Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Modulation of AR by FOXA1
Modulation of AR by FOXO Factors
Perspectives and Conclusion
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2014; 10(6):614-619. doi:10.7150/ijbs.8389 This issue Cite
Review
Modulation of Androgen Receptor by FOXA1 and FOXO1 Factors in Prostate Cancer
Yu Zhao1, Donald J. Tindall1,2,3 ![]() , Haojie Huang1,2,3
, Haojie Huang1,2,3
1. Department of Biochemistry and Molecular Biology, Mayo Clinic College of Medicine, Rochester, MN 55905, USA;
2. Department of Urology, Mayo Clinic College of Medicine, Rochester, MN 55905, USA;
3. Mayo Clinic Cancer Center, Mayo Clinic College of Medicine, Rochester, MN 55905, USA.
Received 2013-12-18; Accepted 2014-1-22; Published 2014-6-5
Abstract
Androgens and the androgen receptor (AR) are essential for growth and differentiation of the normal prostate gland as well as proliferation and survival of prostate cancer (PCa). Increasing evidence suggests that reactivation of the AR plays a pivotal role in disease progression to castration-resistant PCa (CRPC). Forkhead box (FOX) factors exert two distinct effects on AR function in PCa. The A-class of FOX proteins, especially FOXA1, functions as a pioneer factor to facilitate AR transactivation and PCa growth. In contrast, the O-class of FOX proteins such as FOXO1 and FOXO3, which are downstream effectors of the PTEN tumor suppressor, inhibit the transcriptional activity of either full-length AR or constitutively active splice variants of AR in a direct or indirect manner in PCa. FOXO1 also contributes to taxane-mediated inhibition of the AR and CRPC growth. Therefore, FOX family members not only have a tight relationship with AR, but also represent a pivotal group of proteins to be targeted for PCa therapy. The present review focuses primarily on recent advances in the epigenetic, mechanistic and clinical relevant aspects of regulation of the AR by FOXA1 and FOXO1 factors in PCa.
Keywords: Androgens, androgen receptor, FOXA1, prostate cancer
Introduction
Prostate cancer (PCa) is one of the most commonly diagnosed cancers in men. Increasing evidence suggests that its etiology is related to hormones, genetic and epigenetic factors and lifestyle patterns [1]. The androgen receptor (AR) is a nuclear hormone receptor that is activated by binding to androgenic hormones, such as testosterone and dihydrotestosterone (DHT), in the cytoplasm and translocation into the nucleus [2, 3]. AR binds to androgen via its COOH-terminal ligand binding domain (LBD). The NH2-terminal of AR contains the transcriptional activation domain, and this activity is modified by a number of cofactors [4]. Androgens, via the AR, are essential for the growth and survival of androgen-dependent PCa cells [5]. Androgen ablation therapies, which involve surgical or chemical castration and/or androgen antagonists, have been the mainstay of treatment for advanced androgen-dependent PCa since 1941 [6, 7]. However, castration-resistant prostate cancer (CRPC) is a major challenge for PCa therapy in the clinic. In virtually all cases, castration-resistant progression is accompanied by resurgence of prostate-specific antigen (PSA), a canonical AR target gene, indicating that the AR is persistently activated in CRPC. A number of mechanisms underlying AR reactivation under castration conditions have been demonstrated. These include increased AR expression, AR mutation in the LBD, intratumoral (intracrine) synthesis of androgens from inactive precursors, alterations in AR coactivators and expression of constitutively active AR splice variants [8-14]. Here, we review recent findings regarding mechanisms by which the function of AR is modulated by forkhead factors, particularly, FOXA1 and FOXO1 in PCa.
Forkhead box (Fox) transcription factors are a superfamily of proteins. Since the identification of the fork head gene in Drosophila melanogaster, the founding member of this family whose mutation results in the development of a forkhead-like appearance [15], more than 100 structurally related Fox transcription factors have been identified. The defining feature of Fox proteins is the forkhead box, which is also known as the winged helix DNA binding domain [16]. This domain possesses approximately 100 amino acids [16]. The FOX family is divided into 18 subfamilies in humans, such as A-class (FOXA) and O-class (FOXO) of FOX proteins.
