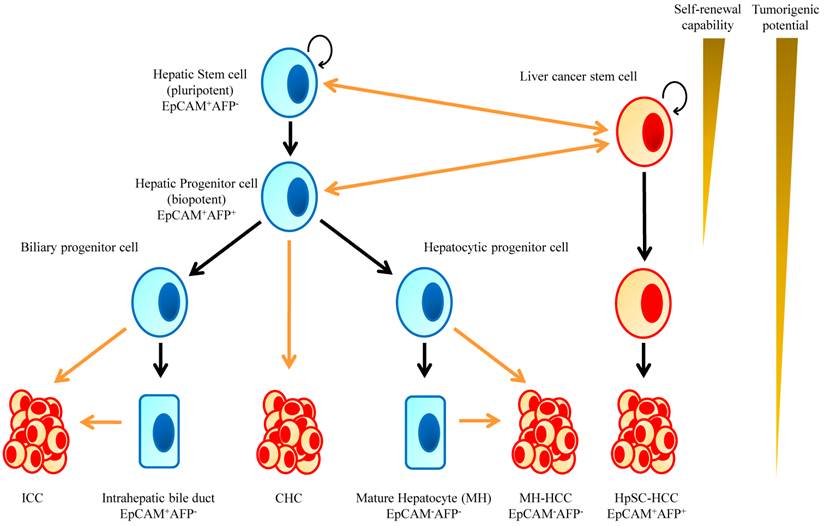
Liver cancer stem cell and primary liver cancer
Hepatocellular carcinoma
The CSC hypothesis is based on the idea that stem cells are present also in cancer tissue and a hierarchy of cells is formed, as is the case with normal tissue. Tumor formation, growth, and propagation are maintained by a minute proportion of cells with stem cell-like properties. Now, CSCs in HCC can be identified by several cell surface antigens including c-kit, CD133, CD90, CD44, OV6, and CD326 (EpCAM), or by selecting the side population (SP) cells by Hoechest dye-staining.
SP cells in HCC cells possess high proliferation potential, tumorigenicity, and anti-apoptotic properties compared with those of non-SP cells [30, 31]. Furthermore, CD133+ cells isolated from HCC cells possess a greater ability to form tumors in vivo and have characteristics similar to those of progenitor cells including the expression of “stemness” genes, the ability to self-renew, and the ability to differentiate into nonhepatocyte-like lineages [32]. In addition, CD133+ HCC cells represented only a minority of the tumor cell population in human HCC specimens, and increased CD133 expression levels were correlated with increased tumor grade, advanced disease stage, shorter overall survival, and higher recurrence rates compared with patients with low CD133 expression [33] . Tumor associated calcium signal transducer 1 (TACSTD1), which encodes a pancarcinoma antigen epithelial cell adhesion molecule (EpCAM), was identified to be an early biomarker of HCC [34, 35]. EpCAM is a direct transcriptional target of the Wnt-β-catenin canonical signaling pathway. EpCAM+alpha-fetoprotein (AFP)+ HCC subtype had features of hepatic stem/progenitor cells, and EpCAM+ HCC cells were correlated with tumor progression and invasiveness. Additionally, this surface molecule is also highly expressed in premalignant hepatic tissues, HPCs and bile duct epithelium, but not in most adult hepatocytes [35-37].
Intrahepatic Cholangiocarcinoma
The origin of intrahepatic cholangiocarcinoma (ICC) is much less defined when compared with HCC. However, since that incidence and mortality of ICCs clarifying the origin of these tumors is important. Recent studies suggest that some ICCs could arise from liver stem cells rather than from mature cholangiocytes [38]. This concept is supported by the identification of a combined hepatocellular cholangiocarcinoma (CHC), which have morphological and phenotypical intermediate features between HCC and ICC [39]. The ability of HPCs to differentiate towards the biliary and the hepatocytic lineages gave rise to the hypothesis that transformed HPCs are the source of origin of intermediate primary liver carcinomas. Some animal models reveal that ICC can originate from HPCs [40, 41]. Furthermore, in a few cases of human ICCs, it has been reported that some tumor cells express specific markers of liver stem cells, indicating a possible stem cell origin [42, 43]. However, there is currently not enough data to make a statement regarding a stem cell origin of ICC and further immunohistochemical characteristics related to the expression of hepatic stem cell markers in ICCs should be elucidated.
Molecular signaling of Liver Cancer Stem Cells
Wnt/β-catenin signaling pathway
The Wnt/β-catenin pathway is an evolutionarily well-conserved pathway to be essential to normal cellular processes such as development, growth, survival, regeneration, and self-renewal [44]. Disruption of Wnt/β-catenin signaling results from both genetic and epigenetic changes is associated with a range of diseases and is frequently found in many cancers, especially colon cancer and HCC. Disrupted Wnt/β-catenin signaling by mutational and non-mutational events is observed in about one third of all HCCs which emphasizes the importance of this pathway in hepatocarcinogenesis [45, 46]. The Wnt pathway diversifies into two main branches, i.e., canonical (β-catenin-dependent) and non-canonical (β-catenin-independent), which play critical roles in specifying cellular fates and movements, respectively, during both embriogenic development and adult tissue regeneration [47-49].
Wnt ligands signal through binding to seven transmembrane Frizzled (Fzd) receptors and single transmembrane lipoprotein receptor-related protein (LRP) 5 or 6 co-receptors [50]. Canonical signaling mediated by ligands such as Wnt3a inhibits a multiprotein degradation complex consisting minimally of axin, adenomatous polyposis coli (APC) and glycogen synthase kinase 3β (GSK3β) [51-54]. This inhibition culminates in nuclear translocation of β-catenin, enabling it to interact with T-cell factor (TCF)/lymphoid enhancer factor (LEF) transcription factors to regulate gene expression [55].
Non-canonical signaling, which is much less defined, is mediated by ligands such as Wnt11 that uses the same Fzd receptors [56]. The Wnt-Fzd complex interacts with heterotrimeric G and Dv1 proteins to activate phospholipase C, which then generates diacylglycerol and inositol-phosphatase from phosphatidyl inositol 4, 5-biphosphate and increase intracellular calcium concentration. The Wnt-Fzd-G protein complex can also stimulate p38 kinase and activate phosphodiesterase 6, which hydrolyzes cyclic GMP and results in the inactivation of protein kinase G and an increase in intracellular calcium. Wnt-mediated increase in intracellular Ca2+ activates protein kinase C and calmodulin-dependent kinase 2 (CamKⅡ). CamKⅡ can activate calcineurin (CAN), and subsequently the NF-AT family of calcineurin-dependent transcription factors, as well as TAK1-NLK kinases. Signaling through the TAK-NLK kinases are proposed to inhibit canonical Wnt signaling. This pathway stimulates the Jun NH2-terminal kinase (JNK), Ca2+/CaMKⅡ and PKC pathways. Both pathways interact with each other, and in some cases, non-canonical signaling antagonizes the canonical pathway [57].
The Wnt receptor, FZD-7 is found to be overexpressed in up to 90% of HCCs [45, 58]. Twenty to 40% of HCCs bear abnormal cytoplasmic and nuclear accumulation of β-catenin [59, 60]. However, not all studies show a correlation between elevated nuclear β-catenin and expression of its transcriptional targets implying that the expression of these target genes is likely to be regulated by alternative signaling mechanisms [59, 60]. While most of the proceeding mutations have not been detected in allelotype analysis, it is salient to note that deletions in the AXIN1 locus (16p) have been described in HCC. Axin1 and β-catenin mutations have also been identified in approximately 25% of HCCs [45, 46, 61-64] , while overexpression of the FZD-7 receptor and glycogen synthase kinase-3 (GSK-3) inactivation can also lead to aberrant β-catenin pathway activation. Elevated expression of Wnt and its downstream mediators was also reported in EpCAM+ liver CSCs [35]. It has been demonstrated that murine hepatic stem/progenitor cells transduced with mutant β-catenin acquired excessive self-renewal capability and tumorigenicity in a similar fashion to BMI1. In addition, the Wnt/β-catenin pathway is activated in both rodent oval cells and OV6-positive tumor cells, and it leads to HCC chemoresistance [65]. These findings indicate that Wnt/β-catenin signaling plays an important role in the maintainance of CSCs.
Recently, the mechanisms leading to malignant transformation of stem/progenitor cells were effectively addressed in pediatric tumors [66]. Hepatoblastoma is a malignant embryonal tumor of the liver, which differs from HCC by distinct morphological patterns reminiscent of hepatoblasts, the bipotent precursors of hepatocytes and cholangiocytes, and of their arrangement in the developing liver. Integrated molecular and genetic studies of hepatoblastoma disclosed two major molecular subclasses of tumors that relate early and late phases of prenatal liver development. It has been suggested that hepatoblastoma might arise from impairment of the normal liver differentiation program associated with excessive Wnt/β-catenin signaling [62]. In addition, the interplay of Wnt/β-catenin and Myc signaling in immature tumors activates a distinct transcriptional program that correlates with tumor aggressiveness. Correlation between stage of hepatic differentiation and clinical manifestation, notably vascular invasion, metastatic spread, and patient survival, was also established [66]. These finding highlight the important role of dysregulated Wnt/β-catenin signaling in the transformation of stem/progenitor cells.
Recently, EpCAM was identified as a direct transcriptional target of Wnt/β-catenin signaling in HCC [35]. Adult hepatocytes are EpCAM-, while the bile duct epithelium is EpCAM+. In addition, expression of EpCAM was observed during fetal liver development, liver regeneration, and liver repair associated with cirrhosis. Moreover, EpCAM is a marker of hepatic progenitors, suggesting that EpCAM+ HCCs are of hepatic progenitor cell origin [67].
The EpCAM signaling can be activated by regulated intramembrane proteolysis (RIP) and shedding of extracellular domain of EpCAM (EpEX) [68]. Sequencial cleavage of EpCAM by tumor-necrosis-factor alpha converting enzyme (TACE/ADAM17) and a gamma-secretase complex containing presenilin 2 (PS-2) result in release of EpEX into the culture medium, and release of an intracellular domain of EpCAM (EpICD) into the cytoplasm. EpICD becomes a part of a large nuclear complex containing transcriptional regulators β-catenin and Lef, both of which are components of Wnt/β-catenin signaling. Four and one-half LIM domain protein 2 (FHL2) is essential for signal transduction by EpCAM. FHL2 further regulates localization and activity of TACE and PS-2. Through its function as a co-activator of β-catenin, FHL2 links EpICD with specific DNA sequences and gene regulation. FHL2 has the potential to serve as a scaffolding protein for various signaling proteins used by EpCAM [69]. A number of EpCAM-regulated target genes have been identified including c-myc and cyclins, and additional genes involved in cell growth and proliferation, cell cycle, and cell death. Upon interference with E-cadherin, EpCAM may increase the availability of its interaction partner β-catenin in the soluble fraction. Cross-talk with the Wnt pathway is possible at the level of β-catenin and Lef-1 interactions with EpICD, and known for induction of the EPCAM promoter by Tcf4. These findings indicated that expression of EpCAM strongly linked with proliferation of stem cells and cancer development by cancer initiating cells after aberrant EpCAM re-expression.
