Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Current ADT acts at the...
Alternative approaches to...
Post-receptor androgen signaling...
Development of post-receptor ADT...
Mounting evidence supports...
Limitations to clinical...
Conclusions
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2014; 10(6):576-587. doi:10.7150/ijbs.8479 This issue Cite
Review
Targeting Androgen Receptor Action for Prostate Cancer Treatment: Does the Post-Receptor Level Provide Novel Opportunities?
Hannelore V. Heemers ![]()
Departments of Urology and Cancer Genetics, Roswell Park Cancer Institute, Buffalo, NY 14263, USA.
Received 2014-1-2; Accepted 2014-1-23; Published 2014-6-1
Abstract
The standard of care for patients who suffer from non-organ confined prostate cancer (CaP) is androgen deprivation therapy (ADT). ADT exploits the reliance of CaP cells on androgen receptor (AR) signaling throughout CaP progression from androgen-stimulated (AS) to castration-recurrent (CR) disease. AR is a member of the nuclear receptor family of ligand-activated transcription factors. Ligand-activated AR relocates from the cytoplasm to the nucleus, where it binds to Androgen Response Elements (AREs) to regulate transcription of target genes that control CaP cell behavior and progression. Current forms of ADT interfere at 2 levels along the AR signaling axis. At the pre-receptor level, ADT limits the availability of ligand for AR, while at the receptor level, ADT interrupts AR-ligand interactions. Both forms of ADT induce remission, but are not curative and, because of extraprostatic actions, are associated with severe side effects. Here, the potential of interference with the molecular regulation of AR-dependent transcription and the action of AR target genes, at the post receptor level, as the foundation for the development of novel, more CaP- specific selective forms of ADT is explored.
Keywords: castration, androgen deprivation therapy, gene expression, transcription, hormones.
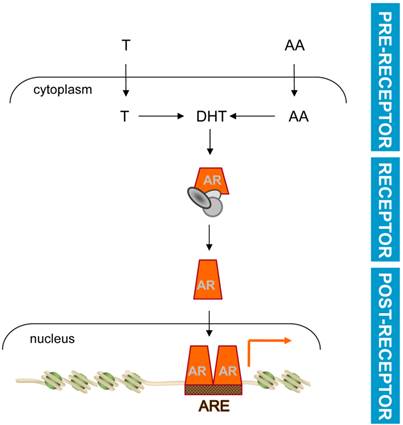
Current ADT acts at the pre-receptor and the receptor level
Ever since the Noble prize-winning findings of Huggins and Hodges [1], now more than 70 years ago, ADT has been the cornerstone for the treatment of advanced CaP. More detailed overviews of available ADT approaches have been described before [2-4] and are the topic of expert contributions to this issue of The International Journal of Biological Sciences. This manuscript will limit itself to a brief historical overview of the evolution of ADT and the perception of the value of androgen signaling as a target for therapy in CaP. Since the recognition that androgen deprivation alleviates symptoms and disease progression in metastatic CaP patients, ADT has focused initially on preventing the synthesis of testosterone, the main androgen in circulation, by testicular Leydig cells [2, 5]. Testosterone is converted into its more bioactive metabolite dihydrotestosterone (DHT) by 5-alpha-reductases in peripheral tissues [6-8]. First surgical castration or oestrogen administration, and then medical castration using GNRH agonists or antagonists, decreased testosterone levels as a source for DHT. This means of androgen ablation can be used by itself or in combination with steroidal (e.g.cyproterone acetate) or non-steroidal (e.g.bicalutamide) anti-androgens. Following the appreciation that a cellular receptor for androgens exists [9-11], the latter compounds were developed to compete with androgens for binding to, and thereby activating, AR [2,4]. These forms of ADT cause CaP remission, but are not curative and almost invariably a “castration-recurrent” (CR) form of CaP emerges during ADT [12]. Steroid production by the adrenal glands is responsible for 5 to 10 percent of androgen precursors in the bloodstream, which are processed further to DHT by a series of enzymatic steps in the periphery. This knowledge served as an impetus to explore whether adrenal androgen synthesis is a therapeutic target in cases in which gonadal androgen synthesis blockade has failed [2,4]. Bilateral adrenalectomy or hypophysectomy represented a major surgical intervention to remove the source or the feedback mechanisms that regulate adrenal androgen synthesis of androgens. Alternatively, drugs such as ketoconazole and aminogluthetimide pharmacologically inhibited adrenal precursor production. Whereas surgeries were complex and associated with considerable morbidity, responses to pharmacological intervention were so short-lived that they were measured in days, and were accompanied also by significant side effects. As a consequence, at that time it was generally accepted that AR had lost its value as a therapeutic target for CR-CaP, and treatment shifted to non-AR targeting therapies such as chemotherapy [13-17].
Around the early 2000s, several major paradigm-shifting findings occurred almost simultaneously, which led androgen signaling to become the prime focus for therapeutic intervention in CaP that has failed primary ADT. A first breakthrough was the appreciation that AR is expressed highly in CR-CaP at levels that are similar to, and may exceed those seen in androgen-stimulated (AS)-CaP or benign prostate epithelium [18,19]. Observations of elevated AR levels in CR-CaP patients samples were recapitulated in CaP model systems [20], where silencing or inactivation of AR inhibited CR-CaP cell proliferation [21,22]. Taken together, these reports pointed to a critical role for AR in the growth of CaP that had recurred during ADT and provided a rationale to start screening for novel and more efficient methods to inhibit AR activity for CR-CaP treatment. Overexpression of AR in AS-CaP cells was sufficient to convert an AR antagonist into an agonist [20], providing both a mechanistic explanation for failure of ADT and a basis for a search for novel, second-generation antiandrogens with stronger affinity for AR than their predecessors using AR-overexpressing model systems. These studies resulted in isolation of enzalutamide (formerly known as MDV-3100), a novel and more potent AR antagonist [23]. At the same time, exploration of the cause(s) for AR overexpression in CR-CaP led investigators to look into the possibility that intratumoral ligands are present that stabilize AR. CR-CaP clinical specimens were shown to contain levels of testosterone that are similar to those found in normal prostate and AS-CaP, and levels of DHT, that albeit lower than those in AS-CaP, were still in the nM range, which is sufficient to activate AR [24-27]. These findings provided evidence for intracrine androgen synthesis in CR-CaP and rekindled interest in the development of more efficient drugs to interfere with intratumoral production of DHT, either from adrenal precursors, or from cholesterol. Ultimately, these efforts led to the redeployment of abiraterone acetate for CR-CaP treatment. Abiraterone acetate is a more specific inhibitor of CYP17A1, a key enzyme involved in the generation of androgens, than ketoconazole, and was first evaluated for CaP treatment in the late 1990s until its clinical development was discontinued, in part due to concerns of significant side effects (reviewed in [4]).