The FOXA subfamily has three members FOXA1, FOXA2, FOXA3. FOXA1 is also known as hepatocyte nuclear factor 3α, HNF-3α. All of these proteins contain a “winged-helix” DNA binding domain (or forkhead domain), consisting of three α-helices, three β-sheets, and two loops or wings [17]. The crystal structure of the forkhead domain suggests that FOXA1 binds to the major groove of DNA. FOXA1 contacts site-specific DNA with the loops in a manner that closely resembles linker histone [18]. FOXA proteins bind to highly compacted or “closed” chromatin [19]. The COOH-terminal domain of FOXA makes these genomic regions more accessible to other transcription factors such as steroid hormone nuclear receptors including AR [20], implying that FOXA proteins can behave as pioneer factors, which engage early transcriptional regulatory events [21-23].
FOXO proteins in humans, which include FOXO1 (FKHR), FOXO3 (FKHRL1 or FOXO3a), FOXO4 (AFX) and FOXO6, are important transcription factors that modulate the expression of genes involved in many cellular functions, such as apoptotic cell death, cell cycle control, DNA damage repair, glucose metabolism, and carcinogenesis[24]. FOXO members contain a conserved DNA binding domain and bind a consensus DNA binding sequence TTGTTTAC at target genes [25, 26]. Binding of the IGF1 receptor (IGF1R) or insulin receptor by their ligands leads to activation of the Phosphatidylinositol 3-kinase (PI3K)/AKT signaling pathway. Activated AKT phosphorylates FOXO proteins at three different Ser/Thr residues (T24, S256 and S319)[27]. FOXO1 can also be phosphorylated by CDK1 and CDK2 at serine 249 in vitro and in vivo [28, 29]. The phosphorylated FOXO proteins are exported from the nucleus to the cytoplasm, sequestered by 14-3-3 chaperone proteins in the cytoplasm [30, 31] and become inactive [31].
Modulation of AR by FOXA1
FOXA1 is a pioneer factor for the AR-mediated transcriptional program [32-34]. It also influences a subset of estrogen receptor (ER) target genes [33, 35, 36]. FOXA1 plays a critical role in the growth and differentiation of a variety of organs, such as prostate, breast, lung and bladder [37-39]. FOXA1 is involved in AR-mediated transcriptional regulation of prostate genes, such as rat probasin and human PSA [40, 41]. FOXA1-binding sites are found in close proximity to AR binding sites in regulatory regions of these genes. The AR and FOXA1 interact through their DNA-binding domains [40].
FOXA1 is a global mediator of AR action in hormone-dependent PCa. High throughput chromatin immunoprecipitation-sequencing (ChIP-seq) has delineated the genome-wide binding sites of AR and FOXA1 (the AR and FOXA1 cistromes) in parental LNCaP-1F5 cells and cells depleted of FOXA1 protein. Similar experiments on AR binding were performed in VCaP cells [42] (Fig. 1). Approximately 70% of AR binding sites are present in junction with FOXA1 binding sites genome-wide in LNCaP cells [43]. Silencing of FOXA1 results in the appearance of over 13,000 new AR-occupied sites. Experiments have demonstrated that FOXA1 can both assist and antagonize AR activity, depending on the cellular context [44].
Converging of FOXA1 and AR-regulated pathways in prostate cancer. Left, FOXA1, as a pioneer factor, opens the closed chromatin, and recruits AR to the binding region of DNA. As a result, AR regulates expression of its target genes to promote PCa cell growth but inhibits PCa cell metastasis. Right, in CRPC, there are two distinct pathways underlying AR regulation of CRPC cell growth. One is that AR itself opens and maintains the chromatin and activates its target genes. The other is that high levels of FOXA1 facilitate AR to become sensitive to low levels of androgens. Thus activated AR transactivates its target genes. Both pathways contribute to CRPC cell growth.