TGF-β family
The TGF-β signaling pathway appears to be most prominent at the interface between development and cancer in liver and gut epithelial cells [70]. Smad signaling has been shown to be pivotal for embryogenic hepatocyte proliferation, as well as in the formation of gastrointestinal cancers [71, 72]. Smad activation is modulated by various receptor- or Smad-interacting proteins that include ubiquitin and small ubiquitin-related modifier (SUMO) ligases, as well as multiple adaptor proteins that include Smad anchor for receptor activation (SARA), Filamin and β2-SPECTRIN. β2-SPECTRIN is crucial for the propagation of TGF-β signaling. Specifically, β2-SPECTRIN associates with Smad3, presenting it to the cytoplasmic domain of the TGF-β Type I receptor complex; followed by heteromeric complex association with Smad4, nuclear translocation and target gene activation [73]. Disruption of β2-SPECTRIN in mice leads to disruption of TGF-β signaling and results in a phenotype similar to Smad2+/-/Smad3+/- mutant mice, mid-gestational death due to gastrointestinal, liver, neural and heart defects, and loss of intrahepatic bile ducts. Interestingly, while the liver lineage is established, hepatocytes are poorly formed and liver architecture is lost with an absence of primitive bile ducts as in the Smad2+/-/Smad3+/- mutants. Moreover, bile duct formation can be induced in liver explants cultures treated with TGF-β [74].
Many studies have reported a reduction of TGF-β receptors in up to 70% of HCC [58, 75]. However, Smad proteins shown to be impaired in other cancers appear to play a minor role in HCC [76, 77]. Yet, TGF-β levels in serum and urine are increased in HCC patients [78, 79]. In addition, up to 40% of HCC have increased TGF-β expression based on immunohistochemical analysis [80, 81]. High TGF-β levels have been correlated with advanced clinical stage of HCC [82, 83]. This dual role of TGF-β signaling in HCC was explained by its effect on the tumor tissue microenvironment and on selective loss of the TGF-β-induced antiproliferative pathway [75]. Tumor cells that have selectively lost their growth-inhibitory responsiveness to TGF-β but retain an otherwise functional TGF-β signaling pathway may exhibit enhanced migration and invasive behavior in response to TGF-β stimulation. TGF-β signaling also has been shown to induce an epithelial to mesenchymal transition (EMT) process in tumor cells. EMT leads to enhanced migration and invasiveness [84]. Recently, loss of ELF, a TGF-β adaptor and signaling molecule, in the liver leads to cancer formation by deregulated hepatocyte proliferation and stimulation of angiogenesis [85]. More recently, it was reported that STAT3/Oct4-positive HCC cells, which have dysfunctional TGF-β signaling, are likely cancer progenitor cells that have the potential to give rise to HCC [86].
Notch pathway
The Notch signaling pathway plays an important role in stem cell self-renewal and differentiation [87-90]. However, other signaling pathways influence whether Notch functions as a tumor suppressor or oncogene in a particular tissue [91]. Notch signaling is activated through four receptors (Notch 1-4) that can interact in a redundant manner with five ligands of the Delta/Jagged family [49, 92]. Ligand binding triggers a γ-sevretase-dependent proteolytic cleavage of Notch receptor and the release of Notch intracellular domain (NCID) to the nucleus [93] , which in turn displaces the co-receptors associated with CSL transcription factors (CBF1 in humans; RBPJ in mice). Activating transcription factors are then recruited and expression of target genes such as Hairy and Enhancer of Split (HES1 and HES5) and Deltex1 is induced [92, 94].
Notch signaling plays a well-defined role in liver embryogenesis and bile duct formation. In addition, Notch family members are involved in angiogenesis and endothelial sprouting [95-97]. Increased expression of genes involved in this pathway has been shown in CD133+ liver cancer cells as compared to CD133-. The activated intracellular form of Notch3, as well as the notch ligand Jagged, is highly expressed in HCC [98-100]. Notch-dependent transformation is associated with extracellular signal-regulated kinase activation downstream of the Ras pathway, which increases Notch mRNA stability and is required for transcription of the Notch target gene, Hes-1 [101, 102]. Conversely, Notch-1 has been reported to function as a tumor suppressor and participate in cross-talk with other signaling pathways such as Ras/Raf/MEK/ERK through the regulation of the PTEN tumor suppressor [103]. Recent evidence indicates that activation of Notch1 signaling increases the expression level of death receptor 5 (DR5) with enhancement of TRAIL-induced apoptosis in vitro and in vivo [104, 105]. Inhibitors of the NOTCH pathway are currently under investigation in clinical trials for treating solid tumors although the effectiveness of NOTCH pathway inhibitors in treating liver cancer remains unclear.
Hedgehog signaling
Conserved from Drosophilia to humans, the Hedgehog (HH) pathway has a central role in embryonic development and adult tissue homeostasis by controlling cell fate specification and pattern formation [49, 106]. The functional importance of this pathway is illustrated by the multiple birth defects and malignancies associated with mutations and/or aberrant activation of the pathway [107, 108]. Three HH ligands Sonic (SHH), Indian (IHH) and Desert (DHH) have been identified in mammals that can bind interchangeably to two related twelve-pass membrane Patched (Ptc) receptors [109] . In the absence of ligand, Ptc antagonizes the pathway by preventing the activity of another transmembrane protein Smoothened (Smo) [110, 111]. Binding of HH ligands to Ptc relieves this inhibition and activates target gene transcription factors (Gli-1, Gli-2, Gli-3) [112, 113]. Like β-catenin, after ligand stimulation, Gli accumulates in the nucleus and induces transcription of genes related to cell cycle and growth including insulin-like growth factor-2 (IGF-2), cyclins, and β-catenin. The different Gli proteins exhibit activating or repressing transcriptional activators depending on proteolytic processing of the full-length proteins. Gli-1 and Gli-2 mainly act as transcriptional activators, while Gli-3 generates a repressor form (Gli3R) in the absence or inhibition of HH signaling [109, 114, 115]. Although functional significance of Gli-3 has been demonstrated by genetic inactivation [116] , the molecular mechanisms that control Gli-3 interactions and targets are largely undefined, whereas the dynamic interplay between Gli-1 and Gli-2 signaling is well documented.
Sonic is the predominant isoform in the liver. Up to 60% of human HCC express Sonic, and concomitant downregulation of Gli-related target genes are found after specific blockade of this pathway [117, 118]. Furthermore, tumorigenic activation of Smo can mediate overexpression of c-myc, a gene known to play an important pathogenic role in liver carcinogenesis [118]. Moreover, recent studies also showed that activation of Hedgehog signaling is critically related to CSCs and EMT features in many types of cancers including colon, gastric, esophagus, hepatic, and other cancers.
BMI1 signaling pathway
BMI1 is a part of the polycomb group genes (PcG) that are highly conserved throughout evolution. BMI acts as an epigenetic chromatin modifier and is known for its contribution to embryonic and stem cell self-renewal programs [119]. It is frequently overexpressed in different cancer types and disruption of BMI1 signaling has been linked to the activation of the hedgehog pathway in some cancers, such as medulloblastoma [120, 121]. Furthermore, BMI1 upregulation is associated with malignant transformation and acquisition of the malignant phenotype in HCC [122]. Aberrant BMI1 expression is reported in many CSC populations and it has been shown to have a critical role in maintaining and propagating the SP population in liver cancer. BMI1 is also highly expressed in CD133+ liver CSCs. The role of BMI1 in liver CSC maintenance is confirmed by ectopic expression of BMI1 in murine hepatic stem/progenitor cells. In these cells, BMI1 and the Wnt/β-catenin pathway regulate the self-renewal of normal or cancer stem cells in liver. Furthermore, BMI1 knockdown in SP cells completely abolished the tumorigenicity of SP cells [32, 123, 124]. Moreover, repression of targets of BMI1 plays a crucial role in the oncogenic transformation of hepatic stem/progenitor cells [125].
In addition to these signaling pathways, signal transducer and activator of transcription 3 (STAT3), mainly activated by IL-6 and its related cytokine, and IL-22 has been shown to play key roles in acute phase response, a protection against liver injury, the promotion of liver regeneration [126]. Furthermore, hyperactive STAT3 signaling results in expansion of oval cell numbers and trigger wound healing, cell migration, and proliferation [127, 128]. This signaling pathway may take part an important role of maintenance of CSCs.
Stem cell signaling network
Multiple studies have suggested that Wnt/β-catenin, Notch, Hedgehog, FGF, and TGF-β/BMP signaling network is implicated in the maintenance of tissue homeostasis by regulating self-renewal of normal stem cells as well as proliferation or differentiation of progenitor cells [129-133]. Especially, it is well established that Wnt/β-catenin and Hedgehog signaling pathways are critical for embryogenic development, as well as in the biology of CSCs and in the acquisition of EMT. Breakage of the signaling network for normal stem cells leads to the transformation to CSC. Alternatively, acquisition of self-renewal potential in progenitor cells due to epigenetic change or genetic alteration of stem cell signaling related genes gives rise to CSC. Detailed analyses on the dysregulation of Wnt/β-catenin, Notch, Hedgehog, FGF, and TGF-β/BMP signaling pathways in CSCs derived from a various type of human tissues or organ should be systematically investigated to better understand CSCs themselves as well as the role they play in carcinogenesis.
miRNA
MiRNAs play critical roles in many biological processes including cancer by directly interacting with specific messenger RNAs (mRNAs) through base pairing and then inhibiting expression of the target genes through a variety of molecular mechanisms. MiRNAs can undergo aberrant regulation during carcinogenesis, and they can act as oncogenes or tumor suppressor genes. Disruption of miRNA expression levels in tumor cells may result from distorted epigenetic regulation of miRNA expression, abnormalities in miRNA processing genes and proteins, and the location of miRNAs at cancer-associated genomic regions. Consequently, abnormal miRNA expression is a ubiquitous feature of solid tumors including HCC [134]. In liver carcinogenesis, miRs have been found to have both tumor suppressive (miR-122, miR-26, miR-223) and oncogenic (miR-130b, miR-221, miR-222) activity [135-139]. Clearly, miRNAs play a critical role in carcinogenesis and oncogenesis. Emerging evidence suggests that certain abnormal miRNA expression induces CSC dysregulation, resulting in unlimited self-renewal and cancer progression. Therefore, miRNA expression is a vital key to CSC dysregulation.
