Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Orchiectomy, adrenalectomy and...
Mass spectrometry, abiraterone...
Perspectives and challenges
Abbreviations
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2014; 10(6):596-601. doi:10.7150/ijbs.8780 This issue Cite
Review
Roles for the Backdoor Pathway of Androgen Metabolism in Prostate Cancer Response to Castration and Drug Treatment
Michael V. Fiandalo, John Wilton, James L. Mohler ![]()
Department of Urology, Roswell Park Cancer Institute, Buffalo, NY 14263, USA.
Received 2014-2-7; Accepted 2014-4-23; Published 2014-6-3
Abstract
Almost all men who present with advanced prostate cancer (CaP) and many men who fail potentially curative therapy are treated with androgen deprivation therapy (ADT). ADT is not curative and CaP recurs as the lethal phenotype. The goal of this review is to describe the evolution of adrenal androgen blockade, how new androgen measurement methods have furthered understanding of androgen metabolism, and how further understanding of the backdoor pathway of androgen metabolism may lead to interventions that extend survival even more.
Keywords: Adrenal androgens, "Backdoor" Pathway, Prostate cancer, Dihydrotestosterone, CYP17A1.
Orchiectomy, adrenalectomy and early adrenal androgen inhibiting agents
Huggins and Hodges demonstrated that CaP was responsive to ADT by orchiectomy; however, orchiectomy alone did not prevent CaP recurrence [1]. A proposed mechanism for failure of ADT was androgen production by the adrenal glands that promoted CaP survival. Huggins and Scott reported that bilateral adrenalectomy after orchiectomy further decreased androgen levels, however, androgen levels were not abolished [2]. Adrenalectomy did not become standard treatment for CaP because patients died due to adrenal insufficiency. Patient survival after adrenalectomy improved after the development of corticosteroid replacement therapy [3, 4]. Despite the addition of corticosteroids, patient survival after adrenalectomy remained poor, either because therapy that addressed testicular and adrenal androgens had no effect or hormone therapy was successful but, the tumor recurred and adrenalectomy fell out of favor [5-8]. The development of pharmaceuticals that could reduce adrenal androgen synthesis led to re-examination of a role for adrenal androgens in advanced CaP.
Aminoglutethimide blocks cytochrome P450, family 11, subfamily A, polypeptide 1 (CYP11A1), which catalyzes the conversion of cholesterol to pregnenolone [9, 10]. Aminoglutethmide was shown to decrease adrenal androgen levels similar to those achieved after bilateral adrenalectomy [11, 12]. Aminoglutethmide was co-administered initially with cortisone [13] and then with hydrocortisone [14]. Aminoglutethimide depleted dehydroepiandrosterone-sulfate (DHEA-sulfate), levels and androstenedione (ASD) levels remained unchanged [15]. ASD in circulation provides CaP substrate to produce either testosterone (T) or androstanedione (5α-dione); both can be converted to dihydrotestosterone (DHT) through either 5α reduction or 17β-hydroxysteroid (17β-HSD) enzymatic activity, respectively [16, 17]. The failure of aminoglutethimide to lower ASD levels and drug toxicity as a result of high aminoglutethimide levels are two factors that lead clinicians from aminoglutethimide to ketoconazole for adrenal androgen synthesis blockade [15].
Ketoconazole is an antifungal [18] that can block androgen synthesis by inhibiting P450-dependent enzymatic activities, like 17,20-lyase activity [19]. At higher concentrations, ketoconazole inhibits 11β-hydroxylase, 20,22-desmolase and 17α-hydroxylase activities. However, 17,20-lyase inhibition is the most relevant clinically [20-22]. Ketoconazole was demonstrated to reduce T [23], ASD and DHEA to undetectable levels in most patients [20, 24, 25]. However, the decline in adrenal androgen levels was not associated with decreased PSA levels or CaP regression [26-28]. Ketoconazole's lack of enzyme specificity, drug toxicities, patient side effects and the need for steroid supplementation warranted development of a more specific adrenal androgen synthesis inhibitor [29-31]. Although ketoconazole was identified as a better adrenal androgen synthesis inhibitor than aminoglutethimide, clinical interest for adrenal androgen inhibition remained low until adrenal androgens were shown to clearly be involved in DHT synthesis.