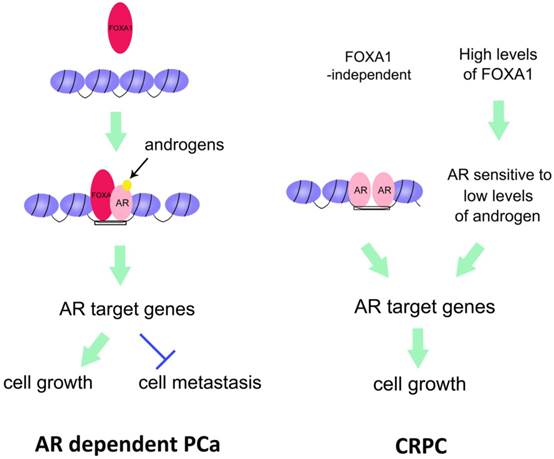
High-throughput analyses revealed a complex relationship between FOXA1, AR and histone markers [45]. These studies include genome-wide distribution data of H3K4 di-methylation (H3K4me2), CCCTC binding factor (CTCF), AR and FOXA1 DNA binding data as well as androgen-regulated gene expression profiling data. Notably, FOXA1 functions not only as a pioneer factor facilitating AR-mediated gene activation, but also as a cofactor of AR-mediated gene repression. A new class of AR-FOXA1 regulated genes has been identified recently [45]. It was found that in a subset of androgen-responsive gene loci, FOXA1 binding sites are located within the CTCF binding sites, which are significantly enriched near the AR/H3K4me2 overlapped regions. Importantly, genes enriched in this class are involved in cancer-related pathways and are downregulated in metastatic PCa in comparison to localized disease. It has been proposed that the activated enhancers occupied by FOXA1 and AR are defined not only by histone markers, but are also related to enhancer-templated expression of non-coding RNA (eRNA) in LNCaP cells [44]. Thus, eRNAs represent a novel and robust indicator of the FOXA1/AR-regulated enhancer in the genome.
FOXA1 can also form a complex with AR and its target genes. NKX3.1, AR, and FOXA1 coordinate with each other, thereby increasing PCa cell survival by directly upregulating RAB3B, a member of the RAB GTPase family [46]. RAB3B is overexpressed in PCa patient specimens, suggesting that RAB3B together with AR, FOXA1, and NKX3.1 are important for PCa progression. These findings suggest that the AR target gene NKX3.1 can coordinate with AR and FOXA1 in a feedback manner in androgen-dependent PCa[46]. FOXA1 regulates two distinct oncogenic processes via different mechanisms [48]. FOXA1 promotes cell growth through the AR pathway. On the other hand, FOXA1 inhibits cell motility and epithelial-to-mesenchymal transition (EMT) through an AR-independent mechanism [25, 47]. This action directly opposes the action of AR signaling. FOXA1 has a metastasis inhibitory function independently of AR in PCa[48] (Fig. 1).
FOXA1 also modulates AR activity in metastatic PCa. High-level nuclear FOXA1 expression is detected in 19% of primary and 89% of metastatic prostate tumors. Increased expression of FOXA1 results in an overactive AR complex that is responsive to low levels of DHT[49] (Fig. 1). As such, high levels of FOXA1 are related to poor prognosis of PCa. Clinical data show that FOXA1expression positively correlates with tumor size, extra prostatic extension, angiolymphatic invasion, AR levels and lymph node metastases at diagnosis, but does not correlate with age, tumor stage, Gleason score, prostatic intraepithelial neoplasia (PIN) lesions, multifocality, perineural association, seminal vesicle invasion, or surgical excision margin status[49]. Importantly, high-level FOXA1 expression is associated with the development of metastatic PCa[49]. Novel recurrent mutations in FOXA1 were identified in 5 of 147 (3.4%) prostate cancers (both untreated localized PCa and CRPC) [50]. Mutated FOXA1 represses androgen/AR signaling, and paradoxically enhances tumor growth [50].
Although previous studies have focused on ligand-dependent AR signaling, a FOXA1-independent function of AR activity has been discovered under androgen deprived conditions [51], where AR persistently occupies a distinct set of genomic loci following androgen deprivation. These androgen-independent AR-occupied regions have constitutively open chromatin structures that lack the canonical androgen response element and are independent of FOXA1 (Fig. 1). FOXA1 acts as an AR cofactor in CRPC cells to drive the G2-to-M phase cell-cycle transit [52]. A mechanistically different, AR-independent role of FOXA1 was shown to drive G1-to-S phase cell-cycle transit in CRPC. FOXA1, without AR, interacts with MYBL2 or CREB1 on the promoters of Cyclin E2 and E2F1. The induced Cyclin E2 and E2F1 drive the G1-to-S cell-cycle transition in CRPC.