Lin28 and let-7 signaling
Lin28 was first characterized in the nematode Caenorhabditis elegans as an important regulator of developmental timing [140, 141]. Recently, Lin28 was used together with OCT4, NANOG and SOX2 to reprogram human somatic fibroblasts to pluripotency [142]. Overexpression of these stem cell factors has been reported to promote oncogenesis by driving self-renewal and proliferation [142]. Moreover, poorly differentiated, aggressive human tumors have recently been shown to have an embryonic stem cell-like gene expression signature; these stem cell factors have also been reported to have roles in tumor progression.
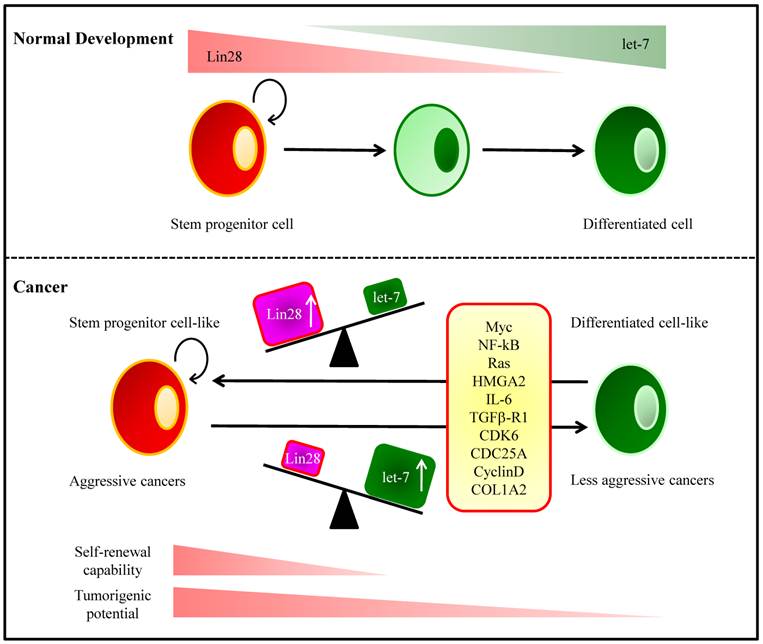
LIN28 and LIN28B are overexpressed in primary human tumors and human cancer cell lines (overall frequency 15%) [143]. The mammalian homologs of lin-28, Lin28 and Lin28b, bind to the terminal loop of the precursors of let-7 family miRNAs and block their processing into mature miRNAs [144, 145]. In HCC, LIN28B-expressing tumors are associated with advanced stage [143]. Moreover, LIN28B-expression was associated with a significantly increased incidence of early recurrence. LIN28 is associated with an advanced disease and poor clinical outcome in HCC [143, 146, 147]. The initiation of hepatocarcinogenesis is linked to chronic inflammation clinically and epidemiologically. The positive feedback loop involving NF-κB, Lin28B, let-7, and IL-6 is required for maintenance of the transformed phenotype and stem cell population [148] (Fig. 2).
Let-7 and Lin28 in Development and Tumorigenesis. (Top) During normal development, the RNA-binding protein Lin28 is highly expressed in stem and progenitor cells. Lin28 blocks processing of let-7 miRNA precursor molecules into mature miRNAs, thereby maintains expression of genes that drive self-renewal and proliferation. As progenitor cells differentiate, Lin28 expression decreases, which allows let-7 processing and increased production of mature let-7 miRNAs. Let-7 miRNAs repress the expression of genes involved in self-renewal resulting in lineage commitment and terminal differentiation. (Bottom) Many molecules contribute to the balancing act of the Lin28/let-7 link in cellular differentiation and tumor progression. An imbalance between Lin28 and let-7 induced by these molecules can result in cellular transformation.
miR-181
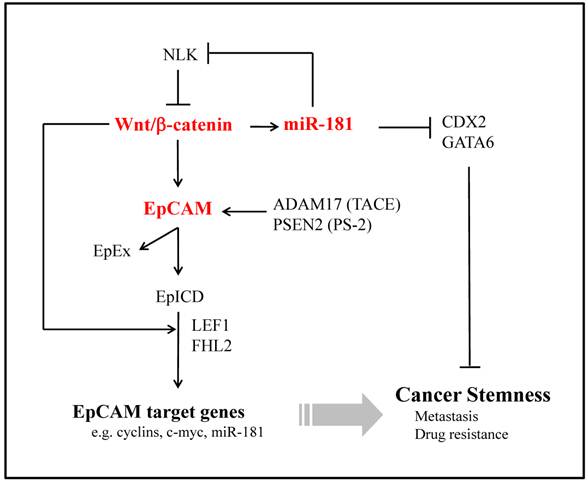
Mir-181 was first characterized in the patients with acute myeloid leukemia as a predictor of prognosis. Recently, we have identified a subset of highly EpCAM+ HCC cells from AFP+ tumors with cancer stem/progenitor cell features [149]. MiR-181 family members are up-regulated in EpCAM+AFP+ HCC cells. Moreover, miR-181 family members were highly expressed in embryogenic livers and isolated hepatic stem cells. Forced expression of miR-181 induces stemness of HCC cells while inhibiting miR-181 results in cell differentiation and inhibition of tumorigenicity. In addition, we identified three targets of miR-181, caudal-related homeobox 2 (CDX2), GATA6 and NLK. CDX2 and GATA6 are transcription factors and link to cancer stemness. NLK is a negative regulator of Wnt/β-catenin signaling. We propose that miR-181 maintains HCC stemness by inhibiting CDX2, GATA6 or NLK. MiR-181 could directly target hepatic transcriptional regulators of differentiation and an inhibitor of Wnt/β-catenin signaling [149]. Therefore, miR-181 and its signaling molecules could be molecular targets for inhibiting CSCs (Fig. 3).
Potential Signalling Pathways of Wnt/β-Catenin, EpCAM and miR-181 Activated in Hepatic Cancer Stem Cells. Upon cleavage by TACE/PS-2, EpICD translocates into the nucleus in a multiprotein complex. Together with FHL2, β-catenin and Lef-1, EpICD contacts DNA at Lef-1 consensus sites. Owing to its ability to inhibit E-cadherin-mediated adhesion, EpCAM provides itself with β-catenin as a central interacting protein.
Therapeutic target of Molecular signaling
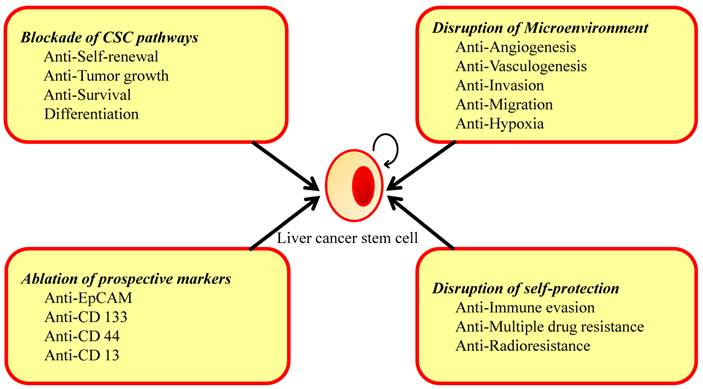
The successful eradication of cancer requires anticancer therapy that affects the differentiated cancer cells and the potential CSC population [150, 151]. At present, conventional anticancer therapies include chemotherapy, radiation and immunotherapy kill rapidly growing differentiated tumor cells, thus reducing tumor mass but potentially leave behind cancer-initiating cells. Therapies that exclusively address the pool of differentiated cancer cells but fail to eradicate the CSC compartment might ultimately result in relapse and the proliferation of therapy-resistant and more aggressive tumor cells. An ideal drug regime would kill differentiated cancer cells and, at the same time, specifically, selectively and quickly target and kill CSCs to avoid toxic side effects for other cell types and to disrupt the self-protection potential of CSCs. Moreover, CSCs clearly have a complex pathogenesis, with the potential for considerable crosstalk and redundancy in signaling pathways, and hence targeting single molecules or pathways may have a limited benefit in treatment. The use of combinations of therapies may be needed to overcome the complex network of signaling pathways, and ultimately inhibit the signaling that controls tumor growth and survival. In addition to the factors in which CSCs possess by themselves, microenvironment surrounding them is important for maintenance, such as angiogenesis, vasculogenesis, and hypoxia. Many new therapeutic strategies targeting CSCs at various stages of differentiation and microenvironment of CSCs have been tried. We will be discussed below (Fig. 4).
Strategies in Eradicate Liver Cancer Stem Cells. CSCs are protected from conventional therapies by changing their microenvironment and self-protection. Specifically targeting any of these areas may lead to the eradication of the CSCs.
Blockage of CSC pathways
Anti-Self-renewal
Targeting key signaling pathways for CSC self-renewal is one approach to therapy [123, 152, 153]. The Wnt/β-catenin signaling pathway is clearly important for the self-renewal and maintenance of stem cells [35]. Several experimental studies have demonstrated a decreased proliferation and an increased apoptosis resulting from inhibiting the Wnt/β-catenin signaling pathway [154]. The Wnt/β-catenin signaling pathway can be inhibited in a number of ways; for example, Dickkopf1 (Dkk1) binds to low-density LRP6 and prevents formation of the Fzd-Wnt-LRP6 complex [155]. A new approach to antagonize Wnt signaling has been the development of small molecules (XAV939) to inhibit the enzyme tankyrase that normally destroys the scaffold protein axin, a crucial component of the β-catenin destruction complex [156]. Furthermore, antibody-based therapeutic approaches targeting EpCAM are currently being developed [157, 158]. EpCAM-directed therapies will be efficacious in eradicating EpCAM-expressing CSCs.
The Hedgehog pathway is another potentially druggable target for CSC eradication. Several small-molecule modulators of Sonic hedgehog signaling have been used to regulate the activity of this pathway in medulloblastoma, basal cell carcinoma, pancreatic cancer, prostate cancer and developmental disorders [159]. In liver cells, suppression of the Sonic Hedgehog pathway by siRNA not only decreased HCC cell proliferation but also chemosensitized the cells to 5-fluorouracil (5-FU) and to the induction of cell apoptosis [160]. Furthermore, in hepatoblastoma, blocking Hh signaling with the antagonist cyclopamine had a strong inhibitory effect on cell proliferation of hepatoblastoma cell lines [161]. Thus, targeting intracellular pathways associated with self-renewal of CSCs remains a viable approach to be extended in the near future.
Anti-Tumor growth and inducing tumor cell differentiation
PTEN plays a role in the expansion of the CD133+ liver CSC population in liver-specific PTEN-deleted mice, which supports PTEN as a promising target in HCC therapy [162]. In addition, TGF-β family proteins have also emerged as key players in promoting the growth of stem cells in their undifferentiated state. A recent investigation revealed normal hepatic stem cells committing to malignant transformation due to aberrant TGF-β and activated IL-6 signaling [74, 163]. Therefore, inhibition of IL-6 signaling may be a potential therapeutic strategy in liver cancer treatment [163, 164].