Alternative approaches to inhibits AR's action on CaP cells are needed
A decade after the initial observations of AR's sustained relevance for treatment of CR-CaP, these insights have led to FDA-approval of 2 novel ADT agents: abiraterone acetate and enzalutamide. Both drugs have shown survival benefits in phase III clinical trials [28-30] and have entered rapidly day-to-day clinical practice, where they are part of a growing arsenal of ADT agents to treat advanced CaP. Despite these successes, it is becoming increasingly clear that CR-CaP remission in response to secondary ADT is relatively short-lived, lasting only a few months. Moreover, the majority of CaP that recurs during treatment with abiraterone acetate or enzalutamide continues to rely on AR action and remains responsive to further manipulation of androgen signaling axis [31-34]. As a consequence, there is great interest in how best to administer these ADT agents and how to enhance the efficacy of current ADT to prolong survival. The impact of timing, sequencing and/or combination of these agents (with other CaP treatments) on CaP progression is under intense investigation. In September of 2013, the National Institute of Health's Clinical Trial site listed 69 and 21 clinical trials addressing these questions for abiraterone acetate and enzalutamide, respectively, in CaP. More and novel compounds are being tested also for second-line ADT. Newer drugs that have fewer side effects (TAK-700) [35, 36] or combine blockade of CYP17A1 with anti-androgen activity and AR degradation (Tok-001) [37] may provide alternatives for abiraterone acetate. Other next-generation antiandrogens such as ARN-509 have passed phase I clinical trials [38]. In addition, considerable efforts are directed towards understanding and targeting the molecular underpinnings of CaP's resistance to second generation ADT. Available evidence indicates that the mechanisms by which AR activity reemerges during second-line ADT are similar to those associated with reawakened androgen signaling upon failure of first-line ADT. Alterations in steroidogenic gene expression, for instance amplification of CYP17A1 expression in CaP cells under abiraterone acetate treatment, indicate active intracrine androgen synthesis despite the selective pressure of pre-receptor ADT [31, 32]. These observations are reminiscent of changes in expression levels of several steroidogenic enzymes that occur under first-line ADT [39]. Previously, substantial interpatient heterogeneity and distinct patterns of coordinated aberrant steroidogenic enzyme expression have been described in CR-CaP tissues [40]. Dysregulated expression profiles of enzymes involved in androgen metabolism in CaP indicates flexibility in steroidogenic pathways that can lead to intratumoral DHT production. Steroidogenic enzymes such 17β-hydroxysteroid dehydrogenase 6 [41] and 3β-hydroxysteroid dehydrogenase type 1 (3βHSD1) [42], previously thought to be of a lesser overall importance, are now gaining momentum as potential new targets for therapy. Moreover, whole exome sequencing approaches are revealing recurring somatic mutations in steroidogenic enzymes in CaP that has failed androgen ablation, and such mutations are present under first-line and second-line ADT [43-45]. Functional consequences of such mutations have been highlighted recently by reports of a gain-of-stability mutation that leads to a gain-of-function in 3βHSD1 [45]. 3βHSD1 catalyzes the initial rate-limiting step in conversion of the adrenal-derived steroid dehydroepiandrosterone (DHEA) to DHT. The mutation does not affect the enzyme's catalytic function, but renders it resistant to degradation. Accumulation of mutant 3βHSD1 accelerates conversion of DHEA to DHT, which is usually very limited, and as such establishes a preference for flux of adrenal precursors through this specific androgen biosynthesis pathway. Events at the AR signaling axis that contribute to failure of ADT are not restricted to the pre-receptor level. In patients failing first-line ADT, an antiandrogen withdrawal syndrome that results from somatic AR mutations is not uncommon when treatment is stopped [46]. Recent literature reports of CaP cells that become resistant to second-line ADT using enzalutamide or ARN-509 because of novel acquired mutations in AR that convert these antagonists into agonists are reminiscent of this [47,48]. The escape from enzalutamide-dependent AR blockade has been attributed also to the induction of the expression of the glucocorticoid receptor (GR), which then acts as an alternate to AR and activates a similar but distinguishable set of target genes and maintains the resistant phenotype [49]. Furthermore, AR variants that lack a ligand binding domain because of splicing or gene rearrangement events and may activate AR target gene expression in androgen independent manner [50] emerge also under newer forms of pre-receptor and receptor level ADT [31,51]. Because of these and related findings, yet other means to target AR for CaP treatment are being explored. Such efforts are directed for instance at identifying and exploiting the differences in the AR protein interactome that is associated with wild-type or mutant AR, development of novel agents that interfere with the ligand-independent activity of AR [52,53], modulation of the degradation rather than synthesis of intratumoral androgens [54], or combined administration of an AR antagonist and an GR antagonist [49]. In the era of precision medicine, it has been proposed to tailor administration of these treatments based on a CaP's expression status of (mutant) AR, pattern of expression or structural integrity of steroidogenic enzymes. Based on available evidence of AR-dependent CaP progression under current ADT and the pattern of disease progression under other targeted therapies, it is likely that AR- and androgen biosynthesis-targeting approaches, will fail eventually while CaP can continue to rely on androgen signaling. One can anticipate CaP to become a chronic disease, where following subsequent rounds of secondary, tertiary and even quaternary ADT truly androgen-independent CaP arises, for which kinases such as Src and Aurora A may emerge as more appropriate targets for therapy [55, 56]. In view of limitations to current ADTs, exploring the possibility of targeting androgen action at a different level of the AR signaling axis to prolong patients' survival and provide alternatives for current AR-targeting therapies is worthwhile.
Post-receptor androgen signaling as a basis for novel selective ADT
Common among current ADT approaches is a focus on preventing ligand availability for AR and/or ligand-AR interaction. These events precede and are required for AR's activity as a ligand-activated transcription factor. Consistent with a previous report [57], these events will be referred to as acting at the pre-receptor (ligand availability) and receptor (AR-ligand interaction) level, respectively, in the androgen signaling axis (Figure 1). Interference with AR's transcriptional output, which ultimately controls CaP cell behavior and progression, may be a viable approach to develop novel forms of ADT that act at the post-receptor level. Conceptually, post-receptor level ADT approaches are attractive because they can block selectively the CaP-specific segment(s) of androgen action that is responsible for progression to lethal CaP. In such a scenario, post-receptor ADT will induce remission while eliminating extra-prostatic side effects on the cardiovascular system, cognition, muscle and bone [58, 59] and will thus improve the quality of life for men receiving ADT. In addition, post-receptor ADT has the advantage that it can pool from a broader range of potential targets for therapy downstream of AR, which implies also more diversity in alternative forms of additional post-receptor ADT should resistance occur to such initial approaches. Development of post-receptor ADT hinges on molecular insights in the transcriptional regulation of AR target genes that are responsible for CaP progression to the lethal stage. One can envision such information to lead to at least 2 novel opportunities for interference with androgen action: first, therapeutically exploiting the molecular machinery responsible for transcription of clinically valuable AR target genes, and second, inhibiting the action of AR target genes that drive CaP progression.
Basic mechanism of androgen action. Testosterone (T) or adrenal androgens (AA) are converted to DHT in CaP cells. Ligand activation induces a conformational change and nuclear translocation of AR. Within the nucleus, AR binds AREs in target genes to regulate their transcription and modulate CaP cell behavior. Pre-receptor level ADT interferes with AR ligand production. Receptor level ADT impedes AR-ligand interaction. Post-receptor level ADT exploits AR-dependent action downstream of AR for therapeutic intervention.
Development of post-receptor ADT requires insights in AR-ARE interaction
Pre-receptor and receptor level ADT were developed before the structure and function of AR had been resolved. The presence of a cellular receptor to which androgens bound was generally accepted [9-11], but reliable antibodies that allowed characterization of AR did not become available until the mid 1980s [60, 61]. The gene encoding AR was first cloned and sequenced in 1988 [62, 63]. Subsequently, AR was found to belong to the nuclear receptor superfamily that harbors also for instance GR. AR consists of the modular structure that is typical for nuclear receptors: an N-terminal domain (NTD) that harbors a ligand-independent transactivation function, a central DNA-binding domain (DBD) responsible for sequence-specific interaction with genomic DNA, and a C-terminal ligand-binding domain (LBD) through which agonists, and antagonists, interact with AR [64, 65]. AR's structure reflects the basic mechanism by which AR is activated to control expression of target genes. Following ligand binding to the LBD, activated AR relocates from the cytoplasm to the nucleus, where its DBD interacts with specific DNA sequences known as Androgen Response Elements (AREs). DNA-bound AR recruits a large number of functionally very diverse proteins, which include components of the basal transcriptional machinery, other transcription factors and coregulators. Combined activity of AR's LBD and NTD and these protein complexes then controls the output from the AR transcriptional complex [65, 66].
The AR's structure-function relationship and transcriptional role have been described before. Essential to AR's regulatory effect on expression of its target genes, the focus of this paper, is the interaction of its DBD with AREs. The AR DBD shows remarkable similarity to that of other nuclear receptors. It consists of 2 Zn fingers; the first is responsible for sequence-specific interaction between AR and AREs, while the second mediates homodimerization between 2 ARs. AR-recruiting AREs consist of 2 repeats of a typical hormone response element halfsite, 5'-TGTTCT-3', separated by 3 bases, to which AR binds as a homodimer [64, 65]. The resulting 15 bp element that makes up an ARE also bares striking resemblance to the genomic binding sites for other nuclear receptors. Although the basis for the specificity by which AR interacts with AREs has not been completely resolved, it is now clear that select residues in AR's second Zn finger and, at least to some extent, the orientation of the 2 half-sites within AREs are determining factors [67].
The first insights into AR-ARE interaction, some of which preceded the formal identification of AR, were derived from studies that used partially purified AR from rodent prostates or recombinant (rat) AR DBD in DNA cellulose competition assays or in vitro footprinting assays. Electrophoretic mobity shift assays and transactivation assays with reporter constructs driven by intact or mutant versions of the putative ARE (containing-regions) were done to functionally validate and characterize the ARE [68]. At that time, molecular insights in AR's interaction with AREs in target genes were limited because of the small number of androgen-regulated genes that were known. Typically, these were genes for which expression had been recognized to be androgen-dependent in rodent prostates, such as the C3 component (C3) of prostatic binding protein (PBP), sex limited protein (slp) or secretory protein (sc). The corresponding AREs were identified predominantly in regions surrounding the gene's transcriptional start sites such as promoters and first introns [69]. Nonetheless, even under these experimental conditions it was becoming increasingly clear that variability exists in the manner in which ARE-driven transcription is regulated by AR. This variation was attributed to non-receptor events, more specifically to sequence composition of the regions adjacent to or in close proximity to ARE [70, 71]. These observations are reflected in the proposition of an Androgen Response Unit, which consists of the ARE(s) and surrounding DNA sequences to which other proteins bind and cooperate with AR to regulate ARE-driven transcription.