Mass spectrometry, abiraterone and the backdoor pathway of androgen metabolism
In 1979, Geller hypothesized that CaP was capable of synthesizing levels of T and DHT sufficient for AR transactivation in medically or surgically castrate men [32]. The conversion of adrenal androgens to T and DHT was shown important for androgen receptor (AR) transactivation and this conversion was postulated to be performed by “peripheral body tissues” [27, 33, 34]. However, these data were acquired using radioimmunoassay and were met with skepticism due to lack of assay standardization and concern for ensuring androgen specificity [35]. However, this issue was resolved in 2004 when prostatic tissue levels of T and DHT in CR-CaP were measured using liquid chromatography tandem mass spectrometry (LC-MS/MS) [36]. LC-MS/MS is more reliable, accurate and allows higher throughput androgen analysis [35, 37, 38]. Tissues must be procured quickly, extracted carefully and LC-MS/MS measurement of tissue androgens remains difficult. LC-MS/MS methods must be adapted for each matrix (whole blood, plasma or serum and tissue from primary, solid organ metastasis or bone marrow metastasis and cell culture media or cultured cells). Measurements need rigorous controlled and the development of internal standards [36, 39]. Limitations of sensitivity require cell samples of about 100,000 that precludes laser microdissection to separate well benign from malignant epithelium and epithelium from microenvironment and analysis of androgen levels in various subcellular components. The presence of intratumoral T and DHT in CaP and CR-CaP have been confirmed by others [40, 41] and Mostagel et al. detected T and DHT in bone marrow metastatic CaP [42]. These studies paved the way for determining how tissue levels of T and DHT are generated either from weak adrenal androgens [43] or from cholesterol, the starting substrate for androgen synthesis [41, 44] or through an alternate backdoor pathway that involves DHT generation without using T as a substrate (Figure 1).
Four androgen metabolism pathways to DHT synthesis. Pathway 1 is the 5α-reduction of T to DHT (dashed lines). Pathway 2 uses adrenal androgens, DHEA or ASD, to synthesize T or 5α-dione that are converted to DHT (faint gray long dashes). Pathway 3 is the cholesterol pathway (small gray dashes). Pathway 4 is the backdoor pathway of DHT synthesis using androstanediol instead of T to generate DHT (outlined in bold). Abiraterone inhibits 17α-hydroxylase and 17,20-lyase of CYP17A1 (relevant to Pathways 2 and 4) whereas TAK-700 or TOK-001 inhibit only 17,20-lyase (relevant to Pathway 2).
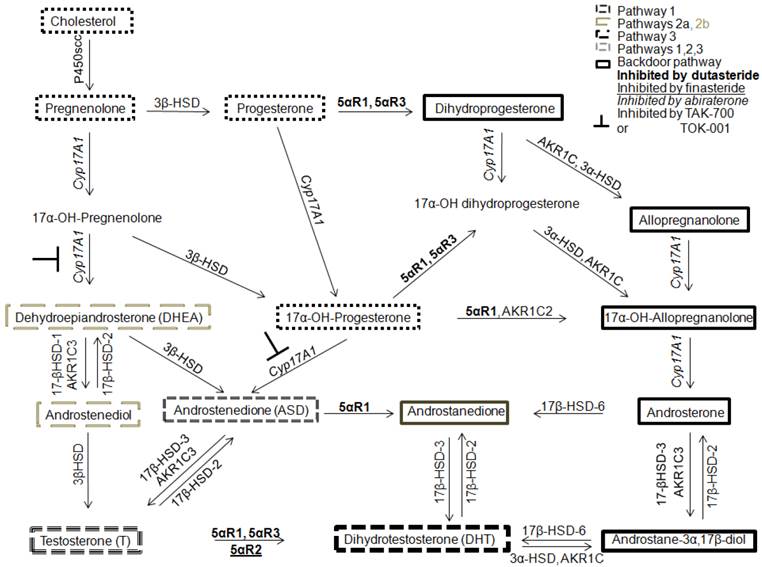
Study of the conversion of adrenal androgens to T or DHT revealed that CYP17A1 is a key enzyme for androgen synthesis in the intratumoral de nova steroid synthesis pathways and CYP17A1 is a central enzyme involved in backdoor pathway metabolism. One potent inhibitor against CYP17A1 is the new FDA-approved drug, abiraterone [31, 45-48]. Abiraterone was investigated first in men with castration-recurrent, metastatic CaP who had failed chemotherapy [49] in whom extension of survival averaged 4 months [50]. Patients who had not undergone chemotherapy [51] had 57% reduction in risk of radiographic progression but the co-primary endpoint of survival had not been reached vs. 8.3 months in the placebo arm. In addition to CYP17A1 inhibition, abiraterone has been shown to inhibit 3β-hydroxysteroid dehydrogenase (3β-HSD), which has been reported to convert DHEA to ASD, which is then 5α-reduced by 5α-reductase 1 to 5α-dione and then DHT (Figure 1) in cell lines and LAPC4 xenografts in laboratory mice. Rodents lack adrenal CYP17A1 expression and consequently the effect of abiraterone must be modulated by inhibition of 3β-HSD [16, 52].