CSC cells, which only make up a small proportion in cancer, have the capability to sustain tumor growth and are more resistant to conventional chemotherapy than other differentiated cancer cells. One approach to treat malignancies is to induce differentiation of the CSC cells. Differentiation therapy could force hepatoma cells to differentiate and lose their self-renewal property. Hepatocyte nuclear factor-4α (HNF4α), a central regulator of differentiated hepatocyte phenotype, suppresses a tumorigenesis and tumor development by inducing HCC differentiation, especially CSC cells [165]. Interferon therapy is effective for eradicating hepatitis viruses and also preventing the development of HCC. Interferon alpha treatment accelerated hepatocytic and biliary differentiation in oval cell lines [166]. Interferon could be applied to the treatment of HCC by targeting CSCs. In addition, oncostatin M (OSM), an interleukin 6-related cytokine known to induce differentiation of hepatoblasts into hepatocytes, could be used to effectively induce differentiation and cell division of dormant EpCAM+ liver CSCs, and the combination of OSM and conventional chemotherapy with 5-FU efficiently eliminates HCC by targeting both CSCs and non-CSCs [167]. These findings indicate that combination of differentiation therapy and conventional chemotherapy may be an effective treatment of HCC.
Liver stem/progenitor cell markers
The identification of CSC markers and their exploitation in targeted chemotherapy is an important research goal. It has been shown that CSCs in HCC can be identified by several cell surface antigens, e.g., CD133, CD90, CD44, OV6, and EpCAM, or by selecting the SP cells by Hoechst dye-staining. Given the phenotypic similarities between CSCs and normal stem cells, it is reasonable to infer that the surface phenotype of CSCs resembles that of normal hepatic stem cells.
Anti-CD133
CD133/prominin-1, a pentaspan membrane glycoprotein, is an important cancer stem cell surface marker in various solid tumor types, including liver [168, 169]. CD133 expressing cells have been suggested to be critical tumorigenic progenitors in HCC, conferring chemoresistance by preferential activation of the AKT/PKB and Bcl-2 cell survival response [170]. The treatment of CD133+ HCC cells with an AKT1 inhibitor, specific to the Akt/PKB pathway, significantly reduced the expression of the survival proteins. In addition, suppression of CD133 by a murine antibody to human CD133 conjugated to a potent cytotoxic drug reduced the proliferation rate of Hep3B cells in vitro and delayed tumor growth in a SCID mouse model [171]. These findings suggest that targeting of CD133 might be a novel therapeutic strategy for liver tumors.
Anti-CD44
CD133+/CD44+ HCC cells were more tumoigenic than those of CD133+/CD44- cells in vivo. A recent study suggested that CSC phenotype could be precisely defined by co-expression of CD133 and CD44 cell surface markers [172]. CD133+/CD44+ HCC cells showed stem cell properties, including extensive proliferation, self-renewal and differentiation into the bulk of cancer cells. In addition, recent studies also revealed that blocking CD44 signaling using an anti-CD44 antibody might be a potential strategy to eradicate liver CSCs and consequently cure those patients [172].
Anti-EpCAM
Currently, several EpCAM-targeting antibodies are in clinical development, which include Catumaxomab (Fresenius Biotech) and Adecatumumab ((MT201) Micromet, Inc.). Clinical trials have been conducted in various cancers, including breast, prostate and colon cancers [157, 158]. In liver cells, RNAi targeting of EpCAM significantly decreased the CSC pool and reduced both tumorigenicity and invasive capacity of CSCs [37, 173]. Since EpCAM expression is a downstream target of Wnt/β-catenin, these results may have implications for development of novel target therapies.
Anti-CD13
Recently, CD13 was identified as a marker for semiquiescent CSCs in human liver cancer cells. In mouse xenograft models, combination of a CD13 inhibitor and 5-FU dramatically reduced tumor volume compared with either agent alone. 5-FU inhibited proliferating CD13+ semiquiescent CSCs, while CD13 inhibition suppressed the self-renewing and tumor-initiating ability of dormant CSCs. These results indicate that combining a CD13 inhibitor with a reactive oxygen species (ROS) -inducing chemo/radiation therapy may improve the treatment of liver cancer [174].
Disruption of Microenvironment
Hypoxia has been identified as a major cause of hypervascularization in HCC, and in patients with HCC, disease free survival is shorter when tumors express high levels of hypoxia-inducible factor-1α (HIF-1α). Hypoxia influences microenvironment in HCC and liver CSCs [175, 176]. Induction of tumor hypoxia combined with chemotherapy by transcatheter transarterial chemoembolization has been widely used in treating unresectable HCC, but tumor response rate is unsatisfactory and only a subgroup of patients benefit from this treatment [177, 178]. HIF-1α may be responsible for in this failure, as suggested by experimental findings obtained in a rat model of primary liver cancer [179]. Therefore, hypoxia-driven clonal selection of apoptosis-resistant tumor cells, together with hypoxia-induced MDR1 expression and angiogenesis, explain why hypoxic tumors are more resistance to conventional anticancer therapy. This justifies the current trials evaluating the use of anti-angiogenic therapy following HCC surgery. Several studies have established that tumor growth and invasion in HCC are dependent on dysregulated angiogenesis [180-182]. There is, therefore, a strong rationale for targeting growth factors that drive angiogenic process as a potential therapeutic strategy for the treatment of HCC. VEGF is a key angiogenic factor, and several agents that target VEGF or VEGFR are currently in development for the treatment of HCC [183, 184]. These agents include the tyrosine kinase inhibitors Vatalanib (PTK787) [185] and Cediranib (AZD2171) [186, 187], and the monoclonal antibody Bevacizumab (Avastin; Genentech, Inc., South San Francisco, CA, USA) [188], and multikinase inhibitors Sorafenib (Nexavar; Bayer HealthCare Pharmaceuticals Inc., Wayne, NJ, USA) [189-194], Sunitinib (Sutent; Pfizer Labs, New York, NY, USA) [195, 196], Brivanib (BMS-582664) [197-199], and Linifanib (ABT-869) [199]. In addition, ligands that bind to the EGFR, such as EGF, have a vital role in both tumor angiogenesis and proliferation, thought to be primarily through activation of the RAF/MEK/ERK and PI3K/AKT/mTOR pathways. Because of their efficacy in other solid tumors and the integral role of growth factors in HCC development and progression, it was hypothesized that agents specifically targeting EGF/EGFR signaling may also be beneficial in HCC. These agents include the tyrosine kinase inhibitors Erotinib [200, 201], Lapatinib (GW572016) [202, 203] and Gefitinib ((ZD1839) Iressa; Astrazeneca Pharmaceuticals LP, Wilmington DE, USA) [204-207], and the monoclonal antibody Cetuximab ((IMC-C255) Erbitux; ImClone LLC, New York, NY, and Bristol-Myers Squibb, Princeton, NJ, USA) [208, 209].
Disruption of self-protection
Anti-immune evasion
The observation that tumors progress in patients with HCC despite the presence of tumor-specific immune responses suggests that development of HCC leads to a number of immunosuppressive mechanism, which are important to be considered when designing immunotherapy protocols. These mechanisms include production of immunosuppressive cytokines such as TGF-β and prostaglandins, impaired antigen-presenting cells, generation of inhibitory macrophages, increase in regulatory T cells and induction of myeloid-derived suppressor cells (MDSC). All these factors provide an environment that promotes angiogenesis, tumor survival and metastasis.
Targeting regulatory T cells has been of great interest to potentially remove the suppression of effecter T cells and enhance tumor-specific immune response. Depletion of regulatory T cells using anti-CD25 monoclonal antibodies or regulatory T cell-inhibiting agents, such as cyclophosphamide, has been shown to have anti-tumor effects in preclinical models [210-213]. In addition, MDSC suppress the cytokine production as well as the cytotoxic capacity of natural killer (NK) cells, playing a critical role in the host defense against cancer, in HCC patients [214]. Impaired NK cells can affect anti-tumor immune responses, which contributes further to tumor escape from both innate and adaptive immune responses in patients with HCC.
Anti-multiple drug resistance
Survival of stem-like cells in response to chemotherapeutic drugs is thought to be governed by the presence of active transmembrane adenosine triphosphate-binding cassette (ABC) transporter family members, such as MDR1, ABCG2 and ABCC2 [215]. It is believed that stem-like SP cells, which are known for their ability to efflux the DNA-binding dye Hoechst 33342, confer resistance to chemotherapeutic drugs, including cisplatin and doxorubicin, through expression high levels of such ABC transporters [216, 217]. In SP cells purified from HCC cell lines, inhibition of MDR1, ABCG2 and ABCC2 reverses their chemoresistance [218]. These cells have been shown to harbor other CSC-like properties, and may be related to the metastatic potential and chemoresistance of HCC [219]. Moreover, it was demonstrated that expression of granulin-epithelin precursor (GEP) and ABCB5 in liver CSCs is associated with chemoresistance and reduced survival times of patients with HCC [175].
Anti-Radioresistance
Several experimental and clinical findings provide evidence that the number of CSCs in a cancer affects its radiocurability. Recurrent tumors after radiotherapy could originate from one surviving CSC, and a permanent local tumor control requires inactivation of all CSCs [220]. Tumor cell hypoxia and tumor cell repopulation are the main factors causing radioresistance. Oxygen mediates the majority of the biological effects of sparsely ionizing radiation, and the response of cells to radiation depends strongly on the availability of oxygen. Various methods to deliver oxygen to cancer tissue have been studied. Enhanced tumor oxygenation has previously been achieved in an animal model using the synthetic heme-based oxygen carrier, albumin-heme which is a recombinant human serum albumin-Fe cyclohexanoil heme (rHSA-FeP). The rHSAFeP is a candidate radiation-enhancing drug, and arterial infusion of rHSA-FeP may serve as a local oxygeneation method that enhances the radiation effect [221].
Future Directions
The rapid development of the CSC field, combined with genome-wide screening techniques, has allowed for the identification of important new CSC markers and pathways, and these discoveries have contributed to one of the most important developments in cancer treatment. However, several important issues still remain to be resolved. Most of the key pathways important to CSCs are also shared by normal stem/progenitor cells and drugs targeting these pathways could have a detrimental effect on normal cells. For example, little is known about CSC directed therapies (e.g. targeting CD133 in CD133+ liver CSCs). Initial results are promising, but its potential short- and long-term side effects of these therapies are unclear. Such therapies will, if not specific for CSCs, lead to tissue and/or organ damage due to the depletion of the reserve/regenerative stem cells. Such treatment with off-target effects lead to acute and irreversible organ failure. Therefore, it is critical in delineating the molecular differences between CSCs and their tissue specific stem cell counterparts, to prevent damage to normal somatic stem cells and to ensure selectively targeting CSCs. This growing knowledge base has the potential to identify candidate genes and pathways that are important for CSC survival and propagation but are not important for normal stem cell function.