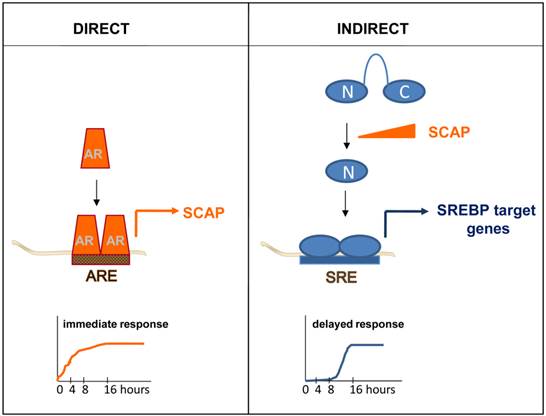
Appreciation of the full breath of this variability in AR-dependent transcription, and its relevance to CaP progression, required identification of more AR target genes. Over the years, individual genes that were androgen-regulated have been examined for the presence of AREs. For instance, segments spanning more than 100kb of the genomic sequence of the androgen-induced gene that encodes Sterol Regulatory Element Binding Protein (SREBP)-cleavage activating protein (SCAP) were cloned in front of a reporter gene. The androgen-responsiveness of the resulting reporter constructs was tested systematically in transactivation studies. Using this approach, an ARE mediating SCAP's androgen-responsiveness was identified and subsequently validated and characterized in intron 8 [72]. Studies like this gradually increased the number of AREs and provided initial evidence that locations of AREs do not have to be restricted to a gene's promoter region or surrounding areas. The development and advances in novel techniques allowed to screen for and identify in a more systemic manner and with increasing genomic-coverage androgen-responsive genes. First subtractive hybridization and differential display approaches, followed by (custom) cDNA array, oligoarray and RNA-Seq assays were applied. Typically, these studies examined RNA obtained from AR-positive CaP model systems that had been cultured in the presence and absence of androgens, or antiandrogens, or in which AR had been silencing or inactivated. These combined studies isolated hundreds of androgen-dependent genes and led to more fully appreciate the spectrum and role of androgen-responsive transcriptome in CaP. Pathway analyses on these gene profiles have identified functions in CaP cell proliferation, survival and differentiation (reviewed in [73]). An obvious limitation in using such profiling approaches as a means to identify ARE-driven genes is that these experiments do not always allow for an easy distinction between direct, ARE-driven, AR target genes and androgen-responsive genes for which androgen regulation is indirect. As a definition, direct target genes are androgen-responsive because of AR interaction with ARE(s) in its regulatory region(s) while indirect target genes become androgen-responsive because of the action of a direct AR target gene. The difference is nicely illustrated when androgen regulation of the gene encoding SCAP is compared to that of SREBP target genes. Androgen exposure leads to rapid increase in the expression of SCAP, which is the result of AR interaction with an ARE within intron 8 [72]. Induction of SCAP expression causes an imbalance in the regulatory protein complexes that normally keep SREBPs from being activated. The resulting proteolytic activation of SREBPs releases a transcriptionally active SREBP fragment that translocates to the cell nucleus where it binds to Sterol Regulatory Elements (SREs) in SREBP target genes. As a consequence, an entire SRE-driven lipogenic program is rendered androgen-responsive. Indirect mechanisms of androgen action, such as SREBPs' control over lipogenic gene expression, is characterized by delayed kinetics (Figure 2) and entails a significant amplification of the initiating androgenic signal to which CaP cells have been exposed [74].
Direct versus indirect androgen action. In a direct mechanism of androgen action, AR interacts with an ARE within a direct AR target gene, for instance the gene encoding SCAP. Androgen regulation of direct AR target genes is characterized by fast kinetics (left panel). Indirect mechanisms of androgen action are the result of androgen dependent changes in the expression of a direct AR target gene. For instance, androgen induction of SCAP results in proteolytic activation of precursor SREBPs. The N-terminal SREBP (N) fragment that is released translocates to the cell nucleus, where it binds SREs and activates a lipogenic program that is executed by SREBP target genes. Indirect mechanisms of androgen action are characterized by a delayed response to the androgenic stimulus (right panel).
Simple kinetics studies can thus provide first mechanistic insights in the manner of androgen regulation of genes of interest and serve as a first screen to tease out direct, ARE-driven genes. While useful, this approach does have some limitations. First, for a significant subset of androgen-dependent genes, the distinction between direct or indirect AR target genes is not made easily using available gene expression profiling assays. Quite often, androgen treatment is done for more than 16 hours, which makes it impossible to determine whether androgen regulation of gene expression is direct or indirect. Alternatively, in cases where cells are treated with androgens for shorter time points, it is likely that the fold androgen regulation of a large fraction of genes is modest and may not be captured fully because of detection or sensitivity limits of the screening assays used. None-the-less, a few more detailed kinetics studies have been done, which generated temporal patterns in which genes are classified as “late” or “early” [75, 76]. It is likely that the “early” gene lists are enriched in direct AR target genes. Second, an indication of direct regulation does not provide information on the existence or location of ARE(s), which is required to assess fully the molecular regulation of AR-ARE interaction.
The androgen-dependent recruitment patterns of AR throughout the genome have been the subject of several studies that combined an AR ChIP approach with high throughput microarray hybridization (ChIP-chip) or deep-sequencing (ChIP-Seq) [77]. In initial experiments, AR binding sites (ARBSs) were determined on a few chromosomes (21 and 22, or 19 and 20), on a restricted number of genomic regions selected by the ENCODE project or on selected promoter or other gene regions. Technology evolved to whole genome AR ChIP-chip and now AR ChIP-Seq, which significantly increased genome coverage to document genomic AR binding patterns (reviewed in [77]). Chip-Seq is rapidly replacing ChIP-chip and has the advantage that is covers the whole genome and is less prone to artefacts because of fewer amplification steps [78].
Collectively, insights from these studies show that the vast majority of ARBSs are located in intergenic and enhancer regions at considerable distance (>10 kb) from transcriptional start sites. ChIP approaches identify the genomic region to which AR binds, but do not pinpoint the precise AR recruitment motif. Sequence motif analysis of the ARBSs have confirmed enrichment for 6 bp and 15 bp motifs that resemble closely ARE half sites and AREs, respectively, in a significant portion of these binding peaks (e.g. [76,77,79,80]). Variability that was observed in the size of the 3 bp spacer between the 2 half sites has led to the proposal of non-canonical AREs [80]. However, mutational analysis and in silico analysis using a position-specific probability matrix based on well-characterized AREs have validated the previously established ARE consensus region and rules of DNA engagement by AR in a sample of selected AR binding peaks [69]. Recruitment of AR to genomic sequences that lack a recognizable ARE may indicate that AR can function as cofactor for other transactivating factors, as has been suggested before [81, 82]. Sequence analyses of ARBSs have demonstrated also enrichment for consensus binding motifs for secondary transcription factors (e.g. FoxA1, GATA-2, NF1, ETS1…) in close proximity to those AREs (e.g. [76,77,79,80]). These findings are in line with the previously proposed concept of an ARU. Conventional ChIP assays have validated interaction of AR and these TFs with their predicted DNA recognition motifs. Characterization of the contribution of secondary TFs to AR-dependent transcription provided further molecular insights in the flexibility, and complexity, of the regulatory process underling AR target gene expression. Physical and functional interaction between these TFs and AR occurred in an ARBS-selective and interdependent manner [79, 80, 83]. These studies yielded unexpected findings, for instance that TFs as FoxA1 appear to prevent AR from binding at certain sites. Deletion of FoxA1 induced changes in AR-dependent cistrome with androgen-dependent recruitment of AR to sites that were otherwise not occupied by AR [84-86]. This finding may be relevant to the clinical situation in view of somatic mutations in FoxA1 that are isolated in CaP patient specimens using NextGen sequencing approaches [44].