Clinical failure of abiraterone has been attributed to several mechanisms involving the backdoor pathway [53] and AR splice variants [54]. Abiraterone inhibition has been shown to elevate CYP17A1 expression levels and induce progesterone accumulation. The over abundant amount of progesterone can compete against abiraterone for CYP17A1 leading to generation of metabolites involved with backdoor metabolism and hence DHT production [53]. Cai et al. demonstrated that abiraterone caused up-regulation of CYP17A1 more than 10-fold in 2 of 5 VCaP xenografts studied that may provide a growth advantage and CYP17A1 inhibition may result in up-regulation of CYP11A1 and AKR1C3; both enzymes are capable of increasing intratumoral de novo androgen synthesis by activating different androgen metabolism pathways [53]. Mostaghel et al. studied two different xenografts derived from the LuCaP xenograft model, LuCaP 23CR and 35CR [54]. When treated with abiraterone, both xenograft cell lines showed increased CYP17A1 and17β-HSD-3 (2 enzymes mediating conversion of adrenal androgen intermediates to T in the prostate and testis respectively [55, 56]), full length AR, and AR splice variants expression. The major differences between the xenografts were the mechanism and time of recurrence. LuCaP 23CR recurred earlier and this earlier recurrence was independent of intracrine production of T and DHT. In contrast, LuCaP 35CR recurred later and had statistically significant increases in T and a trend toward significant increases in DHT. Greater complexity was observed in the expression of oxidative genes mediating back conversion of 5androstane-3α,17β-diol (androstanediol) to DHT. The authors suggested inhibitors of other critical components of the steroid metabolism pathway, such as 3β-HSD1 or 5α-reductase type 1 or 2 could offset adaptive up-regulation of CYP17A1 [54].
Other investigators suggested that dose escalation may overcome the CYP17A1 overexpression [49] or that targeting AR splice variants in conjunction with treatment with abiraterone might be beneficial [54]. Mosteghel et al. lament the fact that “serum samples adequate to assess circulating androgen levels were not available for analysis in this study”, which may have provided insight into mechanisms of resistance to CYP17A1 inhibition with abiraterone [54]. Li et al. postulate that increasing abiraterone concentration also may inhibit 3β-HSD simultaneously with counteracting the effects of over expression of CYP17A1 [52].
New CYP17A1 inhibitors are under development to address the clinical flaws associated with abiraterone. Two inhibitors in clinical trials currently are TOK-001 and TAK-700. TOK-001 is a novel CYP17A1 inhibitor that has completed a phase 1 clinical trial, ARMOR1 [57]. TOK-001 was tolerated well and demonstrated anti-tumor activity. Purushottamachar et al. provided evidence that TOK-001 not only inhibits CYP17A1 activity but has AR antagonistic capabilities similar to bicalutamide [47, 58]. TAK-700 was designed to inhibit specifically the 18,20-lyase activity of CYP17A1, which focused the drug to target specifically adrenal androgen synthesis. TAK-700 demonstrated specific CYP17A1 18,20-lyase activity, was well tolerated by patients and exhibited anti-tumoral effects in phase 1 and phase 2 clinical trials [59]. However, the phase 3 post-chemotherapy clinical trial, ELM-PC 5 (C21005), was stopped because patients showed disease progression in the TAK-700 + prednisone arm and the study would not be able to reach its primary endpoint.
Perspectives and challenges
Huggins introduced the first strategies for depleting serum testicular and adrenal androgen levels as a treatment for CaP in the 1940s and 1950s. However, 60-70 years after the initial works of Huggins and despite newer adrenal androgen inhibitors, which are more specific, safer and more effective at targeting adrenal androgen synthesis than their predecessors, CaP still recurs and patient survival remains disappointing. Hence, it appears that depletion of serum levels of testicular and adrenal androgens are not sufficient to deplete tumor tissue levels.