In addition, CSCs clearly have a complex pathogenesis, with the potential for considerable crosstalk and redundancy in signaling pathways, and hence targeting single molecules or pathways may have a limited benefit in treatment. Use of combinations of therapies may be needed to overcome the complex network of signaling pathways, and ultimately inhibit the signaling that controls tumor growth and survival. However, use of a combination regimen can lead to tolerability and drug-drug interaction problems, and hence an alternative approach is to use molecularly targeted agents that have multiple modes of action. It is useful to understand which combination regimen is the most effective for inhibiting CSC survival and propagation with the least impact on normal stem cell function. When a sufficient number of CSC markers become available and an ideal combination therapy identify, CSC-specific therapies might be developed that spare healthy stem cells and thus reduce side effects and retain regenerative tissue capacities. Discoveries made in the CSC field will feed back into other areas of stem cell research because many marker gene products found in CSCs are shares with the normal stem cell population. It is also expected that a better understanding of the processes that control autonomous growth, differentiation and cell migration will contribute to novel regenerative-medicine-based treatments that will revolutionize therapeutic strategies and bring renewed hope to cancer patients.
Acknowledgements
We thank NIH Fellows Editorial Board for editorial assistance of manuscript. This study was supported by grant (Z01-BC 010313) from the Intramural Research Program of the Center for Cancer Research of the National Cancer Institute.
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. el-Serag HB. Epidemiology of hepatocellular carcinoma. Clin Liver Dis. 2001;5:87-107
2. Eguchi S, Kanematsu T, Arii S. et al. Recurrence-free survival more than 10 years after liver resection for hepatocellular carcinoma. Br J Surg. 2011;98:552-557
3. Huang J, Yan L, Cheng Z. et al. A randomized trial comparing radiofrequency ablation and surgical resection for HCC conforming to the Milan criteria. Ann Surg. 2010;252:903-912
4. Wang W, Shi J, Xie WF. Transarterial chemoembolization in combination with percutaneous ablation therapy in unresectable hepatocellular carcinoma: a meta-analysis. Liver Int. 2010;30:741-749
5. Marelli L, Stigliano R, Triantos C. et al. Treatment outcomes for hepatocellular carcinoma using chemoembolization in combination with other therapies. Cancer Treat Rev. 2006;32:594-606
6. Michalopoulos GK. Liver regeneration. J Cell Physiol. 2007;213:286-300
7. Michalopoulos GK, DeFrances MC. Liver regeneration. Science. 1997;276:60-66
8. Riehle KJ, Dan YY, Campbell JS. et al. New concepts in liver regeneration. J Gastroenterol Hepatol. 2011;26(Suppl 1):S203-S212
9. Russo FP, Parola M. Stem and progenitor cells in liver regeneration and repair. Cytotherapy. 2011;13:135-144
10. Lechler T, Fuchs E. Asymmetric cell divisions promote stratification and differentiation of mammalian skin. Nature. 2005;437:275-280
11. Steiner JW, Perz ZM, Taichman LB. Cell population dynamics in the liver. A review of quantitative morphological techniques applied to the study of physiological and pathological growth. Exp Mol Pathol. 1966;5:146-181
12. Alison MR. Liver stem cells: implications for hepatocarcinogenesis. Stem Cell Rev. 2005;1:253-260
13. Fausto N. Liver regeneration and repair: hepatocytes, progenitor cells, and stem cells. Hepatology. 2004;39:1477-1487
14. Falkowski O, An HJ, Ianus IA. et al. Regeneration of hepatocyte 'buds' in cirrhosis from intrabiliary stem cells. J Hepatol. 2003;39:357-364
15. Marshall A, Rushbrook S, Davies SE. et al. Relation between hepatocyte G1 arrest, impaired hepatic regeneration, and fibrosis in chronic hepatitis C virus infection. Gastroenterology. 2005;128:33-42
16. Lowes KN, Brennan BA, Yeoh GC. et al. Oval cell numbers in human chronic liver diseases are directly related to disease severity. Am J Pathol. 1999;154:537-541
17. Kofman AV, Morgan G, Kirschenbaum A. et al. Dose- and time-dependent oval cell reaction in acetaminophen-induced murine liver injury. Hepatology. 2005;41:1252-1261
18. Yang S, Koteish A, Lin H. et al. Oval cells compensate for damage and replicative senescence of mature hepatocytes in mice with fatty liver disease. Hepatology. 2004;39:403-411
19. Wang Y, Yao HL, Cui CB. et al. Paracrine signals from mesenchymal cell populations govern the expansion and differentiation of human hepatic stem cells to adult liver fates. Hepatology. 2010;52:1443-1454
20. Turner R, Lozoya O, Wang Y. et al. Human hepatic stem cell and maturational liver lineage biology. Hepatology. 2011;53:1035-1045
21. Brill S, Holst P, Sigal S. et al. Hepatic progenitor populations in embryonic, neonatal, and adult liver. Proc Soc Exp Biol Med. 1993;204:261-269
22. Navarro-Alvarez N, Soto-Gutierrez A, Kobayashi N. Hepatic stem cells and liver development. Methods Mol Biol. 2010;640:181-236
23. Alison MR, Lovell MJ. Liver cancer: the role of stem cells. Cell Prolif. 2005;38:407-421
24. Shafritz DA, Oertel M, Menthena A. et al. Liver stem cells and prospects for liver reconstitution by transplanted cells. Hepatology. 2006;43(Suppl 1):S89-S98
25. Theise ND, Yao JL, Harada K. et al. Hepatic 'stem cell' malignancies in adults: four cases. Histopathology. 2003;43:263-271
26. Yao Z, Mishra L. Cancer stem cells and hepatocellular carcinoma. Cancer Biol Ther. 2009;8:1691-1698
27. Durnez A, Verslype C, Nevens F. et al. The clinicopathological and prognostic relevance of cytokeratin 7 and 19 expression in hepatocellular carcinoma. A possible progenitor cell origin. Histopathology. 2006;49:138-151
28. Roskams T. Liver stem cells and their implication in hepatocellular and cholangiocarcinoma. Oncogene. 2006;25:3818-3822
29. Zimmermann A. Hepatoblastoma with cholangioblastic features ('cholangioblastic hepatoblastoma') and other liver tumors with bimodal differentiation in young patients. Med Pediatr Oncol. 2002;39:487-491
30. Forbes SJ, Alison MR. Side population (SP) cells: taking center stage in regeneration and liver cancer? Hepatology. 2006;44:23-26
31. Chiba T, Kita K, Zheng YW. et al. Side population purified from hepatocellular carcinoma cells harbors cancer stem cell-like properties. Hepatology. 2006;44:240-251
32. Ma S, Chan KW, Hu L. et al. Identification and characterization of tumorigenic liver cancer stem/progenitor cells. Gastroenterology. 2007;132:2542-2556
33. Song W, Li H, Tao K. et al. Expression and clinical significance of the stem cell marker CD133 in hepatocellular carcinoma. Int J Clin Pract. 2008;62:1212-1218
34. Kim JW, Ye Q, Forgues M. et al. Cancer-associated molecular signature in the tissue samples of patients with cirrhosis. Hepatology. 2004;39:518-527
35. Yamashita T, Budhu A, Forgues M. et al. Activation of hepatic stem cell marker EpCAM by Wnt-beta-catenin signaling in hepatocellular carcinoma. Cancer Res. 2007;67:10831-10839
36. Yamashita T, Forgues M, Wang W. et al. EpCAM and alpha-fetoprotein expression defines novel prognostic subtypes of hepatocellular carcinoma. Cancer Res. 2008;68:1451-1461
37. Yamashita T, Ji J, Budhu A. et al. EpCAM-positive hepatocellular carcinoma cells are tumor-initiating cells with stem/progenitor cell features. Gastroenterology. 2009;136:1012-1024
38. Zhou H, Rogler LE, Teperman L. et al. Identification of hepatocytic and bile ductular cell lineages and candidate stem cells in bipolar ductular reactions in cirrhotic human liver. Hepatology. 2007;45:716-724
39. Tanaka S, Yamamoto T, Tanaka H. et al. Potentiality of combined hepatocellular and intrahepatic cholangiocellular carcinoma originating from a hepatic precursor cell: Immunohistochemical evidence. Hepatol Res. 2005;32:52-57
40. Lee JH, Rim HJ, Sell S. Heterogeneity of the "oval-cell" response in the hamster liver during cholangiocarcinogenesis following Clonorchis sinensis infection and dimethylnitrosamine treatment. J Hepatol. 1997;26:1313-1323
41. Steinberg P, Steinbrecher R, Radaeva S. et al. Oval cell lines OC/CDE 6 and OC/CDE 22 give rise to cholangio-cellular and undifferentiated carcinomas after transformation. Lab Invest. 1994;71:700-709
42. Komuta M, Spee B, Vander Borght S. et al. Clinicopathological study on cholangiolocellular carcinoma suggesting hepatic progenitor cell origin. Hepatology. 2008;47:1544-1556
43. Zhang F, Chen XP, Zhang W. et al. Combined hepatocellular cholangiocarcinoma originating from hepatic progenitor cells: immunohistochemical and double-fluorescence immunostaining evidence. Histopathology. 2008;52:224-232
44. Branda M, Wands JR. Signal transduction cascades and hepatitis B and C related hepatocellular carcinoma. Hepatology. 2006;43:891-902
45. Merle P, de la Monte S, Kim M, Herrmann M. et al. Functional consequences of frizzled-7 receptor overexpression in human hepatocellular carcinoma. Gastroenterology. 2004;127:1110-1122
46. Ishizaki Y, Ikeda S, Fujimori M. et al. Immunohistochemical analysis and mutational analyses of beta-catenin, Axin family and APC genes in hepatocellular carcinomas. Int J Oncol. 2004;24:1077-1083
47. Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. 2004;20:781-810
48. Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature. 2005;434:843-850
49. Cerdan C, Bhatia M. Novel roles for Notch, Wnt and Hedgehog in hematopoesis derived from human pluripotent stem cells. Int J Dev Biol. 2010;54:955-963