To date, thousands of ARBSs have been identified, and it is unclear whether all of these are equally important or even actively involved in AR-dependent transcription. Integration of complementary androgen-dependent gene expression profiles and ARBS patterns is being used to isolate the ARBSs associated with active AR-regulated transcription. An arbitrary cut-off distance between the location of the ARBS (for instance 25 kb) and the transcriptional start site(s) of an androgen-responsive gene is accepted often to indicate functional involvement of AR-recruiting regions in regulation of gene expression [76]. The overlap or close proximity of the ARBS peaks with epigenetic histone marks that have been associated with active AR-dependent transcription and/or the presence or RNA polymerase II at those sites serves to strengthen this assumption [76]. True validation of functional involvement of AR in regulation of androgen-dependence of these genes would require a form of homologous recombination in which AREs or TF binding sites are mutated selectively, but this seems not feasible in light of the large number of ARBSs and androgen-regulated genes identified already. In addition, such an approach would not address functional interactions between ARBSs that occur over long distances, involve chromosome looping and are of pivotal importance to androgen dependence of the affected genes [87]. As technology is continuously being refined, it is expected that the complex interplay between proteins recruited to cooperative regulatory elements within AR target genes will provide answers to these questions by application of for instance ChIA-PET [88]. Similarly, expansion of AR-centered studies to include also the contribution of AR-associated coregulators, which have been demonstrated to differentially and selectively regulate androgen-responsiveness of subsets of AR target genes [89-91], will provide further insights in the flexibility in regulation and composition of the AR transcriptional complex at individual ARBSs.
Mounting evidence supports feasibility of developing post-receptor level ADT
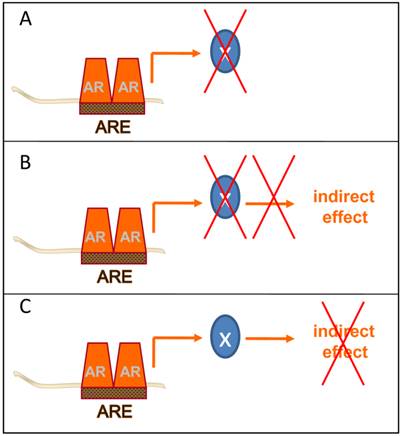
Ultimately, the goal of these AR-centered systems approaches-driven studies is to identify novel and clinically valuable targets downstream of AR that are actionable for therapeutic intervention. To isolate these targets, several groups have taken the approach to further integrate gene expression and genomic ARBS patterns with data from gene expression profiles derived from clinical CaP specimens. Cross-matching these diverse datasets aims to prioritize CaP target development to AR target genes that are enriched or depleted in CaP patient specimens and are critical to CaP progression. While still in its infancy, this method has yielded already several attractive targets and possible opportunities to interfere therapeutically at different points at the post-receptor level of the AR signaling axis (Figure 3).
Post-receptor level ADT takes different forms. Several methods of post-receptor ADT can be conceived. A few examples are provided. A. Inhibition of the immediate action of the direct AR target gene X with a key role in CaP progression. B. Inhibition of the action of the direct AR target gene X prevents activity of an indirect mechanism of androgen action with a critical role in CaP progression. C. Inhibition of the activity of an indirect mechanism of androgen action that drives CaP progression downstream of AR.
Interference with the immediate action of a direct AR target gene
Calcium/calmodulin-dependent protein kinase kinase 2 (CAMKK2) was identified as a direct AR target gene following integration of AR ChIP-Seq and gene expression profiling of CaP cell lines before and after androgen treatment. When AR target gene expression profiling was evaluated against multiple independent gene expression data sets from clinical prostate (cancer) specimens, CAMKK2 was consistently overexpressed in CaP. AR ChIP using chromatin from CaP patient specimens validated AR binding to CAMKK2 regulatory regions [76]. CAMKK2 plays a central role in the cellular metabolism, which has been identified as a seminal AR-dependent cell process in CaP [74, 92]. An inhibitor for CAMKK2, STO-609, has been developed and is readily available. Silencing of CAMKK2 expression or pharmacological inhibition of its action using STO-609 decreased proliferation of CaP cells and reduced growth of CR-CaP in xenograft models. Remarkably, no measurable effects of the CAMKK2 inhibitor on normal prostate size or cytoplasmic volume of prostatic epithelial cells were noted. On the other hand, castration resulted in loss of prostate size and atrophy of luminal epithelial cells [76]. Greater selective effects on cancer tissues by targeting the AR target gene CAMKK2 than by complete inhibition of AR action support the possibility to develop more specific post-receptor level ADT.
Interference with the action of a direct AR target gene to inhibit an indirect mechanism of androgen action
As discussed above, androgen induction of the direct AR target gene SCAP leads to coordinated androgen control over SREBP target genes [74]. SREBP is a key lipogenic transcription factor and central regulator of cellular lipid metabolism. SREBP controls expression level of genes involved in both fatty acid and cholesterol synthesis. Increased lipogenesis is a hallmark of many human cancers, including CaP, where SCAP overexpression and SREBP activation have been demonstrated. Increased lipogenesis in CaP correlates with poor prognosis and is considered an attractive target for novel therapies (reviewed in [74, 92]).
Compounds that prevent SREBP-dependent lipid synthesis may yield also a selective post-receptor ADT. Small molecules that modulate activity of the SCAP/SREBP pathway have been discovered [93, 94]. Most recently, fatostatin has been identified as a novel agent that inhibits SREBP action by targeting SCAP [94]. In a mouse model for obesity, where SREBP action is also actively studied for therapeutic intervention, fatostatin did not affect food intake, did not lead to toxicity and caused a minor reduction only in overall weight. Fatostatin did, however, lower serum glucose, cholesterol and LDL levels and lowered hepatic fat accumulation. The latter was accompanied by decreased expression of SREBP target genes including fatty acid synthase and 3-hydroxy-3-methylglutaryl-CoA reductase [94]. Noteworthy, these SRE-driven genes are overexpressed in CaP [74]. Effective shutdown of the fraction of androgen action mediated by SCAP may be particularly effective in preventing CaP progression as androgen synthesis from cholesterol may play a crucial role in emergence of CR-CaP [95, 96]. Interestingly, fatostatin suppressed serum-independent growth of DU145 CaP cells [94].
Interference with an indirect mechanism of androgen action
Yet an alternative approach to block clinically valuable androgen action is to interfere selectively with androgen-responsive gene expression that results from an indirect mechanism of androgen action. Recently, we have identified a novel indirect mode of AR action, in which androgen exposure activates the secondary transcription factor Serum Response Factor (SRF) [97, 98]. The SRF target gene signature that is rendered androgen-responsive because of this signaling event, was the first to be enriched in CaP compared to benign prostate and is sufficient to distinguish between benign and malignant prostate. SRF-dependent androgen action is associated with aggressive CaP and biochemical recurrence after radical prostatectomy and is active in AS- as well as CR-CaP. What makes this interesting from the perspective of therapeutic intervention is that androgen-responsiveness is conveyed to SRF via androgen activation of the small GTPase RhoA [99]. Activated RhoA recruits RhoA effectors to transfer signaling to downstream targets such as SRF. Actin polymerization resulting from RhoA effector action induces translocation of the critical SRF cofactor MKL1 to the cell nucleus where it is required for SRF dependent transcription [98, 99]. Inhibitors for RhoA effectors such as RhoA associated kinase are available and have been tested already in clinical trials for atherosclerosis and glaucoma (clinicaltrials.gov), and could be moved forward in CaP. In addition, CCG-1423, a small molecule that interferes with interaction between SRF and MKL1 has been tested in preclinical CaP model systems, where it blunts CaP cell proliferation and invasion [100, 101].
Limitations to clinical application of post-receptor level ADT
These examples demonstrate the preclinical promise of post-receptor level ADT using several agents, some of which have been shown to be well-tolerated in clinical trials. What are the most important limitations when considering successfully translating these findings from preclinical CaP models into the clinic?
First and most obviously, targeting AR function at the post-receptor level will be valuable for CaP treatment only if such an approach is directed against androgen action that is relevant to clinical CaP. Many studies have described in detail the transcriptional programs under control of AR, but used preclinical CaP models only, with a few selected AR binding sites ARBSs tested only in tissue [73-76, 79, 80, 102-107]. Application of the resulting cell-line derived AR target gene signatures to expression profiles from patient specimens has suggested loss of AR signaling in localized CaP compared to benign prostate [98, 108], and diminished AR activity in CR-CaP relative to AS-CaP [107, 108]. These findings contrast with observations from functional studies and the success of novel ADTs, which indicate that cell lines do not adequately model the AR transcriptional program in clinical CaP. This may be related in part to the experimental conditions chosen to isolate AR-dependent gene signatures from cell line models (discussed in [73]). On the other hand, it is also possible that the AR-dependent cistrome derived from CaP model systems is fundamentally different from that in clinical specimens. Only over the last year has it become technically feasible to obtain whole-genome datasets with high sensitivity for both AR recruitment to DNA and transcriptional changes associated with those recruitment events directly from clinical samples. For the first time, the existence of an in vivo restricted set of AR target genes and a tissue-specific transcriptional network in clinical CR-CaP samples that is not present in cell line models has been demonstrated [83]. These findings indicate context-dependence of AR function, which has significant implications for our understanding and evaluation of the role of AR in clinical CaP. These observations for CR-CaP are reminiscent also of previous reports on the expression level of cell line-derived AR target genes in AS-CaP. When evaluated at baseline and at different time points after neoadjuvant ADT, expression of many of these genes was not altered in radical prostatectomy samples from men with localized CaP [26], which suggests that similar context-dependence also may apply to AS-CaP.