Modulation of AR by FOXO Factors
FOXO1 is deleted in a significant portion of human PCa cell lines, xenografts and clinical specimens [53], suggesting that FOXO1 functions as a tumor suppressor in the prostate. In agreement with this concept, FOXO1 inhibits androgen/androgen receptor-mediated gene expression and suppresses PCa cell proliferation. These inhibitory effects of FOXO1 on the transcriptional activity of AR are attenuated by IGF1 through the AKT pathway [54]. Interestingly, a positive feedback circuit working locally in an autocrine/intracrine manner has been described. Ligand-bound AR up-regulates IGF1 receptor expression in PCa cells, presumably resulting in higher IGF1 signaling output and further enhancing functions of the receptor itself through inhibition of FOXO1[54]. Notably, there exists another feedback loop. AR can bind to the FOXO1 promoter and repress its expression. Chromodomain helicase DNA-binding protein 1 (CHD1) is required for efficient recruitment of AR to responsive promoters and regulates expression of known AR-responsive tumor suppressor genes, including FOXO1, NKX3.1, and PPAR γ[55].
Mutagenesis analysis revealed that a region in FOXO1, from amino acids 150 to 655, which contains the forkhead box and COOH-terminal transactivation domain, is required for AR inhibition. Mammalian two-hybrid and glutathione-S-transferase pull-down assays demonstrated that the inhibition of AR activity by FoxO1 involves the interference of androgen-induced interaction of the N- and C-termini of the AR and the recruitment of p160 coactivators to the AR N-terminus and to the androgen response elements of AR target genes[56].
In addition to inhibiting androgen-dependent activation, FOXO1 also inhibits androgen-independent activation of the AR in C4-2 CRPC cells [57]. In agreement with this observation, FOXO1 binds directly to and inhibits the transcriptional activity of AR-V7, an AR NH2-terminal splicing variant that is constitutively active in the absence of androgens [58]. The transcriptional activity of AR-V7 is inhibited by the AKT inhibitors (LY294002, Wortmanin, and AKT inhibitor II) in both LNCaP and PC3 PCa cell lines. Mechanistic analyses demonstrated that the effect of AKT inhibitors is mediated by the PI3K-AKT-FOXO1 signaling axis. Accordingly, the constitutive activity of AR-V7 is largely reduced under conditions of PTEN reactivation.
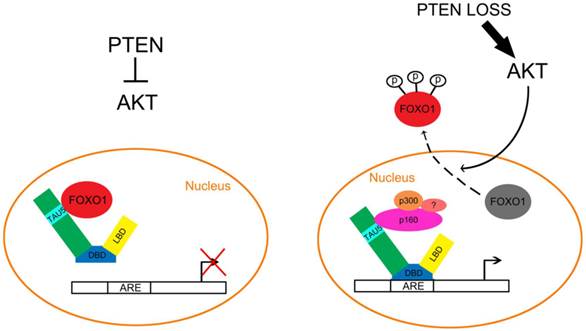
The transcription activation unit 5 (TAU5) motif in the AR NH2-terminal domain (NTD) interacts with AR coactivators such as the p160 family protein SRC-1[59]. The TAU5 motif is important for androgen independent activation of the AR [60]. FOXO1 binds directly to TAU5 and inhibits the constitutive activation of AR splice variants including AR-V2, V4, V5 and V7 [61]. While expression of SRC-1 enhances the transcriptional activity of AR variants, FOXO1 inhibits the SRC-1-induced increase in AR variant transcriptional activity by competing with the binding of SRC-1 to the TAU5 motif in the NTD [61]. Importantly, AR variants-mediated expression of endogenous AR target genes is also inhibited by endogenous FOXO1 in 22Rv1 cells [61]. Based upon these findings, we envisage a model whereby activation of PTEN results in inactivation of AKT, thereby promoting the localization of FOXO1 in the nucleus, binding to the TAU5 motif and inhibiting the androgen-independent activation of the full-length AR and AR splice variants (Fig. 2). However, loss of PTEN activates AKT, which in turn results in nuclear exclusion of FOXO1 and abolishes FOXO1-mediated inhibition of AR, thereby driving castration-resistant progression of PCa (Fig. 2).