50. Wu CH, Nusse R. Ligand receptor interactions in the Wnt signaling pathway in Drosophila. J Biol Chem. 2002;277:41762-41769
51. Lepourcelet M, Chen YN, France DS. et al. Small-molecule antagonists of the oncogenic Tcf/beta-catenin protein complex. Cancer Cell. 2004;5:91-102
52. Ben-Ze'ev A, Geiger B. Differential molecular interactions of beta-catenin and plakoglobin in adhesion, signaling and cancer. Curr Opin Cell Biol. 1998;10:629-639
53. Huber AH, Weis WI. The structure of the beta-catenin/E-cadherin complex and the molecular basis of diverse ligand recognition by beta-catenin. Cell. 2001;105:391-402
54. Giles RH, van Es JH, Clevers H. Caught up in a Wnt storm: Wnt signaling in cancer. Biochim Biophys Acta. 2003;1653:1-24
55. Kohn AD, Moon RT. Wnt and calcium signaling: beta-catenin-independent pathways. Cell Calcium. 2005;38:439-446
56. Kokolus K, Nemeth MJ. Non-canonical Wnt signaling pathways in hematopoiesis. Immunol Res. 2010;46:155-164
57. Kuhl M. Non-canonical Wnt signaling in Xenopus: regulation of axis formation and gastrulation. Semin Cell Dev Biol. 2002;13:243-249
58. Breuhahn K, Longerich T, Schirmacher P. Dysregulation of growth factor signaling in human hepatocellular carcinoma. Oncogene. 2006;25:3787-3800
59. Inagawa S, Itabashi M, Adachi S. et al. Expression and prognostic roles of beta-catenin in hepatocellular carcinoma: correlation with tumor progression and postoperative survival. Clin Cancer Res. 2002;8:450-456
60. Joo M, Lee HK, Kang YK. Expression of beta-catenin in hepatocellular carcinoma in relation to tumor cell proliferation and cyclin D1 expression. J Korean Med Sci. 2003;18:211-217
61. Laurent-Puig P, Legoix P, Bluteau O. et al. Genetic alterations associated with hepatocellular carcinomas define distinct pathways of hepatocarcinogenesis. Gastroenterology. 2001;120:1763-1773
62. Taniguchi K, Roberts LR, Aderca IN. et al. Mutational spectrum of beta-catenin, AXIN1, and AXIN2 in hepatocellular carcinomas and hepatoblastomas. Oncogene. 2002;21:4863-4871
63. Park JY, Park WS, Nam SW. et al. Mutations of beta-catenin and AXIN I genes are a late event in human hepatocellular carcinogenesis. Liver Int. 2005;25:70-76
64. Satoh S, Daigo Y, Furukawa Y. et al. AXIN1 mutations in hepatocellular carcinomas, and growth suppression in cancer cells by virus-mediated transfer of AXIN1. Nat Genet. 2000;24:245-250
65. Yang W, Yan HX, Chen L. et al. Wnt/beta-catenin signaling contributes to activation of normal and tumorigenic liver progenitor cells. Cancer Res. 2008;68:4287-4295
66. Cairo S, Armengol C, De Reynies A. et al. Hepatic stem-like phenotype and interplay of Wnt/beta-catenin and Myc signaling in aggressive childhood liver cancer. Cancer Cell. 2008;14:471-484
67. Schmelzer E, Zhang L, Bruce A. et al. Human hepatic stem cells from fetal and postnatal donors. J Exp Med. 2007;204:1973-1987
68. Munz M, Baeuerle PA, Gires O. The emerging role of EpCAM in cancer and stem cell signaling. Cancer Res. 2009;69:5627-5629
69. Maetzel D, Denzel S, Mack B. et al. Nuclear signalling by tumour-associated antigen EpCAM. Nat Cell Biol. 2009;11:162-171
70. Mishra L, Derynck R, Mishra B. Transforming growth factor-beta signaling in stem cells and cancer. Science. 2005;310:68-71
71. Massague J, Blain SW, Lo RS. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell. 2000;103:295-309
72. Chang H, Brown CW, Matzuk MM. Genetic analysis of the mammalian transforming growth factor-beta superfamily. Endocr Rev. 2002;23:787-823
73. Tang Y, Katuri V, Dillner A. et al. Disruption of transforming growth factor-beta signaling in ELF beta-spectrin-deficient mice. Science. 2003;299:574-577
74. Amin R, Mishra L. Liver stem cells and tgf-Beta in hepatic carcinogenesis. Gastrointest Cancer Res. 2008;2(4 Suppl):S27-S30
75. Ikegami T. Transforming growth factor-beta signaling and liver cancer stem cell. Hepatol Res. 2009;39:847-849
76. Yakicier MC, Irmak MB, Romano A. et al. Smad2 and Smad4 gene mutations in hepatocellular carcinoma. Oncogene. 1999;18:4879-4883
77. Longerich T, Breuhahn K, Odenthal M. et al. Factors of transforming growth factor beta signalling are co-regulated in human hepatocellular carcinoma. Virchows Arch. 2004;445:589-596
78. Kim HG, Chung YH, Song BC. et al. Expression of transforming growth factor beta-1 in chronic hepatitis and hepatocellular carcinoma associated with hepatitis C virus infection. Korean J Intern Med. 2000;15:165-170
79. Tsai JF, Jeng JE, Chuang LY. et al. Elevated urinary transforming growth factor-beta1 level as a tumour marker and predictor of poor survival in cirrhotic hepatocellular carcinoma. Br J Cancer. 1997;76:244-250
80. Bedossa P, Peltier E, Terris B. et al. Transforming growth factor-beta 1 (TGF-beta 1) and TGF-beta 1 receptors in normal, cirrhotic, and neoplastic human livers. Hepatology. 1995;21:760-766
81. Abou-Shady M, Baer HU, Friess H. et al. Transforming growth factor betas and their signaling receptors in human hepatocellular carcinoma. Am J Surg. 1999;177:209-215
82. Lu Y, Wu LQ, Li CS. et al. Expression of transforming growth factors in hepatocellular carcinoma and its relations with clinicopathological parameters and prognosis. Hepatobiliary Pancreat Dis Int. 2008;7:174-178
83. Ikeguchi M, Iwamoto A, Taniguchi K. et al. The gene expression level of transforming growth factor-beta (TGF-beta) as a biological prognostic marker of hepatocellular carcinoma. J Exp Clin Cancer Res. 2005;24:415-421
84. van Zijl F, Zulehner G, Petz M. et al. Epithelial-mesenchymal transition in hepatocellular carcinoma. Future Oncol. 2009;5:1169-1179
85. Baek HJ, Lim SC, Kitisin K. et al. Hepatocellular cancer arises from loss of transforming growth factor beta signaling adaptor protein embryonic liver fodrin through abnormal angiogenesis. Hepatology. 2008;48:1128-1137
86. Yuan F, Zhou W, Zou C. et al. Expression of Oct4 in HCC and modulation to wnt/beta-catenin and TGF-beta signal pathways. Mol Cell Biochem. 2010;343:155-162
87. Reya T, Morrison SJ, Clarke MF. et al. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105-111
88. Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science. 1999;284:770-776
89. Androutsellis-Theotokis A, Leker RR, Soldner F. et al. Notch signalling regulates stem cell numbers in vitro and in vivo. Nature. 2006;442:823-826
90. Tien AC, Rajan A, Bellen HJ. A Notch updated. J Cell Biol. 2009;184:621-629
91. Weng AP, Aster JC. Multiple niches for Notch in cancer: context is everything. Curr Opin Genet Dev. 2004;14:48-54
92. Bray SJ. Notch signalling: a simple pathway becomes complex. Nat Rev Mol Cell Biol. 2006;7:678-689
93. De Strooper B, Annaert W, Cupers P. et al. A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature. 1999;398:518-522
94. Davis RL, Turner DL. Vertebrate hairy and Enhancer of split related proteins: transcriptional repressors regulating cellular differentiation and embryonic patterning. Oncogene. 2001;20:8342-8357
95. Benedito R, Roca C, Sorensen I. et al. The notch ligands Dll4 and Jagged1 have opposing effects on angiogenesis. Cell. 2009;137:1124-1135
96. Siekmann AF, Lawson ND. Notch signalling and the regulation of angiogenesis. Cell Adh Migr. 2007;1:104-106
97. Suchting S, Freitas C, le Noble F. et al. The Notch ligand Delta-like 4 negatively regulates endothelial tip cell formation and vessel branching. Proc Natl Acad Sci U S A. 2007;104:3225-3230
98. Giovannini C, Lacchini M, Gramantieri L. et al. Notch3 intracellular domain accumulates in HepG2 cell line. Anticancer Res. 2006;26:2123-2127
99. Gramantieri L, Giovannini C, Lanzi A. et al. Aberrant Notch3 and Notch4 expression in human hepatocellular carcinoma. Liver Int. 2007;27:997-1007
100. Gao J, Song Z, Chen Y. et al. Deregulated expression of Notch receptors in human hepatocellular carcinoma. Dig Liver Dis. 2008;40:114-121
101. Fitzgerald K, Harrington A, Leder P. Ras pathway signals are required for notch-mediated oncogenesis. Oncogene. 2000;19:4191-4198
102. Stockhausen MT, Sjolund J, Axelson H. Regulation of the Notch target gene Hes-1 by TGFalpha induced Ras/MAPK signaling in human neuroblastoma cells. Exp Cell Res. 2005;310:218-228
103. Chappell WH, Green TD, Spengeman JD. et al. Increased protein expression of the PTEN tumor suppressor in the presence of constitutively active Notch-1. Cell Cycle. 2005;4:1389-1395
104. Qi R, An H, Yu Y. et al. Notch1 signaling inhibits growth of human hepatocellular carcinoma through induction of cell cycle arrest and apoptosis. Cancer Res. 2003;63:8323-8329
105. Wang C, Qi R, Li N. et al. Notch1 signaling sensitizes tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis in human hepatocellular carcinoma cells by inhibiting Akt/Hdm2-mediated p53 degradation and up-regulating p53-dependent DR5 expression. J Biol Chem. 2009;284:16183-16190
106. McMahon AP, Ingham PW, Tabin CJ. Developmental roles and clinical significance of hedgehog signaling. Curr Top Dev Biol. 2003;53:1-114
107. Bai LY, Chiu CF, Lin CW. et al. Differential expression of Sonic hedgehog and Gli1 in hematological malignancies. Leukemia. 2008;22:226-228
108. Villavicencio EH, Walterhouse DO, Iannaccone PM. The sonic hedgehog-patched-gli pathway in human development and disease. Am J Hum Genet. 2000;67:1047-1054
109. Ingham PW, McMahon AP. Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 2001;15:3059-3087
110. Lum L, Beachy PA. The Hedgehog response network: sensors, switches, and routers. Science. 2004;304:1755-1759