Androgen metabolism pathways are biologically and technically challenging to study. The biological downsides include enzyme or pathway redundancy, such that if one arm of the DHT synthesis pathway is inhibited, another arm or set of enzymes are activated to overcome the initial enzyme or pathway blockade. The literature is confused by the lack of standard nomenclature for androgen metabolism enzymes, the many enzymes that catalyze the same conversion and the presence of many isozymes for most androgen metabolism enzymes. Another challenge to studying androgen metabolism enzymes is enzymatic activity is difficult to measure. Enzyme activity cannot be determined reliably from message or protein levels. Enzymes are sensitive to pH and cofactor availability, and both are difficult to measure accurately in vivo. Enzyme assays are conducted in vitro using these uncertain pH cofactor levels that make in vitro assays even more artificial. One reliable method is to measure substrate to product conversion using LC-MS/MS but LC-MS/MS is technically challenging.
The available in vitro and in vivo model systems for studying androgen metabolism may reflect the human situation poorly. CaP cell lines used for modeling CaP are cultured in FBS that contains 0.03 nM T [60], which is close to serum levels of T in the castrated human male. These CaP cell lines, which include the commonly labeled androgen-sensitive LNCaP and LAPC4, have adapted to a castration-like environment to grow in culture and therefore may not be as androgen sensitive as necessary to model the clinical situation. Perhaps CaP cell lines should be trained using more physiologically relevant levels of androgens before subjecting these cells to an androgen-deprived environment using double charcoal stripped serum.
Studying androgen metabolism in vivo is complicated by the mouse host and the xenografts. Mice produce very low levels of adrenal androgens; the castration environment is dissimilar to the human high levels of circulating adrenal androgens [61-63]. Therefore, the mouse model to mimic human castration should be humanized by introducing and maintaining stable adrenal androgen levels prior to study. The CaP xenografts implanted into mice respond differently to castration. For example, the growth of LNCaP and LAPC4 xenografts slow after castration but neither xenograft regresses completely. These models may be less appropriate [64] because both xenografts respond to ADT with slowed growth and not tumor regression. The CWR22 tumor model is a more physiologically relevant model since the CWR22 model mimics the changes in human CaP after ADT. The CWR22 tumor model is an androgen-dependent human CaP xenograft that is propagated subcutaneously in nude mice. The CWR22 is a better xenograft model than LNCaP or LAPC4 because the CWR22 model regresses after ADT. After CWR22 tumor regresses, the tumor recurs 4-5 months after ADT [65-68]. Additionally, recurrent CWR22 has AR protein expression levels and AR-dependent or AR-independent gene expression profiles that are similar to androgen-stimulated CWR22 [69-75]. Our laboratory has used the CWR22 model to model AR transactivation by testicular androgens synthesized by intracrine metabolism [76, 77].
However, the studies by Cai and Mostaghel et al. support the notion that no matter how well we design or redesign our inhibitors, tumors are capable of rendering new agents ineffective. For example, both studies provide evidence that the androgen metabolism pathways can reconfigure themselves to overcome DHT inhibition and produce DHT by activating arms of the androgen metabolism pathways not affected by the inhibitors. In addition to androgen metabolism reconfiguration, the tumor overcomes abiraterone by increasing AR splice variant expression, rendering abiraterone inhibition ineffective. A better understanding of these pathways may allow their interruption using multi-functional inhibitors, such as TOK-001 or ARN-509 [57, 78] or optimal combination of single target agents that affect multiple androgen synthesis pathways at one time [79].