Inactivation of the PTEN/FOXO1 pathway promotes aberrant activation of the AR in prostate cancer. Left, in PTEN positive PCa cells, AKT is inactivated and FOXO1 cannot be phosphorylated by AKT. As a result, the unphosphorylated FOXO1 remains in the nucleus and binds directly to the TAU5 domain of AR, thereby inhibiting androgen-dependent and independent activation of the AR. Right, in PTEN-inactivated PCa cells, activation of AKT leads to FOXO1 phosphorylation and nuclear exclusion, which permits the interaction of AR with coactivators including p160 family proteins such as p160 and p300, thereby favoring androgen-dependent and independent activation of the AR in PCa cells.
Taxol chemotherapy is often used to treat CRPC patients. Paclitaxel treatment of 22Rv1 CRPC cells induces nuclear accumulation of FOXO1, increases the association of FOXO1 with AR proteins in the nucleus, and decreases expression of AR target genes including PSA, NKX3.1 and TMPRSS2[62]. Increased levels of FOXO1 proteins are detected in the promoter of the PSA gene in cells treated with paclitaxel. Furthermore, FOXO1 knockdown with small interfering RNA attenuates the inhibitory effect of paclitaxel on AR transcriptional activity, expression of PSA and NKX3.1, and cell survival [62]. Paclitaxel treatment induces increased expression and nuclear localization of FOXO1 in 22Rv1 CRPC cells, similar to that observed in breast and ovarian cancer cells [63-65]. Importantly, Taxol-induced inhibition of the AR is mediated by the nuclear factor FOXO1 [62]. Taxol-induced inhibition of the AR also suggests a connection between the microtubule-dependent trafficking of the AR and the clinical efficacy of Taxol[66].
Other members of the FOXO subfamily, such as FOXO3, can also modulate AR expression in an indirect manner in PCa cells. It has been hypothesized that AKT phosphorylation of FOXO3 is inhibited by isoflavone, thereby affecting the down-regulation of both AR and its target gene PSA [67]. It was demonstrated that isoflavone blocks FOXO3 binding to the promoter of AR but increases FOXO3a binding to the p27KIP1 promoter [67]. This mechanism alters p27KIP1 and AR expression, inhibits cell proliferation, and induces cell apoptosis in both androgen-sensitive and -insensitive PCa cells [67]. FOXO3 can induce AR expression at both the protein and mRNA levels. FOXO3 can induce AR promoter activity via binding to the AR promoter region from 1290 to 1297 (5'-TTGTTTCA-3'). Under normal growth conditions, blocking PI3K/AKT signals by LY294002 (PI3K inhibitor) causes LNCaP cell cycle arrest in G1 phase rather than apoptosis [68, 69].
Perspectives and Conclusion
Taken together, these findings suggest that increased FOXA1 results in an overactive AR complex, which is responsive to low levels of DHT in CRPC. As such, high levels FOXA1 are related to poor prognosis of PCa. On the other hand, AR also has a FOXA1-independent function to open and maintain chromatin structure. Thus, these AR bound DNA regions do not require FOXA1 for relaxing the chromatin. Apart from FOXA1, FOXO1 blocks AR activity via both direct and indirect mechanisms in PCa. FOXO1 can bind to AR and affect its transcriptional activity. FOXO1 can mediate the signaling that controls the activity of AR in PCa. However, FOXO3 upregulates AR expression via binding to the promoter of AR. Therefore, FOXO factors and AR serve as important links between metabolism, the PTEN/AKT pathway and nuclear receptor activation in PCa. Targeting the FOXA and FOXO factors with drugs may offer selective means of affecting AR activity in CRPC patients.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Choi SW, Friso S. Epigenetics: A New Bridge between Nutrition and Health. Adv Nutr. 2010;1:8-16
2. Elbi C, Walker DA, Romero G, Sullivan WP, Toft DO, Hager GL. et al. Molecular chaperones function as steroid receptor nuclear mobility factors. Proc Natl Acad Sci U S A. 2004;101:2876-81