111. Alcedo J, Noll M. Hedgehog and its patched-smoothened receptor complex: a novel signalling mechanism at the cell surface. Biol Chem. 1997;378:583-590
112. Koebernick K, Pieler T. Gli-type zinc finger proteins as bipotential transducers of Hedgehog signaling. Differentiation. 2002;70:69-76
113. Aza-Blanc P, Kornberg TB. Ci: a complex transducer of the hedgehog signal. Trends Genet. 1999;15:458-462
114. Dai P, Akimaru H, Tanaka Y. et al. Sonic Hedgehog-induced activation of the Gli1 promoter is mediated by GLI3. J Biol Chem. 1999;274:8143-8152
115. Wang B, Fallon JF, Beachy PA. Hedgehog-regulated processing of Gli3 produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell. 2000;100:423-434
116. Litingtung Y, Dahn RD, Li Y. et al. Shh and Gli3 are dispensable for limb skeleton formation but regulate digit number and identity. Nature. 2002;418:979-983
117. Sicklick JK, Li YX, Jayaraman A. et al. Dysregulation of the Hedgehog pathway in human hepatocarcinogenesis. Carcinogenesis. 2006;27:748-757
118. Huang S, He J, Zhang X. et al. Activation of the hedgehog pathway in human hepatocellular carcinomas. Carcinogenesis. 2006;27:1334-1340
119. Park IK, Qian D, Kiel M. et al. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature. 2003;423:302-305
120. Valk-Lingbeek ME, Bruggeman SW, van Lohuizen M. Stem cells and cancer; the polycomb connection. Cell. 2004;118:409-418
121. Leung C, Lingbeek M, Shakhova O. et al. Bmi1 is essential for cerebellar development and is overexpressed in human medulloblastomas. Nature. 2004;428:337-341
122. Sasaki M, Ikeda H, Itatsu K. et al. The overexpression of polycomb group proteins Bmi1 and EZH2 is associated with the progression and aggressive biological behavior of hepatocellular carcinoma. Lab Invest. 2008;88:873-882
123. Chiba T, Zheng YW, Kita K. et al. Enhanced self-renewal capability in hepatic stem/progenitor cells drives cancer initiation. Gastroenterology. 2007;133:937-950
124. Chiba T, Miyagi S, Saraya A. et al. The polycomb gene product BMI1 contributes to the maintenance of tumor-initiating side population cells in hepatocellular carcinoma. Cancer Res. 2008;68:7742-7749
125. Chiba T, Seki A, Aoki R. et al. Bmi1 promotes hepatic stem cell expansion and tumorigenicity in both Ink4a/Arf-dependent and -independent manners in mice. Hepatology. 2010;52:1111-1123
126. Gao B. Cytokines, STATs and liver disease. Cell Mol Immunol. 2005;2:92-100
127. Yeoh GC, Ernst M, Rose-John S. et al. Opposing roles of gp130-mediated STAT-3 and ERK-1/ 2 signaling in liver progenitor cell migration and proliferation. Hepatology. 2007;45:486-494
128. Lam SP, Luk JM, Man K. et al. Activation of interleukin-6-induced glycoprotein 130/signal transducer and activator of transcription 3 pathway in mesenchymal stem cells enhances hepatic differentiation, proliferation, and liver regeneration. Liver Transpl. 2010;16:1195-1206
129. Hing HK, Sun X, Artavanis-Tsakonas S. Modulation of wingless signaling by Notch in Drosophila. Mech Dev. 1994;47:261-268
130. Maloof JN, Whangbo J, Harris JM. et al. A Wnt signaling pathway controls hox gene expression and neuroblast migration in C. elegans. Development. 1999;126:37-49
131. Hooper JE. Distinct pathways for autocrine and paracrine Wingless signalling in Drosophila embryos. Nature. 1994;372:461-464
132. Katoh M. WNT signaling pathway and stem cell signaling network. Clin Cancer Res. 2007;13:4042-4045
133. Katoh M. Network of WNT and Other Regulatory Signaling Cascades in Pluripotent Stem Cells and Cancer Stem Cells. Curr Pharm Biotechnol. 2011;12:160-170
134. Budhu A, Jia HL, Forgues M. et al. Identification of metastasis-related microRNAs in hepatocellular carcinoma. Hepatology. 2008;47:897-907
135. Coulouarn C, Factor VM, Andersen JB. et al. Loss of miR-122 expression in liver cancer correlates with suppression of the hepatic phenotype and gain of metastatic properties. Oncogene. 2009;28:3526-3536
136. Pineau P, Volinia S, McJunkin K. et al. miR-221 overexpression contributes to liver tumorigenesis. Proc Natl Acad Sci U S A. 2010;107:264-269
137. Ma S, Tang KH, Chan YP. et al. miR-130b Promotes CD133(+) liver tumor-initiating cell growth and self-renewal via tumor protein 53-induced nuclear protein 1. Cell Stem Cell. 2010;7:694-707
138. Wong QW, Lung RW, Law PT. et al. MicroRNA-223 is commonly repressed in hepatocellular carcinoma and potentiates expression of Stathmin1. Gastroenterology. 2008;135:257-269
139. Wong QW, Ching AK, Chan AW. et al. MiR-222 overexpression confers cell migratory advantages in hepatocellular carcinoma through enhancing AKT signaling. Clin Cancer Res. 2010;16:867-875
140. Ambros V, Horvitz HR. Heterochronic mutants of the nematode Caenorhabditis elegans. Science. 1984;226:409-416
141. Moss EG, Lee RC, Ambros V. The cold shock domain protein LIN-28 controls developmental timing in C. elegans and is regulated by the lin-4 RNA. Cell. 1997;88:637-646
142. Yamanaka S. Pluripotency and nuclear reprogramming. Philos Trans R Soc Lond B Biol Sci. 2008;363:2079-2087
143. Viswanathan SR, Powers JT, Einhorn W. et al. Lin28 promotes transformation and is associated with advanced human malignancies. Nat Genet. 2009;41:843-848
144. Viswanathan SR, Daley GQ, Gregory RI. Selective blockade of microRNA processing by Lin28. Science. 2008;320:97-100
145. Chang TC, Zeitels LR, Hwang HW. et al. Lin-28B transactivation is necessary for Myc-mediated let-7 repression and proliferation. Proc Natl Acad Sci U S A. 2009;106:3384-3389
146. Wang YC, Chen YL, Yuan RH. et al. Lin-28B expression promotes transformation and invasion in human hepatocellular carcinoma. Carcinogenesis. 2010;31:1516-1522
147. Guo Y, Chen Y, Ito H. et al. Identification and characterization of lin-28 homolog B (LIN28B) in human hepatocellular carcinoma. Gene. 2006;384:51-61
148. Iliopoulos D, Hirsch HA, Struhl K. An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell. 2009;139:693-706
149. Ji J, Yamashita T, Budhu A. et al. Identification of microRNA-181 by genome-wide screening as a critical player in EpCAM-positive hepatic cancer stem cells. Hepatology. 2009;50:472-480
150. Klonisch T, Wiechec E, Hombach-Klonisch S. et al. Cancer stem cell markers in common cancers - therapeutic implications. Trends Mol Med. 2008;14:450-460
151. Dingli D, Michor F. Successful therapy must eradicate cancer stem cells. Stem Cells. 2006;24:2603-2610
152. O'Brien CA, Kreso A, Jamieson CH. Cancer stem cells and self-renewal. Clin Cancer Res. 2010;16:3113-3120
153. Alison MR, Lim SM, Nicholson LJ. Cancer stem cells: problems for therapy? J Pathol. 2010;223:148-162
154. Zeng G, Apte U, Cieply B. et al. siRNA-mediated beta-catenin knockdown in human hepatoma cells results in decreased growth and survival. Neoplasia. 2007;9:951-959
155. Li Y, Lu W, King TD. et al. Dkk1 stabilizes Wnt co-receptor LRP6: implication for Wnt ligand-induced LRP6 down-regulation. PLoS One. 2010;5:e11014
156. Huang SM, Mishina YM, Liu S. et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature. 2009;461:614-620
157. Kurtz JE, Dufour P. Adecatumumab: an anti-EpCAM monoclonal antibody, from the bench to the bedside. Expert Opin Biol Ther. 2010;10:951-958
158. Gires O, Bauerle PA. EpCAM as a target in cancer therapy. J Clin Oncol. 2010;28:e239-240
159. Stanton BZ, Peng LF. Small-molecule modulators of the Sonic Hedgehog signaling pathway. Mol Biosyst. 2010;6:44-54
160. Wang Q, Huang S, Yang L. et al. Down-regulation of Sonic hedgehog signaling pathway activity is involved in 5-fluorouracil-induced apoptosis and motility inhibition in Hep3B cells. Acta Biochim Biophys Sin (Shanghai). 2008;40:819-829
161. Eichenmuller M, Gruner I, Hagl B. et al. Blocking the hedgehog pathway inhibits hepatoblastoma growth. Hepatology. 2009;49:482-490
162. Rountree CB, Ding W, He L. et al. Expansion of CD133-expressing liver cancer stem cells in liver-specific phosphatase and tensin homolog deleted on chromosome 10-deleted mice. Stem Cells. 2009;27:290-299
163. Tang Y, Kitisin K, Jogunoori W. et al. Progenitor/stem cells give rise to liver cancer due to aberrant TGF-beta and IL-6 signaling. Proc Natl Acad Sci U S A. 2008;105:2445-2450
164. Lee TK, Castilho A, Ma S. et al. Liver cancer stem cells: implications for a new therapeutic target. Liver Int. 2009;29:955-965
165. Yin C, Lin Y, Zhang X. et al. Differentiation therapy of hepatocellular carcinoma in mice with recombinant adenovirus carrying hepatocyte nuclear factor-4alpha gene. Hepatology. 2008;48:1528-1539
166. Lim R, Knight B, Patel K. et al. Antiproliferative effects of interferon alpha on hepatic progenitor cells in vitro and in vivo. Hepatology. 2006;43:1074-1083
167. Yamashita T, Honda M, Nio K. et al. Oncostatin m renders epithelial cell adhesion molecule-positive liver cancer stem cells sensitive to 5-Fluorouracil by inducing hepatocytic differentiation. Cancer Res. 2010;70:4687-4697
168. Suetsugu A, Nagaki M, Aoki H. et al. Characterization of CD133+ hepatocellular carcinoma cells as cancer stem/progenitor cells. Biochem Biophys Res Commun. 2006;351:820-824
169. Yin S, Li J, Hu C. et al. CD133 positive hepatocellular carcinoma cells possess high capacity for tumorigenicity. Int J Cancer. 2007;120:1444-1450
170. Ma S, Lee TK, Zheng BJ. et al. CD133+ HCC cancer stem cells confer chemoresistance by preferential expression of the Akt/PKB survival pathway. Oncogene. 2008;27:1749-1758