Abbreviations
17β-HSD: 17β-hydroxysteroid dehydrogenase; 5α-dione: 5α-androstanedione; 3β-HSD: 3β-hydroxysteroid dehydrogenase; ADT: androgen deprivation therapy; AND: androsterone; AR: androgen receptor; androstanediol: 5androstane-3α,17β-diol; ASD: androstenedione; CaP: prostate cancer; CR-CaP: castration recurrent prostate cancer; CYP11A1: cytochrome P450, family 11, subfamily A, polypeptide 1; CYP17A1: cytochrome p450, family 17, subfamily A polypeptide 1; DHEA: dehydroepiandrosterone; DHEA-sulfate: dehydroepiandrosterone-sulfate; DHT: dihydrotestosterone; LC-MS/MS: liquid chromatography tandem mass spectrometry; LHRH: luteinizing hormone releasing hormone; T: testosterone.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Huggins C, Hodges CV. Studies on prostatic cancer: I. The effect of castration, of estrogen and androgen injection on serum phsphatases in metastatic carcinoma of prostate. Cancer Res. 1941;1:293-297
2. Huggins C, Scott WW. Bilateral adrenalectomy in prostatic cancer: clinical features and urinary excretion of 17-ketosteroids and estrogen. Ann Surg. 1945;122:1031-1041
3. Thorn GW, Forsham PH, Frawley TF. et al. The clinical usefulness of ACTH and cortisone. N Engl J Med. 1950;242:865-872
4. Huggins C, Bergenstal DM. Surgery of the adrenals. J Am Med Assoc. 1951;147:101-106
5. Harrison JH, Thorn GW, Jenkins D. Total adrenalectomy for reactivated carcinoma of the prostate. N Engl J Med. 1953;248:86-92
6. Bubley GJ, Balk SP. Treatment of androgen-independent prostate cancer. Oncologist. 1996;1:30-35
7. Geller J. Rationale for blockade of adrenal as well as testicular androgens in the treatment of advanced prostate cancer. Semin Oncol. 1985;12:28-35
8. Hendry WF. Adrenalectomy and hypophysectomy in disseminated prostatic cancer. In: (ed.) Castro J. The Treatment of Prostatic Hypertrophy and Neoplasia. Netherlands: Springer. 1974:171-196
9. Dexter RN, Fishman LM, Ney RL, Liddle GW. Inhibition of adrenal corticosteroid synthesis by aminoglutethimide: studies of the mechanism of action. J Clin Endocrinol Metab. 1967;27:473-480
10. Gunter JH, Sarkar PL, Lubik AA, Nelson CC. New players for advanced prostate cancer and the rationalisation of insulin-sensitising medication. Int J Cell Biol. 2013;2013:834684
11. Worgul TJ, Santen RJ, Samojlik E. et al. Clinical and biochemical effect of aminoglutethimide in the treatment of advanced prostatic carcinoma. J Urol. 1983;129:51-55
12. Block M, Trump D, Rose DP, Cummings KB, Hogan TF. Evaluation of aminoglutethimide in stage D prostate cancer: an assessment of efficacy and toxicity in patients with tumors refractory to hormonal therapy. Cancer Treat Rep. 1984;68:719-722
13. Ponder BA, Shearer RJ, Pocock RD. et al. Response to aminoglutethimide and cortisone acetate in advanced prostatic cancer. Br J Cancer. 1984;50:757-763
14. Dowsett M, Shearer RJ, Ponder BA, Malone P, Jeffcoate SL. The effects of aminoglutethimide and hydrocortisone, alone and combined, on androgen levels in post-orchiectomy prostatic cancer patients. Br J Cancer. 1988;57:190-192
15. Ahmann FR, Crawford ED, Kreis W, Levasseur Y. Adrenal steroid levels in castrated men with prostatic carcinoma treated with aminoglutethimide plus hydrocortisone. Cancer Res. 1987;47:4736-4739
16. Chang KH, Li R, Papari-Zareei M. et al. Dihydrotestosterone synthesis bypasses testosterone to drive castration-resistant prostate cancer. Proc Natl Acad Sci U S A. 2011;108:13728-13733
17. Mohler JL, Gregory CW, Ford OH 3rd. et al. The androgen axis in recurrent prostate cancer. Clin Cancer Res. 2004;10:440-448
18. Odds FC, Milne LJ, Gentles JC, Ball EH. The activity in vitro and in vivo of a new imidazole antifungal, ketoconazole. J Antimicrob Chemother. 1980;6:97-104
19. Loose DS, Kan PB, Hirst MA, Marcus RA, Feldman D. Ketoconazole blocks adrenal steroidogenesis by inhibiting cytochrome P450-dependent enzymes. J Clin Invest. 1983;71:1495-1499
20. De Coster R, Caers I, Coene MC, Amery W, Beerens D, Haelterman C. Effects of high dose ketoconazole therapy on the main plasma testicular and adrenal steroids in previously untreated prostatic cancer patients. Clin Endocrinol (Oxf). 1986;24:657-664