Second, it is important to realize that the spectrum of AR target genes may evolve considerably during CaP progression. A growing body of evidence derived from isogenic CaP model systems indicates that the transcriptional program under direct control of AR and the AR-interacting proteome undergoes a shift during the transition from AS-CaP to CR-CaP [83, 106, 109]. This evolution is likely related to the amplification and overexpression of AR, the generation of AR variants that lack ligand-binding domain, and the introduction of mutations in AR that occur while CaP is subjected to ADT [47, 66, 110]. A systematic and unbiased comparison of genomic sites that are bound by wild-type, mutated or variant forms of AR has yet to be performed. The nature of the tissue microenvironment, which changes during disease progression, may have an important influence of AR function. It may be necessary to adjust post-receptor level ADT during CaP progression to address accurately such variation in AR activity.
Third, most, if not all, studies that have reported on AR target gene signatures in clinical CaP have overlooked the critically important fact that CaP is a heterogeneous disease. The majority of prostates that are removed by radical prostatectomy for CaP treatment with curative intent harbor multiple topographically separate and clonally distinct foci of CaP. These intraprostatic CaPs, of which 5 to 7 are present per radical prostatectomy specimen [111, 112], are independent clones that can develop at different times or simultaneously, harbor unique genetic mutations and are characterized by varying biological aggressiveness [111, 113-117]. Whether each of the individual CaPs conduct androgen metabolism in the same way or respond to different forms of ADT similarly is not known. The patterns of ARBSs and AR target gene expression between intraprostatic CaPs have not been investigated. Whether the individual CaPs are the same somatically as the patient is genomically, and the impact of mutations in regulators of androgen action on response to ADT remains to be resolved. In contrast to the multiclonal nature of localized CaP, analyses of disseminated, separate CaP samples from patients that succumbed to lethal metastatic CaP have indicated a monoclonal origin [118]. These findings suggest that a single intraprostatic CaP is responsible for development of aggressive and lethal disease, and that biological heterogeneity among different intraprostatic CaPs underlies variability in response to ADT. Heterogeneity among the 5-7 individual AS-CaP foci in the average radical prostatectomy specimens may need to be taken into account in the decision how best to administer post-receptor level ADT.
Conclusions
Ongoing investigation of the mechanisms by which AR exerts its effects on CaP cells is uncovering considerable variability and flexibility in the manner in which androgen-dependent gene expression is regulated. A growing body of evidence suggests that discrete signaling events downstream of ligand-activated AR may represent attractive targets for post-receptor level ADT. Further validation of AR-dependent signaling that show promise for therapeutic intervention in preclinical models using patient specimens and direct application of systems biology approaches to intraprostatic CaPs may lead to novel androgen ablation approaches that selectively prevent only those androgen-dependent events that drive CaP progression to the lethal stage.
Abbreviations
ADT: androgen deprivation therapy; AR: androgen receptor; ARBS: androgen receptor binding site; ARE: androgen response element; ARU: androgen response unit; AS: androgen-stimulated; C3: C3 component of prostatic binding protein; CAMKK2: calcium/calmodulin-dependent protein protein kinase 2; CaP: prostate cancer; ChIA-PET: chromatin interaction analysis by paired-end tag sequencing; ChIP: chromatin immunoprecipitation; ChIP-chip: ChIP on chip; ChIP-Seq: ChIP sequencing; CR: castration-recurrent; DBD: DNA-binding domain; DHEA: dehydroepiandrosterone; DHT: dihydrotestosterone; GNRH: gonadotrophin releasing hormone;GR: glucocorticoid receptor, FDA: food and drug administration; 3βHSD1: 3β-hydroxysteroid dehydrogenase type 1; LBD: ligand-binding domain; LDL: low density lipoprotein; MKL1: megakaryoblastic leukemia (translocation) 1; NTD: N-terminal domain, PBP: prostatic binding protein; SC: secretory component; SCAP: SREBP cleavage activating protein; SLP: sex-limiting protein; SRE: sterol-regulatory element; SREBP: sterol-regulatory element binding protein; SRF: serum response factor, TF: transcription factor
Acknowledgements
Supported by the Department of Defense Prostate Cancer Research Program and NCI grant P30 CA016056.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Huggins C, Hodges CV. Studies on prostatic cancer. I. The effect of castration, of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. Cancer Res. 1941;1:293-7
2. Denmeade SR, Isaacs JT. A history of prostate cancer treatment. Nat Rev Cancer. 2002;2:389-96 doi:10.1038/nrc801
3. Miyamoto H, Messing EM, Chang C. Androgen deprivation therapy for prostate cancer: current status and future prospects. Prostate. 2004;61:332-53 doi:10.1002/pros.20115
4. Lim AC, Attard G. Improved therapeutic targeting of the androgen receptor: rational drug design improves survival in castration-resistant prostate cancer. Curr Drug Targets. 2013;14:408-19
5. Schmidt LJ, Tindall DJ. Androgen receptor: past, present and future. Curr Drug Targets. 2013;14:401-7
6. Russell DW, Wilson JD.Steroid 5 alpha-reductase. two genes/two enzymes. Annu Rev Biochem. 1994;63:25-61
7. Uemura M, Tamura K, Chung S, Honma S, Okuyama A, Nakamura Y, Nakagawa H. Novel 5 alpha-steroid reductase (SRD5A3, type-3) is overexpressed in hormone-refractory prostate cancer. Cancer Sci. 2008;99:81-6
8. Godoy A, Kawinski E, Li Y, Oka D, Alexiev B, Azzouni F, Titus MA, Mohler JL.5α-reductase type 3 expression in human benign, malignant tissues. a comparative analysis during prostate cancer progression. Prostate. 2011;71:1033-46 doi: 10.1002/pros.21318
9. Anderson KM, Liao S. Selective retention of dihydrotestosterone by prostatic nuclei. Nature. 1968;219:277-9
10. Bruchovsky N, Wilson JD. The intranuclear binding of testosterone and 5-alpha-androstan-17-beta-ol-3-one by rat prostate. J Biol Chem. 1968;243:5953-60
11. Mainwaring WI. A soluble androgen receptor in the cytoplasm of rat prostate. J Endocrinol. 1969;45:531-41
12. Mohler JL. Castration-recurrent prostate cancer is not androgen-independent. Adv Exp Med Biol. 2008;617:223-34 doi:10.1007/978-0-387-69080-3_21
13. Tannock IF, Osoba D, Stockler MR, Ernst DS, Neville AJ, Moore MJ, Armitage GR, Wilson JJ, Venner PM, Coppin CM, Murphy KC. Chemotherapy with mitoxantrone plus prednisone or prednisone alone for symptomatic hormone-resistant prostate cancer: a Canadian randomized trial with palliative end points. J Clin Oncol. 1996Jun;14(6):1756-64
14. Kantoff PW, Halabi S, Conaway M, Picus J, Kirshner J, Hars V, Trump D, Winer EP, Vogelzang NJ. Hydrocortisone with or without mitoxantrone in men with hormone-refractory prostate cancer: results of the cancer and leukemia group B 9182 study. J Clin Oncol. 1999;17:2506-13
15. Hudes GR, Nathan F, Khater C, Haas N, Cornfield M, Giantonio B, Greenberg R, Gomella L, Litwin S, Ross E, Roethke S, McAleer C. Phase II trial of 96-hour paclitaxel plus oral estramustine phosphate in metastatic hormone-refractory prostate cancer. J Clin Oncol. 1997;15:3156-63