3. Roy AK, Lavrovsky Y, Song CS, Chen S, Jung MH, Velu NK. et al. Regulation of androgen action. Vitam Horm. 1999;55:309-52
4. Askew EB, Minges JT, Hnat AT, Wilson EM. Structural features discriminate androgen receptor N/C terminal and coactivator interactions. Mol Cell Endocrinol. 2012;348:403-10
5. Heinlein CA, Chang C. Androgen receptor in prostate cancer. Endocr Rev. 2004;25:276-308
6. Huggins C, Hodges CV. Studies on prostatic cancer: I. The effect of castration, of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. 1941. J Urol. 2002;168:9-12
7. Scherr D, Swindle PW, Scardino PT. National Comprehensive Cancer Network guidelines for the management of prostate cancer. Urology. 2003;61:14-24
8. Debes JD, Tindall DJ. Mechanisms of androgen-refractory prostate cancer. N Engl J Med. 2004;351:1488-90
9. Feldman BJ, Feldman D. The development of androgen-independent prostate cancer. Nat Rev Cancer. 2001;1:34-45
10. Grossmann ME, Huang H, Tindall DJ. Androgen receptor signaling in androgen-refractory prostate cancer. J Natl Cancer Inst. 2001;93:1687-97
11. Titus MA, Schell MJ, Lih FB, Tomer KB, Mohler JL. Testosterone and dihydrotestosterone tissue levels in recurrent prostate cancer. Clin Cancer Res. 2005;11:4653-7
12. Dehm SM, Schmidt LJ, Heemers HV, Vessella RL, Tindall DJ. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 2008;68:5469-77
13. Guo Z, Yang X, Sun F, Jiang R, Linn DE, Chen H. et al. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res. 2009;69:2305-13
14. Hu R, Dunn TA, Wei S, Isharwal S, Veltri RW, Humphreys E. et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009;69:16-22
15. Weigel D, Jurgens G, Kuttner F, Seifert E, Jackle H. The homeotic gene fork head encodes a nuclear protein and is expressed in the terminal regions of the Drosophila embryo. Cell. 1989;57:645-58
16. Lehmann OJ, Sowden JC, Carlsson P, Jordan T, Bhattacharya SS. Fox's in development and disease. Trends Genet. 2003;19:339-44
17. Hannenhalli S, Kaestner KH. The evolution of Fox genes and their role in development and disease. Nat Rev Genet. 2009;10:233-40
18. Clark KL, Halay ED, Lai E, Burley SK. Co-crystal structure of the HNF-3/fork head DNA-recognition motif resembles histone H5. Nature. 1993;364:412-20
19. Cirillo LA, McPherson CE, Bossard P, Stevens K, Cherian S, Shim EY. et al. Binding of the winged-helix transcription factor HNF3 to a linker histone site on the nucleosome. Embo J. 1998;17:244-54
20. Cirillo LA, Lin FR, Cuesta I, Friedman D, Jarnik M, Zaret KS. Opening of compacted chromatin by early developmental transcription factors HNF3 (FoxA) and GATA-4. Mol Cell. 2002;9:279-89
21. Kaestner KH. The FoxA factors in organogenesis and differentiation. Curr Opin Genet Dev. 2010;20:527-32
22. Belikov S, Astrand C, Wrange O. FoxA1 binding directs chromatin structure and the functional response of a glucocorticoid receptor-regulated promoter. Mol Cell Biol. 2009;29:5413-25
23. Sekiya T, Muthurajan UM, Luger K, Tulin AV, Zaret KS. Nucleosome-binding affinity as a primary determinant of the nuclear mobility of the pioneer transcription factor FoxA. Genes Dev. 2009;23:804-9
24. Huang H, Tindall DJ. Dynamic FoxO transcription factors. J Cell Sci. 2007;120:2479-87
25. Furuyama T, Nakazawa T, Nakano I, Mori N. Identification of the differential distribution patterns of mRNAs and consensus binding sequences for mouse DAF-16 homologues. Biochem J. 2000;349:629-34
26. Gilley J, Coffer PJ, Ham J. FOXO transcription factors directly activate bim gene expression and promote apoptosis in sympathetic neurons. J Cell Biol. 2003;162:613-22
27. Huang H, Tindall DJ. Dynamic FoxO transcription factors. J Cell Sci. 2007;120:2479-87
28. Huang H, Regan KM, Lou Z, Chen J, Tindall DJ. CDK2-dependent phosphorylation of FOXO1 as an apoptotic response to DNA damage. Science. 2006;314:294-7