21. Havlin KA, Trump DL. Aminoglutethimide: theoretical considerations and clinical results in advanced prostate cancer. Cancer Treat Res. 1988;39:83-96
22. Rajfer J, Sikka SC, Rivera F, Handelsman DJ. Mechanism of inhibition of human testicular steroidogenesis by oral ketoconazole. J Clin Endocrinol Metab. 1986;63:1193-1198
23. Pont A, Williams PL, Azhar S. et al. Ketoconazole blocks testosterone synthesis. Arch Intern Med. 1982;142:2137-2140
24. Pont A, Williams PL, Loose DS. et al. Ketoconazole blocks adrenal steroid synthesis. Ann Intern Med. 1982;97:370-372
25. Trachtenberg J, Halpern N, Pont A. Ketoconazole: a novel and rapid treatment for advanced prostatic cancer. J Urol. 1983;130:152-153
26. Harris KA, Weinberg V, Bok RA, Kakefuda M, Small EJ. Low dose ketoconazole with replacement doses of hydrocortisone in patients with progressive androgen independent prostate cancer. J Urol. 2002;168:542-545
27. Trump DL, Havlin KH, Messing EM, Cummings KB, Lange PH, Jordan VC. High-dose ketoconazole in advanced hormone-refractory prostate cancer: endocrinologic and clinical effects. J Clin Oncol. 1989;7:1093-1098
28. Keizman D, Huang P, Carducci MA, Eisenberger MA. Contemporary experience with ketoconazole in patients with metastatic castration-resistant prostate cancer: clinical factors associated with PSA response and disease progression. Prostate. 2012;72:461-467
29. Kim W, Ryan CJ. Androgen receptor directed therapies in castration-resistant metastatic prostate cancer. Curr Treat Options Oncol. 2012;13:189-200
30. De Coster R, Wouters W, Bruynseels J. P450-dependent enzymes as targets for prostate cancer therapy. J Steroid Biochem Mol Biol. 1996;56:133-143
31. Njar VC, Hector M, Hartmann RW. 20-amino and 20,21-aziridinyl pregnene steroids: development of potent inhibitors of 17 alpha-hydroxylase/C17,20-lyase (P450 17). Bioorg Med Chem. 1996;4:1447-1453
32. Geller J, Albert J, Nachtsheim D, Loza D, Lippman S. Steroid levels in cancer of the prostate--markers of tumor differentiation and adequacy of anti-androgen therapy. Prog Clin Biol Res. 1979;33:103-111
33. Harper ME, Pike A, Peeling WB, Griffiths K. Steroids of adrenal origin metabolized by human prostatic tissue both in vivo and in vitro. J Endocrinol. 1974;60:117-125
34. Geller J, Albert J, Loza D, Geller S, Stoeltzing W, de la Vega D. DHT concentrations in human prostate cancer tissue. J Clin Endocrinol Metab. 1978;46:440-444
35. Titus M, Tomer KB. Androgen quantitation in prostate cancer tissue using liquid chromatography tandem mass spectrometry. Methods Mol Biol. 2011;776:47-57
36. Titus MA, Schell MJ, Lih FB, Tomer KB, Mohler JL. Testosterone and dihydrotestosterone tissue levels in recurrent prostate cancer. Clin Cancer Res. 2005;11:4653-4657
37. Wang C, Shiraishi S, Leung A. et al. Validation of a testosterone and dihydrotestosterone liquid chromatography tandem mass spectrometry assay: Interference and comparison with established methods. Steroids. 2008;73:1345-1352
38. Lih FB, Titus MA, Mohler JL, Tomer KB. Atmospheric pressure photoionization tandem mass spectrometry of androgens in prostate cancer. Anal Chem. 2010;82:6000-6007
39. Wilton J, Titus M, Efstathiou E, Fetterly G, Mohler J. Androgenic biomarker profiling in human matrices and cell culture samples using high throughput, electrospray tandem mass spectrometry with column-switching for the analysis of five androgens in human prostatic tissue and other matrices. Prostate. 2014 in press
40. Locke JA, Guns ES, Lubik AA. et al. Androgen levels increase by intratumoral de novo steroidogenesis during progression of castration-resistant prostate cancer. Cancer Res. 2008;68:6407-6415
41. Montgomery RB, Mostaghel EA, Vessella R. et al. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res. 2008;68:4447-4454
42. Mostaghel EA, Page ST, Lin DW. et al. Intraprostatic androgens and androgen-regulated gene expression persist after testosterone suppression: therapeutic implications for castration-resistant prostate cancer. Cancer Res. 2007;67:5033-5041