16. Hudes GR, Greenberg R, Krigel RL, Fox S, Scher R, Litwin S, Watts P, Speicher L, Tew K, Comis R. Phase II study of estramustine and vinblastine, two microtubule inhibitors, in hormone-refractory prostate cancer. J Clin Oncol. 1992;10:1754-61
17. Pienta KJ, Redman B, Hussain M, Cummings G, Esper PS, Appel C, Flaherty LE. Phase II evaluation of oral estramustine and oral etoposide in hormone-refractory adenocarcinoma of the prostate. J Clin Oncol. 1994;12:2005-12
18. van der Kwast TH, Schalken J, Ruizeveld de Winter JA, van Vroonhoven CC, Mulder E, Boersma W, Trapman J. Androgen receptors in endocrine-therapy-resistant human prostate cancer. Int J Cancer. 1991;48:189-93
19. Ford OH 3rd, Gregory CW, Kim D, Smitherman AB, Mohler JL. Androgen receptor gene amplification and protein expression in recurrent prostate cancer. J Urol. 2003;170:1817-21
20. Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, Rosenfeld MG, Sawyers CL. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10:33-9
21. Zegarra-Moro OL, Schmidt LJ, Huang H, Tindall DJ. Disruption of androgen receptor function inhibits proliferation of androgen-refractory prostate cancer cells. Cancer Res. 2002;62:1008-13
22. Cheng H, Snoek R, Ghaidi F, Cox ME, Rennie PS. Short hairpin RNA knockdown of the androgen receptor attenuates ligand-independent activation and delays tumor progression. Cancer Res. 2006;66:10613-20
23. Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V. et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324:787-90 doi:1168175 [pii] 10.1126/science.1168175
24. Mohler JL, Gregory CW, Ford OH 3rd, Kim D, Weaver CM, Petrusz P. et al. The androgen axis in recurrent prostate cancer. Clin Cancer Res. 2004;10:440-8
25. Titus MA, Schell MJ, Lih FB, Tomer KB, Mohler JL. Testosterone and dihydrotestosterone tissue levels in recurrent prostate cancer. Clin Cancer Res. 2005;11:4653-7 doi:10.1158/1078-0432.CCR-05-0525
26. Mostaghel EA, Page ST, Lin DW, Fazli L, Coleman IM, True LD. et al. Intraprostatic androgens and androgen-regulated gene expression persist after testosterone suppression: therapeutic implications for castration-resistant prostate cancer. Cancer Res. 2007;67:5033-41 doi:10.1158/0008-5472.CAN-06-3332
27. Montgomery RB, Mostaghel EA, Vessella R, Hess DL, Kalhorn TF, Higano CS. et al. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res. 2008;68:4447-54 doi:68/11/4447 [pii]10.1158/0008-5472.CAN-08-0249
28. de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L. et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364:1995-2005 doi:10.1056/NEJMoa1014618
29. Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K. et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012;367:1187-97 doi:10.1056/NEJMoa1207506. 10.1016/j.ccr.2009.11.006
30. Ryan CJ, Smith MR, de Bono JS, Molina A, Logothetis CJ, de Souza P. et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med. 2013;368:138-48 doi:10.1056/NEJMoa1209096
31. Mostaghel EA, Marck BT, Plymate SR, Vessella RL, Balk S, Matsumoto AM. et al. Resistance to CYP17A1 inhibition with abiraterone in castration-resistant prostate cancer: induction of steroidogenesis and androgen receptor splice variants. Clin Cancer Res. 2011;17:5913-25 doi:1078-0432.CCR-11-0728 [pii] 10.1158/1078-0432.CCR-11-0728
32. Cai C, Balk SP. Intratumoral androgen biosynthesis in prostate cancer pathogenesis and response to therapy. Endocr Relat Cancer. 2011;18:R175-82 doi:10.1530/ERC-10-0339
33. Richards J, Lim AC, Hay CW, Taylor AE, Wingate A, Nowakowska K. et al. Interactions of abiraterone, eplerenone, and prednisolone with wild-type and mutant androgen receptor: a rationale for increasing abiraterone exposure or combining with MDV3100. Cancer Res. 2012;72:2176-82 doi:10.1158/0008-5472.CAN-11-3980
34. Nelson WG, Yegnasubramanian S. Resistance emerges to second-generation antiandrogens in prostate cancer. Cancer Discov. 2013;3:971-4 doi: 10.1158/2159-8290.CD-13-0405
35. Yamaoka M, Hara T, Hitaka T, Kaku T, Takeuchi T, Takahashi J. et al. Orteronel (TAK-700), a novel non-steroidal 17,20-lyase inhibitor: effects on steroid synthesis in human and monkey adrenal cells and serum steroid levels in cynomolgus monkeys. J Steroid Biochem Mol Biol. 2012;129:115-28 doi:10.1016/j.jsbmb.2012.01.001
36. Dreicer R, Agus D, MacVicar G, Wang J, MacLean D, Stadler W. Safety, pharmacokinetics, and efficacy of TAK-700 in metastatic castration-resistant prostate cancer: A phase I/II, open-label study. J Clin Oncol. 2010;28:3084
37. Bruno RD, Vasaitis TS, Gediya LK, Purushottamachar P, Godbole AM, Ates-Alagoz Z. et al. Synthesis and biological evaluations of putative metabolically stable analogs of VN/124-1 (TOK-001): head to head anti-tumor efficacy evaluation of VN/124-1 (TOK-001) and abiraterone in LAPC-4 human prostate cancer xenograft model. Steroids. 2011;76:1268-79 doi:10.1016/j.steroids.2011.06.002
38. Rathkopf DE, Morris MJ, Fox JJ, Danila DC, Slovin SF, Hager JH. et al. Phase I study of ARN-509, a novel antiandrogen, in the treatment of castration-resistant prostate cancer. J Clin Oncol. 2013;31:3525-30 doi:10.1200/JCO.2013.50.1684
39. Stanbrough M, Bubley GJ, Ross K, Golub TR, Rubin MA, Penning TM. et al. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res. 2006;66:2815-25 doi:10.1158/0008-5472.CAN-05-4000
40. Mitsiades N, Sung CC, Schultz N, Danila DC, He B, Eedunuri VK. et al. Distinct patterns of dysregulated expression of enzymes involved in androgen synthesis and metabolism in metastatic prostate cancer tumors. Cancer Res. 2012;72:6142-52 doi:10.1158/0008-5472.CAN-12-1335
41. Mohler JL, Titus MA, Bai S, Kennerley BJ, Lih FB, Tomer KB. et al. Activation of the androgen receptor by intratumoral bioconversion of androstanediol to dihydrotestosterone in prostate cancer. Cancer Res. 2011;71:1486-96 doi:10.1158/0008-5472.CAN-10-1343
42. Li R, Evaul K, Sharma KK, Chang KH, Yoshimoto J, Liu J. et al. Abiraterone inhibits 3beta-hydroxysteroid dehydrogenase: a rationale for increasing drug exposure in castration-resistant prostate cancer. Clin Cancer Res. 2012;18:3571-9 doi:10.1158/1078-0432.CCR-12-0908
43. Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP. et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239-43 doi:10.1038/nature11125
44. Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat JP. et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet. 2012;44:685-9 doi:10.1038/ng.2279
45. Chang KH, Li R, Kuri B, Lotan Y, Roehrborn CG, Liu J. et al. A gain-of-function mutation in DHT synthesis in castration-resistant prostate cancer. Cell. 2013;154:1074-84 doi:10.1016/j.cell.2013.07.029
46. Small EJ, Halabi S, Dawson NA, Stadler WM, Rini BI, Picus J, Gable P, Torti FM, Kaplan E, Vogelzang NJ.Antiandrogen withdrawal alone or in combination with ketoconazole in androgen-independent prostate cancer patients. a phase III trial (CALGB 9583). J Clin Oncol. 2004;22:1025-33
47. Korpal M, Korn JM, Gao X, Rakiec DP, Ruddy DA, Doshi S, Yuan J, Kovats SG, Kim S, Cooke VG, Monahan JE, Stegmeier F, Roberts TM, Sellers WR, Zhou W, Zhu P. An F876L mutation in androgen receptor confers genetic and phenotypic resistance to MDV3100 (enzalutamide). Cancer Discov. 2013;3:1030-43
48. Joseph JD, Lu N, Qian J, Sensintaffar J, Shao G, Brigham D, Moon M, Maneval EC, Chen I, Darimont B, Hager JH. A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer Discov. 2013;3:1020-9
49. Arora VK, Schenkein E, Murali R, Subudhi SK, Wongvipat J, Balbas MD, Shah N, Cai L, Efstathiou E, Logothetis C, Zheng D, Sawyers CL. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell. 2013;155:1309-22