171. Smith LM, Nesterova A, Ryan MC. et al. CD133/prominin-1 is a potential therapeutic target for antibody-drug conjugates in hepatocellular and gastric cancers. Br J Cancer. 2008;99:100-109
172. Zhu Z, Hao X, Yan M, Yao M, Ge C, Gu J. et al. Cancer stem/progenitor cells are highly enriched in CD133+CD44+ population in hepatocellular carcinoma. Int J Cancer. 2010;126:2067-2078
173. Terris B, Cavard C, Perret C. EpCAM, a new marker for cancer stem cells in hepatocellular carcinoma. J Hepatol. 2010;52:280-281
174. Haraguchi N, Ishii H, Mimori K. et al. CD13 is a therapeutic target in human liver cancer stem cells. J Clin Invest. 2010;120:3326-3339
175. Lau CK, Yang ZF, Ho DW. et al. An Akt/hypoxia-inducible factor-1alpha/platelet-derived growth factor-BB autocrine loop mediates hypoxia-induced chemoresistance in liver cancer cells and tumorigenic hepatic progenitor cells. Clin Cancer Res. 2009;15:3462-3471
176. Rosmorduc O, Housset C. Hypoxia: a link between fibrogenesis, angiogenesis, and carcinogenesis in liver disease. Semin Liver Dis. 2010;30:258-270
177. Liu L, Ren ZG, Shen Y. et al. Influence of hepatic artery occlusion on tumor growth and metastatic potential in a human orthotopic hepatoma nude mouse model: relevance of epithelial-mesenchymal transition. Cancer Sci. 2010;101:120-128
178. Wu XZ, Xie GR, Chen D. Hypoxia and hepatocellular carcinoma: The therapeutic target for hepatocellular carcinoma. J Gastroenterol Hepatol. 2007;22:1178-1182
179. Patsenker E, Popov Y, Stickel F. et al. Pharmacological inhibition of integrin alphavbeta3 aggravates experimental liver fibrosis and suppresses hepatic angiogenesis. Hepatology. 2009;50:1501-1511
180. Semela D, Dufour JF. Angiogenesis and hepatocellular carcinoma. J Hepatol. 2004;41:864-880
181. Folkman J. Fundamental concepts of the angiogenic process. Curr Mol Med. 2003;3:643-651
182. Roberts LR, Gores GJ. Emerging drugs for hepatocellular carcinoma. Expert Opin Emerg Drugs. 2006;11:469-487
183. Siegel AB, Cohen EI, Ocean A. et al. Phase II trial evaluating the clinical and biologic effects of bevacizumab in unresectable hepatocellular carcinoma. J Clin Oncol. 2008;26:2992-2998
184. Whittaker S, Marais R, Zhu AX. The role of signaling pathways in the development and treatment of hepatocellular carcinoma. Oncogene. 2010;29:4989-5005
185. Wood JM, Bold G, Buchdunger E. et al. PTK787/ZK 222584, a novel and potent inhibitor of vascular endothelial growth factor receptor tyrosine kinases, impairs vascular endothelial growth factor-induced responses and tumor growth after oral administration. Cancer Res. 2000;60:2178-2189
186. Wedge SR, Kendrew J, Hennequin LF. et al. AZD2171: a highly potent, orally bioavailable, vascular endothelial growth factor receptor-2 tyrosine kinase inhibitor for the treatment of cancer. Cancer Res. 2005;65:4389-4400
187. Drevs J, Siegert P, Medinger M. et al. Phase I clinical study of AZD2171, an oral vascular endothelial growth factor signaling inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2007;25:3045-3054
188. Presta LG, Chen H, O'Connor SJ. et al. Humanization of an anti-vascular endothelial growth factor monoclonal antibody for the therapy of solid tumors and other disorders. Cancer Res. 1997;57:4593-4599
189. Adnane L, Trail PA, Taylor I. et al. Sorafenib (BAY 43-9006, Nexavar), a dual-action inhibitor that targets RAF/MEK/ERK pathway in tumor cells and tyrosine kinases VEGFR/PDGFR in tumor vasculature. Methods Enzymol. 2006;407:597-612
190. Eisen T, Ahmad T, Flaherty KT. et al. Sorafenib in advanced melanoma: a Phase II randomised discontinuation trial analysis. Br J Cancer. 2006;95:581-586
191. Wilhelm SM, Carter C, Tang L. et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64:7099-7109
192. Wilhelm S, Carter C, Lynch M. et al. Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat Rev Drug Discov. 2006;5:835-844
193. Carlomagno F, Anaganti S, Guida T. et al. BAY 43-9006 inhibition of oncogenic RET mutants. J Natl Cancer Inst. 2006;98:326-334
194. Lierman E, Folens C, Stover EH. et al. Sorafenib is a potent inhibitor of FIP1L1-PDGFRalpha and the imatinib-resistant FIP1L1-PDGFRalpha T674I mutant. Blood. 2006;108:1374-1376
195. Mendel DB, Laird AD, Xin X. et al. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res. 2003;9:327-337
196. Chow LQ, Eckhardt SG. Sunitinib: from rational design to clinical efficacy. J Clin Oncol. 2007;25:884-896
197. Cai ZW, Zhang Y, Borzilleri RM. et al. Discovery of brivanib alaninate ((S)-((R)-1-(4-(4-fluoro-2-methyl-1H-indol-5-yloxy)-5-methylpyrrolo[2,1-f] [1,2,4]triazin-6-yloxy)propan-2-yl)2-aminopropanoate), a novel prodrug of dual vascular endothelial growth factor receptor-2 and fibroblast growth factor receptor-1 kinase inhibitor (BMS-540215). J Med Chem. 2008;51:1976-1980
198. Huynh H, Ngo VC, Fargnoli J. et al. Brivanib alaninate, a dual inhibitor of vascular endothelial growth factor receptor and fibroblast growth factor receptor tyrosine kinases, induces growth inhibition in mouse models of human hepatocellular carcinoma. Clin Cancer Res. 2008;14:6146-6153
199. Albert DH, Tapang P, Magoc TJ. et al. Preclinical activity of ABT-869, a multitargeted receptor tyrosine kinase inhibitor. Mol Cancer Ther. 2006;5:995-1006
200. Grunwald V, Hidalgo M. Developing inhibitors of the epidermal growth factor receptor for cancer treatment. J Natl Cancer Inst. 2003;95:851-867
201. Thomas MB, Morris JS, Chadha R. et al. Phase II trial of the combination of bevacizumab and erlotinib in patients who have advanced hepatocellular carcinoma. J Clin Oncol. 2009;27:843-850
202. Johnston SR, Leary A. Lapatinib: a novel EGFR/HER2 tyrosine kinase inhibitor for cancer. Drugs Today (Barc). 2006;42:441-453
203. Ramanathan RK, Belani CP, Singh DA. et al. A phase II study of lapatinib in patients with advanced biliary tree and hepatocellular cancer. Cancer Chemother Pharmacol. 2009;64:777-783
204. Matsuo M, Sakurai H, Saiki I. ZD1839, a selective epidermal growth factor receptor tyrosine kinase inhibitor, shows antimetastatic activity using a hepatocellular carcinoma model. Mol Cancer Ther. 2003;2:557-561
205. Okano J, Matsumoto K, Nagahara T. et al. Gefitinib and the modulation of the signaling pathways downstream of epidermal growth factor receptor in human liver cancer cells. J Gastroenterol. 2006;41:166-176
206. Ueda S, Basaki Y, Yoshie M. et al. PTEN/Akt signaling through epidermal growth factor receptor is prerequisite for angiogenesis by hepatocellular carcinoma cells that is susceptible to inhibition by gefitinib. Cancer Res. 2006;66:5346-5353
207. Hopfner M, Sutter AP, Huether A. et al. Targeting the epidermal growth factor receptor by gefitinib for treatment of hepatocellular carcinoma. J Hepatol. 2004;41:1008-1016
208. Huether A, Hopfner M, Sutter AP. et al. Erlotinib induces cell cycle arrest and apoptosis in hepatocellular cancer cells and enhances chemosensitivity towards cytostatics. J Hepatol. 2005;43:661-669
209. Zhu AX. Systemic therapy of advanced hepatocellular carcinoma: how hopeful should we be? Oncologist. 2006;11:790-800
210. Shimizu J, Yamazaki S, Sakaguchi S. Induction of tumor immunity by removing CD25+CD4+ T cells: a common basis between tumor immunity and autoimmunity. J Immunol. 1999;163:5211-5218
211. Shimizu K, Fields RC, Giedlin M. et al. Systemic administration of interleukin 2 enhances the therapeutic efficacy of dendritic cell-based tumor vaccines. Proc Natl Acad Sci U S A. 1999;96:2268-2273
212. Golgher D, Jones E, Powrie F. et al. Depletion of CD25+ regulatory cells uncovers immune responses to shared murine tumor rejection antigens. Eur J Immunol. 2002;32:3267-3275
213. Ghiringhelli F, Larmonier N, Schmitt E. et al. CD4+CD25+ regulatory T cells suppress tumor immunity but are sensitive to cyclophosphamide which allows immunotherapy of established tumors to be curative. Eur J Immunol. 2004;34:336-344
214. Hoechst B, Voigtlaender T, Ormandy L. et al. Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology. 2009;50:799-807
215. Donnenberg VS, Donnenberg AD. Multiple drug resistance in cancer revisited: the cancer stem cell hypothesis. J Clin Pharmacol. 2005;45:872-877
216. Hirschmann-Jax C, Foster AE, Wulf GG. et al. A distinct "side population" of cells with high drug efflux capacity in human tumor cells. Proc Natl Acad Sci U S A. 2004;101:14228-14233
217. Zhou S, Schuetz JD, Bunting KD. et al. The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat Med. 2001;7:1028-1034
218. Li B, Ye T, Zhao L, Li DH. et al. Effects of multidrug resistance, antisense RNA on the chemosensitivity of hepatocellular carcinoma cells. Hepatobiliary Pancreat Dis Int. 2006;5:552-559
219. Cheung ST, Cheung PF, Cheng CK. et al. Granulin-epithelin precursor and ATP-dependent binding cassette (ABC)B5 regulate liver cancer cell chemoresistance. Gastroenterology. 2010;140:344-355
220. Baumann M, Krause M, Hill R. Exploring the role of cancer stem cells in radioresistance. Nat Rev Cancer. 2008;8:545-554
221. Horinouchi H, Yamamoto H, Komatsu T. et al. Enhanced radiation response of a solid tumor with the artificial oxygen carrier 'albumin-heme'. Cancer Sci. 2008;99:1274-1278
Author contact
![]() Corresponding author: Xin Wei Wang, National Cancer Institute, 37 Convent Drive, Building 37, Room 3044A, Bethesda, MD 20892-4258. E-mail: xw3ugov; fax: 301-496-0497
Corresponding author: Xin Wei Wang, National Cancer Institute, 37 Convent Drive, Building 37, Room 3044A, Bethesda, MD 20892-4258. E-mail: xw3ugov; fax: 301-496-0497