43. Stanbrough M, Bubley GJ, Ross K. et al. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res. 2006;66:2815-2825
44. Leon CG, Locke JA, Adomat HH. et al. Alterations in cholesterol regulation contribute to the production of intratumoral androgens during progression to castration-resistant prostate cancer in a mouse xenograft model. Prostate. 2010;70:390-400
45. Attard G, Reid AH, A'Hern R. et al. Selective inhibition of CYP17 with abiraterone acetate is highly active in the treatment of castration-resistant prostate cancer. J Clin Oncol. 2009;27:3742-3748
46. Jarman M, Barrie SE, Llera JM. The 16,17-double bond is needed for irreversible inhibition of human cytochrome p45017alpha by abiraterone (17-(3-pyridyl)androsta-5, 16-dien-3beta-ol) and related steroidal inhibitors. J Med Chem. 1998;41:5375-5381
47. Purushottamachar P, Khandelwal A, Vasaitis TS, Bruno RD, Gediya LK, Njar VC. Potent anti-prostate cancer agents derived from a novel androgen receptor down-regulating agent. Bioorg Med Chem. 2008;16:3519-3529
48. Attard G, Reid AH, Yap TA. et al. Phase I clinical trial of a selective inhibitor of CYP17, abiraterone acetate, confirms that castration-resistant prostate cancer commonly remains hormone driven. J Clin Oncol. 2008;26:4563-4571
49. de Bono JS, Logothetis CJ, Molina A. et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364:1995-2005
50. Scher HI. Evaluation of circulating tumor cell (CTC) enumeration as an efficacy response biomarker of overall survivial (OS) in metastatic castration-resistant prostate cancer (mCRPC); Planned final analysis (FA) of Cou-AA-301, a randomized double-blind, placebo-controlled phase III study of abiraterone acetate (AA) plus low-dose prednisone (P) post docetaxel. Journal of Clinical Oncology. 2011 29
51. Ryan CJ, Smith MR, de Bono JS. et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med. 2013;368:138-148
52. Li R, Evaul K, Sharma KK. et al. Abiraterone inhibits 3beta-hydroxysteroid dehydrogenase: a rationale for increasing drug exposure in castration-resistant prostate cancer. Clin Cancer Res. 2012;18:3571-3579
53. Cai C, Chen S, Ng P. et al. Intratumoral de novo steroid synthesis activates androgen receptor in castration-resistant prostate cancer and is upregulated by treatment with CYP17A1 inhibitors. Cancer Res. 2011;71:6503-6513
54. Mostaghel EA, Marck BT, Plymate SR. et al. Resistance to CYP17A1 inhibition with abiraterone in castration-resistant prostate cancer: induction of steroidogenesis and androgen receptor splice variants. Clin Cancer Res. 2011;17:5913-5925
55. Geissler WM, Davis DL, Wu L. et al. Male pseudohermaphroditism caused by mutations of testicular 17 beta-hydroxysteroid dehydrogenase 3. Nat Genet. 1994;7:34-39
56. Mohler JL, Titus MA, Bai S. et al. Activation of the androgen receptor by intratumoral bioconversion of androstanediol to dihydrotestosterone in prostate cancer. Cancer Res. 2011;71:1486-1496
57. Taplin ME. Abstract CT-07: ARMOR1: Safety of galeterone (TOK-001) in a Phase 1 clinical trial in chemotherapy naïve patients with castration resistant prostate cancer (CRPC). Cancer Res. 2012 72
58. Purushottamachar P, Godbole AM, Gediya LK. et al. Systematic structure modifications of multitarget prostate cancer drug candidate galeterone to produce novel androgen receptor down-regulating agents as an approach to treatment of advanced prostate cancer. J Med Chem. 2013;56:4880-4898
59. Petrylak D. A phase I/II study of safety and efficacy of orteronel (TAK-700), an oral, investigational, nonsteroidal 17,20-lyase inhibitor, with docetaxel and prednisone (DP) in metastatic castration-resistant prostate cancer (mCRPC): Updated phase II results. Journal of Clinical Oncology. 2013 31
60. Cheng J, Wu Y, Mohler JL, Ip C. The transcriptomics of de novo androgen biosynthesis in prostate cancer cells following androgen reduction. Cancer Biol Ther. 2010;9:1033-1042
61. van Weerden WM, van Kreuningen A, Elissen NM, de Jong FH, van Steenbrugge GJ, Schroder FH. Effects of adrenal androgens on the transplantable human prostate tumor PC-82. Endocrinology. 1992;131:2909-2913