50. Dehm SM, Tindall DJ. Alternatively spliced androgen receptor variants. Endocr Relat Cancer. 2011;18:R183-96 doi:ERC-11-0141 [pii] 10.1530/ERC-11-0141
51. Liu LL, Xie N, Sun S, Plymate S, Mostaghel E, Dong X. Mechanisms of the androgen receptor splicing in prostate cancer cells. Oncogene. 2013 doi: 10.1038/onc.2013.284
52. Andersen RJ, Mawji NR, Wang J, Wang G, Haile S, Myung JK, Watt K, Tam T, Yang YC, Bañuelos CA, Williams DE, McEwan IJ, Wang Y, Sadar MD. Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor. Cancer Cell. 2010;17:535-46 doi: 10.1016/j.ccr.2010.04.027
53. Myung JK, Banuelos CA, Fernandez JG, Mawji NR, Wang J, Tien AH, Yang YC, Tavakoli I, Haile S, Watt K, McEwan IJ, Plymate S, Andersen RJ, Sadar MD. An androgen receptor N-terminal domain antagonist for treating prostate cancer. J Clin Invest. 2013;123:2948-60 doi: 10.1172/JCI66398
54. Grosse L, Pâquet S, Caron P, Fazli L, Rennie PS, Bélanger A, Barbier O. Androgen glucuronidation: an unexpected target for androgen deprivation therapy, with prognosis and diagnostic implications. Cancer Res. 2013;73:6963-71 doi: 10.1158/0008-5472.CAN-13-1462
55. Mendiratta P, Mostaghel E, Guinney J, Tewari AK, Porrello A, Barry WT. et al. Genomic strategy for targeting therapy in castration-resistant prostate cancer. J Clin Oncol. 2009;27:2022-9 doi:JCO.2008.17.2882 [pii] 10.1200/JCO.2008.17.2882
56. Mosquera JM, Beltran H, Park K, MacDonald TY, Robinson BD, Tagawa ST, Perner S, Bismar TA, Erbersdobler A, Dhir R, Nelson JB, Nanus DM, Rubin MA. Concurrent AURKA and MYCN gene amplifications are harbingers of lethal treatment-related neuroendocrine prostate cancer. Neoplasia. 2013;15:1-10
57. Ryan CJ, Tindall DJ. Androgen receptor rediscovered: the new biology and targeting the androgen receptor therapeutically. J Clin Oncol. 2011;29:3651-8 doi:JCO.2011.35.2005 [pii]10.1200/JCO.2011.35.2005
58. Hellerstedt BA, Pienta KJ. The truth is out there: an overall perspective on androgen deprivation. Urol Oncol. 2003;21:272-81 doi:S1078143903000462 [pii]
59. Higano CS. Side effects of androgen deprivation therapy: monitoring and minimizing toxicity. Urology. 2003;61:32-8 doi:S009042950202397X [pii]
60. Liao S, Witte D. Autoimmune anti-androgen-receptor antibodies in human serum. Proc Natl Acad Sci U S A. 1985;82:8345-8
61. Young CY, Murthy LR, Prescott JL, Johnson MP, Rowley DR, Cunningham GR, Killian CS, Scardino PT, VonEschenbach A, Tindall DJ. Monoclonal antibodies against the androgen receptor: recognition of human and other mammalian androgen receptors. Endocrinology. 1988;123:601-10
62. Chang CS, Kokontis J, Liao ST. Molecular cloning of human and rat complementary DNA encoding androgen receptors. Science. 1988;240:324-6
63. Lubahn DB, Joseph DR, Sullivan PM, Willard HF, French FS, Wilson EM. Cloning of human androgen receptor complementary DNA and localization to the X chromosome. Science. 1988;240:327-30
64. Heinlein CA, Chang C. Androgen receptor in prostate cancer. Endocr Rev. 2004;25:276-308
65. Heemers HV, Tindall DJ. Androgen receptor (AR) coregulators: a diversity of functions converging on and regulating the AR transcriptional complex. Endocr Rev. 2007;28:778-808 doi:er.2007-0019 [pii] 10.1210/er.2007-0019
66. Yadav N, Heemers HV. Androgen action in the prostate gland. Minerva Urol Nefrol. 2012;64:35-49 doi:R19121948 [pii]
67. Verrijdt G, Tanner T, Moehren U, Callewaert L, Haelens A, Claessens F. The androgen receptor DNA-binding domain determines androgen selectivity of transcriptional response. Biochem Soc Trans. 2006;34:1089-94
68. De BR, Bollen R, Claessens F. Identification and characterization of androgen response elements. Methods Mol Biol. 2011;776:81-93 doi: 10.1007/978-1-61779-243-4_6
69. Denayer S, Helsen C, Thorrez L, Haelens A, Claessens F. The rules of DNA recognition by the androgen receptor. Mol Endocrinol. 2010;24:898-913
70. Robins DM, Scheller A, Adler AJ. Specific steroid response from a nonspecific DNA element. J Steroid Biochem Mol Biol. 1994;49:251-5
71. Roche PJ, Hoare SA, Parker MG. A consensus DNA-binding site for the androgen receptor. Mol Endocrinol. 1992;6:2229-35
72. Heemers H, Verrijdt G, Organe S, Claessens F, Heyns W, Verhoeven G. et al. Identification of an androgen response element in intron 8 of the sterol regulatory element-binding protein cleavage-activating protein gene allowing direct regulation by the androgen receptor. J Biol Chem. 2004;279:30880-7 doi:10.1074/jbc.M401615200 M401615200 [pii]
73. Dehm SM, Tindall DJ. Molecular regulation of androgen action in prostate cancer. J Cell Biochem. 2006;99:333-44 doi:10.1002/jcb.20794
74. Heemers HV, Verhoeven G, Swinnen JV. Androgen activation of the sterol regulatory element-binding protein pathway: Current insights. Mol Endocrinol. 2006;20:2265-77 doi:me.2005-0479 [pii] 10.1210/me.2005-0479
75. Segawa T, Nau ME, Xu LL, Chilukuri RN, Makarem M, Zhang W. et al. Androgen-induced expression of endoplasmic reticulum (ER) stress response genes in prostate cancer cells. Oncogene. 2002;21:8749-58 doi:10.1038/sj.onc.1205992
76. Massie CE, Lynch A, Ramos-Montoya A, Boren J, Stark R, Fazli L. et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. The EMBO journal. 2011;30:2719-33 doi:10.1038/emboj.2011.158
77. Horie-Inoue K, Inoue S. Genome-wide integrated analyses of androgen receptor signaling in prostate cancer based on high-throughput technology. Curr Drug Targets. 2013;4:472-80
78. Massie CE, Mills IG. Global identification of androgen response elements. Methods Mol Biol. 2011;776:255-73 doi:10.1007/978-1-61779-243-4_15
79. Massie CE, Adryan B, Barbosa-Morais NL, Lynch AG, Tran MG, Neal DE. et al. New androgen receptor genomic targets show an interaction with the ETS1 transcription factor. EMBO Rep. 2007;8:871-8 doi:7401046 [pii] 10.1038/sj.embor.7401046
80. Wang Q, Li W, Liu XS, Carroll JS, Janne OA, Keeton EK. et al. A hierarchical network of transcription factors governs androgen receptor-dependent prostate cancer growth. Mol Cell. 2007;27:380-92 doi:S1097-2765(07)00373-5 [pii] 10.1016/j.molcel.2007.05.041
81. Baniwal SK, Khalid O, Sir D, Buchanan G, Coetzee GA, Frenkel B. Repression of Runx2 by androgen receptor (AR) in osteoblasts and prostate cancer cells: AR binds Runx2 and abrogates its recruitment to DNA. Mol Endocrinol. 2009;23:1203-14 doi: 10.1210/me.2008-0470
82. Norris JD, Chang CY, Wittmann BM, Kunder RS, Cui H, Fan D, Joseph JD, McDonnell DP. The homeodomain protein HOXB13 regulates the cellular response to androgens. Mol Cell. 2009;36:405-16 doi: 10.1016/j.molcel.2009.10.020
83. Sharma NL, Massie CE, Ramos-Montoya A, Zecchini V, Scott HE, Lamb AD. et al. The androgen receptor induces a distinct transcriptional program in castration-resistant prostate cancer in man. Cancer Cell. 2013;23:35-47 doi:10.1016/j.ccr.2012.11.010
84. Sahu B, Laakso M, Ovaska K, Mirtti T, Lundin J, Rannikko A. et al. Dual role of FoxA1 in androgen receptor binding to chromatin, androgen signalling and prostate cancer. EMBO J. 2011;30:3962-76 doi:10.1038/emboj.2011.328
85. Sahu B, Laakso M, Pihlajamaa P, Ovaska K, Sinielnikov I, Hautaniemi S. et al. FoxA1 specifies unique androgen and glucocorticoid receptor binding events in prostate cancer cells. Cancer Res. 2013Mar;73(5):1570-80 doi:10.1158/0008-5472.CAN-12-2350
86. Wang D, Garcia-Bassets I, Benner C, Li W, Su X, Zhou Y, Qiu J, Liu W, Kaikkonen MU, Ohgi KA, Glass CK, Rosenfeld MG, Fu XD. Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature. 2011;474:390-4 doi: 10.1038/nature10006