62. Bielohuby M, Herbach N, Wanke R. et al. Growth analysis of the mouse adrenal gland from weaning to adulthood: time- and gender-dependent alterations of cell size and number in the cortical compartment. Am J Physiol Endocrinol Metab. 2007;293:E139-146
63. Mohler J, Titus M. Tissue levels of androgens in castration-recurrent prostate cancer. In: (ed.) Mohler JL, Tindall D. Androgen Action in Prostate Cancer. New York, NY: Springer Science. 2009:175-185
64. Craft N, Shostak Y, Carey M, Sawyers CL. A mechanism for hormone-independent prostate cancer through modulation of androgen receptor signaling by the HER-2/neu tyrosine kinase. Nat Med. 1999;5:280-285
65. Nagabhushan M, Miller CM, Pretlow TP. et al. CWR22: the first human prostate cancer xenograft with strongly androgen-dependent and relapsed strains both in vivo and in soft agar. Cancer Res. 1996;56:3042-3046
66. Pretlow TG, Wolman SR, Micale MA. et al. Xenografts of primary human prostatic carcinoma. J Natl Cancer Inst. 1993;85:394-398
67. Tan J, Sharief Y, Hamil KG. et al. Dehydroepiandrosterone activates mutant androgen receptors expressed in the androgen-dependent human prostate cancer xenograft CWR22 and LNCaP cells. Mol Endocrinol. 1997;11:450-459
68. Wainstein MA, He F, Robinson D. et al. CWR22: androgen-dependent xenograft model derived from a primary human prostatic carcinoma. Cancer Res. 1994;54:6049-6052
69. Kim D, Gregory CW, Smith GJ, Mohler JL. Immunohistochemical quantitation of androgen receptor expression using color video image analysis. Cytometry. 1999;35:2-10
70. Kim D, Gregory CW, French FS, Smith GJ, Mohler JL. Androgen receptor expression and cellular proliferation during transition from androgen-dependent to recurrent growth after castration in the CWR22 prostate cancer xenograft. Am J Pathol. 2002;160:219-226
71. Gregory CW, Hamil KG, Kim D. et al. Androgen receptor expression in androgen-independent prostate cancer is associated with increased expression of androgen-regulated genes. Cancer Res. 1998;58:5718-5724
72. Mohler JL, Morris TL, Ford OH 3rd, Alvey RF, Sakamoto C, Gregory CW. Identification of differentially expressed genes associated with androgen-independent growth of prostate cancer. Prostate. 2002;51:247-255
73. Mousses S, Wagner U, Chen Y. et al. Failure of hormone therapy in prostate cancer involves systematic restoration of androgen responsive genes and activation of rapamycin sensitive signaling. Oncogene. 2001;20:6718-6723
74. Gregory CW, Johnson RT Jr, Presnell SC, Mohler JL, French FS. Androgen receptor regulation of G1 cyclin and cyclin-dependent kinase function in the CWR22 human prostate cancer xenograft. J Androl. 2001;22:537-548
75. Gregory CW, Kim D, Ye P. et al. Androgen receptor up-regulates insulin-like growth factor binding protein-5 (IGFBP-5) expression in a human prostate cancer xenograft. Endocrinology. 1999;140:2372-2381
76. Gregory CW, He B, Johnson RT. et al. A mechanism for androgen receptor-mediated prostate cancer recurrence after androgen deprivation therapy. Cancer Res. 2001;61:4315-4319
77. Gregory CW, Johnson RT Jr, Mohler JL, French FS, Wilson EM. Androgen receptor stabilization in recurrent prostate cancer is associated with hypersensitivity to low androgen. Cancer Res. 2001;61:2892-2898
78. Clegg NJ, Wongvipat J, Joseph JD. et al. ARN-509: a novel antiandrogen for prostate cancer treatment. Cancer Res. 2012;72:1494-1503
79. Mohler J. Concept and viability of androgen annihilation for advanced prostate cancer. Cancer Research. 2014 in press
Author contact
![]() Corresponding author: James L. Mohler MD. Department of Urology, Roswell Park Cancer Institute, Elm and Carlton Streets, Buffalo, NY 14263. Telephone: (716) 845-8433 Fax: (716) 845-3300 Email: James.Mohlerorg.
Corresponding author: James L. Mohler MD. Department of Urology, Roswell Park Cancer Institute, Elm and Carlton Streets, Buffalo, NY 14263. Telephone: (716) 845-8433 Fax: (716) 845-3300 Email: James.Mohlerorg.