87. Wang Q, Carroll JS, Brown M. Spatial and temporal recruitment of androgen receptor and its coactivators involves chromosomal looping and polymerase tracking. Mol Cell. 2005;19:631-42
88. Li G, Fullwood MJ, Xu H, Mulawadi FH, Velkov S, Vega V, Ariyaratne PN, Mohamed YB, Ooi HS, Tennakoon C, Wei CL, Ruan Y, Sung WK. ChIA-PET tool for comprehensive chromatin interaction analysis with paired-end tag sequencing. Genome Biol. 2010;11(R):22. doi: 10.1186/gb-2010-11-2-r22
89. Marshall TW, Link KA, Petre-Draviam CE, Knudsen KE. Differential requirement of SWI/SNF for androgen receptor activity. J Biol Chem. 2003;278:30605-13
90. Heemers HV, Regan KM, Schmidt LJ, Anderson SK, Ballman KV, Tindall DJ. Androgen modulation of coregulator expression in prostate cancer cells. Mol Endocrinol. 2009;23:572-83 doi:10.1210/me.2008-0363
91. Ianculescu I, Wu DY, Siegmund KD, Stallcup MR. Selective roles for cAMP response element-binding protein binding protein and p300 protein as coregulators for androgen-regulated gene expression in advanced prostate cancer cells. J Biol Chem. 2012;287:4000-13 doi: 10.1074/jbc.M111.300194
92. Swinnen JV, Heemers H, van de Sande T, de Schrijver E, Brusselmans K, Heyns W, Verhoeven G. Androgens, lipogenesis and prostate cancer. J Steroid Biochem Mol Biol. 2004;92:273-9
93. Grand-Perret T, Bouillot A, Perrot A, Commans S, Walker M, Issandou M. SCAP ligands are potent new lipid-lowering drugs. Nat Med. 2001;7:1332-8
94. Kamisuki S, Mao Q, Abu-Elheiga L, Gu Z, Kugimiya A, Kwon Y, Shinohara T, Kawazoe Y, Sato S, Asakura K, Choo HY, Sakai J, Wakil SJ, Uesugi M. A small molecule that blocks fat synthesis by inhibiting the activation of SREBP. Chem Biol. 2009;16:882-92
95. Locke JA, Guns ES, Lubik AA, Adomat HH, Hendy SC, Wood CA. et al. Androgen levels increase by intratumoral de novo steroidogenesis during progression of castration-resistant prostate cancer. Cancer Res. 2008;68:6407-15 doi:10.1158/0008-5472.CAN-07-5997
96. Locke JA, Guns ES, Lehman ML, Ettinger S, Zoubeidi A, Lubik A. et al. Arachidonic acid activation of intratumoral steroid synthesis during prostate cancer progression to castration resistance. Prostate. 2010;70:239-51 doi:10.1002/pros.21057
97. Heemers HV, Regan KM, Dehm SM, Tindall DJ. Androgen induction of the androgen receptor coactivator four and a half LIM domain protein-2: evidence for a role for serum response factor in prostate cancer. Cancer Res. 2007;67:10592-9 doi:67/21/10592 [pii]10.1158/0008-5472.CAN-07-1917
98. Heemers HV, Schmidt LJ, Sun Z, Regan KM, Anderson SK, Duncan K. et al. Identification of a clinically relevant androgen-dependent gene signature in prostate cancer. Cancer Res. 2011;71:1978-88 doi:0008-5472.CAN-10-2512 [pii] 10.1158/0008-5472.CAN-10-2512
99. Schmidt LJ, Duncan K, Yadav N, Regan KM, Verone AR, Lohse CM. et al. RhoA as a mediator of clinically relevant androgen action in prostate cancer cells. Mol Endocrinol. 2012;26:716-35 doi:10.1210/me.2011-1130
100. Evelyn CR, Wade SM, Wang Q, Wu M, Iñiguez-Lluhí JA, Merajver SD, Neubig RR. CCG-1423: a small-molecule inhibitor of RhoA transcriptional signaling. Mol Cancer Ther. 2007;6:2249-60
101. Bell JL, Haak AJ, Wade SM, Kirchhoff PD, Neubig RR, Larsen SD. Optimization of novel nipecotic bis(amide) inhibitors of the Rho/MKL1/SRF transcriptional pathway as potential anti-metastasis agents. Bioorg Med Chem Lett. 2013;23:3826-32 doi: 10.1016/j.bmcl.2013.04.080
102. Velasco AM, Gillis KA, Li Y, Brown EL, Sadler TM, Achilleos M. et al. Identification and validation of novel androgen-regulated genes in prostate cancer. Endocrinology. 2004;145:3913-24 doi:10.1210/en.2004-0311 en.2004-0311 [pii]
103. Ngan S, Stronach EA, Photiou A, Waxman J, Ali S, Buluwela L. Microarray coupled to quantitative RT-PCR analysis of androgen-regulated genes in human LNCaP prostate cancer cells. Oncogene. 2009;28:2051-63 doi:onc200968 [pii] 10.1038/onc.2009.68
104. Bolton EC, So AY, Chaivorapol C, Haqq CM, Li H, Yamamoto KR. Cell- and gene-specific regulation of primary target genes by the androgen receptor. Genes Dev. 2007;21:2005-17 doi:21/16/2005 [pii] 10.1101/gad.1564207
105. Jia L, Berman BP, Jariwala U, Yan X, Cogan JP, Walters A. et al. Genomic androgen receptor-occupied regions with different functions, defined by histone acetylation, coregulators and transcriptional capacity. PLoS One. 2008;3:e3645. doi:10.1371/journal.pone.0003645
106. Wang Q, Li W, Zhang Y, Yuan X, Xu K, Yu J. et al. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell. 2009;138:245-56 doi:S0092-8674(09)00517-0 [pii] 10.1016/j.cell.2009.04.056
107. Yu J, Mani RS, Cao Q, Brenner CJ, Cao X, Wang X. et al. An integrated network of androgen receptor, polycomb, and TMPRSS2-ERG gene fusions in prostate cancer progression. Cancer Cell. 2010;17:443-54 doi:S1535-6108(10)00109-1 [pii] 10.1016/j.ccr.2010.03.018
108. Tomlins SA, Mehra R, Rhodes DR, Cao X, Wang L, Dhanasekaran SM. et al. Integrative molecular concept modeling of prostate cancer progression. Nat Genet. 2007;39:41-51 doi:ng1935 [pii] 10.1038/ng1935
109. Decker KF, Zheng D, He Y, Bowman T, Edwards JR, Jia L. Persistent androgen receptor-mediated transcription in castration-resistant prostate cancer under androgen-deprived conditions. Nucleic Acids Res. 2012;40:10765-79 doi:10.1093/nar/gks888
110. Fiandalo MV, Wu W, Mohler JL. The role of intracrine androgen metabolism, androgen receptor and apoptosis in the survival and recurrence of prostate cancer during androgen deprivation therapy. Curr Drug Targets. 2013;14:420-40
111. Aihara M, Wheeler TM, Ohori M, Scardino PT. Heterogeneity of prostate cancer in radical prostatectomy specimens. Urology. 1994;43:60-6
112. Murphy GP, Busch C, Abrahamsson PA, Epstein JI, McNeal JE, Miller GJ. et al. Histopathology of localized prostate cancer. Consensus Conference on Diagnosis and Prognostic Parameters in Localized Prostate Cancer. Stockholm, Sweden, May 12-13, 1993. Scand J Urol Nephrol Suppl. 1994;162:7-42
113. Miller GJ, Cygan JM. Morphology of prostate cancer: the effects of multifocality on histological grade, tumor volume and capsule penetration. J Urol. 1994;152:1709-13
114. Ruijter ET, van de Kaa CA, Schalken JA, Debruyne FM, Ruiter DJ. Histological grade heterogeneity in multifocal prostate cancer. Biological and clinical implications. J Pathol. 1996;180:295-9
115. Cheng L, Song SY, Pretlow TG, Abdul-Karim FW, Kung HJ, Dawson DV. et al. Evidence of independent origin of multiple tumors from patients with prostate cancer. J Natl Cancer Inst. 1998;90:233-7
116. Macintosh CA, Stower M, Reid N, Maitland NJ. Precise microdissection of human prostate cancers reveals genotypic heterogeneity. Cancer Res. 1998;58:23-8
117. Cheng L, Bostwick DG, Li G, Wang Q, Hu N, Vortmeyer AO. et al. Allelic imbalance in the clonal evolution of prostate carcinoma. Cancer. 1999;85:2017-22
118. Liu W, Laitinen S, Khan S, Vihinen M, Kowalski J, Yu G. et al. Copy number analysis indicates monoclonal origin of lethal metastatic prostate cancer. Nat Med. 2009;15:559-65 doi:10.1038/nm.1944
Author contact
![]() Corresponding author: Dr. Hannelore V. Heemers, Roswell Park Cancer Institute, Departments of Urology and Cancer Genetics, Center for Pharmacology and Genetics, room L2-314, Elm & Carlton Streets, Buffalo, NY 14263, phone: 716-8457644, email: hannelore.heemersorg.
Corresponding author: Dr. Hannelore V. Heemers, Roswell Park Cancer Institute, Departments of Urology and Cancer Genetics, Center for Pharmacology and Genetics, room L2-314, Elm & Carlton Streets, Buffalo, NY 14263, phone: 716-8457644, email: hannelore.heemersorg.