29. Liu P, Kao TP, Huang H. CDK1 promotes cell proliferation and survival via phosphorylation and inhibition of FOXO1 transcription factor. Oncogene. 2008;27:4733-44
30. Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS. et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857-68
31. Brunet A, Kanai F, Stehn J, Xu J, Sarbassova D, Frangioni JV. et al. 14-3-3 transits to the nucleus and participates in dynamic nucleocytoplasmic transport. J Cell Biol. 2002;156:817-28
32. Gao N, Zhang J, Rao MA, Case TC, Mirosevich J, Wang Y. et al. The role of hepatocyte nuclear factor-3 alpha (Forkhead Box A1) and androgen receptor in transcriptional regulation of prostatic genes. Mol Endocrinol. 2003;17:1484-507
33. Lupien M, Eeckhoute J, Meyer CA, Wang Q, Zhang Y, Li W. et al. FoxA1 translates epigenetic signatures into enhancer-driven lineage-specific transcription. Cell. 2008;132:958-70
34. Jia L, Landan G, Pomerantz M, Jaschek R, Herman P, Reich D. et al. Functional enhancers at the gene-poor 8q24 cancer-linked locus. PLoS Genet. 2009;5:e1000597
35. Carroll JS, Liu XS, Brodsky AS, Li W, Meyer CA, Szary AJ. et al. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell. 2005;122:33-43
36. Eeckhoute J, Carroll JS, Geistlinger TR, Torres-Arzayus MI, Brown M. A cell-type-specific transcriptional network required for estrogen regulation of cyclin D1 and cell cycle progression in breast cancer. Genes Dev. 2006;20:2513-26
37. Lee CS, Friedman JR, Fulmer JT, Kaestner KH. The initiation of liver development is dependent on Foxa transcription factors. Nature. 2005;435:944-7
38. Friedman JR, Kaestner KH. The Foxa family of transcription factors in development and metabolism. Cell Mol Life Sci. 2006;63:2317-28
39. Kaestner KH. The FoxA factors in organogenesis and differentiation. Curr Opin Genet Dev. 2010;20:527-32
40. Gao N, Zhang J, Rao MA, Case TC, Mirosevich J, Wang Y. et al. The role of hepatocyte nuclear factor-3 alpha (Forkhead Box A1) and androgen receptor in transcriptional regulation of prostatic genes. Mol Endocrinol. 2003;17:1484-507
41. Mirosevich J, Gao N, Gupta A, Shappell SB, Jove R, Matusik RJ. Expression and role of Foxa proteins in prostate cancer. Prostate. 2006;66:1013-28
42. Sahu B, Laakso M, Ovaska K, Mirtti T, Lundin J, Rannikko A. et al. Dual role of FoxA1 in androgen receptor binding to chromatin, androgen signalling and prostate cancer. EMBO J. 2011;30:3962-76
43. Sahu B, Laakso M, Ovaska K, Mirtti T, Lundin J, Rannikko A. et al. Dual role of FoxA1 in androgen receptor binding to chromatin, androgen signalling and prostate cancer. Embo J. 2011;30:3962-76
44. Wang D, Garcia-Bassets I, Benner C, Li W, Su X, Zhou Y. et al. Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature. 2011;474:390-4
45. Taslim C, Chen Z, Huang K, Huang TH, Wang Q, Lin S. Integrated analysis identifies a class of androgen-responsive genes regulated by short combinatorial long-range mechanism facilitated by CTCF. Nucleic Acids Res. 2012;40:4754-64
46. Tan PY, Chang CW, Chng KR, Wansa KD, Sung WK, Cheung E. Integration of regulatory networks by NKX3-1 promotes androgen-dependent prostate cancer survival. Mol Cell Biol. 2012;32:399-414
47. Song Y, Washington MK, Crawford HC. Loss of FOXA1/2 is essential for the epithelial-to-mesenchymal transition in pancreatic cancer. Cancer Res. 2010;70:2115-25
48. Jin HJ, Zhao JC, Ogden I, Bergan RC, Yu J. Androgen receptor-independent function of FoxA1 in prostate cancer metastasis. Cancer Res. 2013;73:3725-36
49. Jain RK, Mehta RJ, Nakshatri H, Idrees MT, Badve SS. High-level expression of forkhead-box protein A1 in metastatic prostate cancer. Histopathology. 2011;58:766-72
50. Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP. et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239-43
51. Decker KF, Zheng D, He Y, Bowman T, Edwards JR, Jia L. Persistent androgen receptor-mediated transcription in castration-resistant prostate cancer under androgen-deprived conditions. Nucleic Acids Res. 2012;40:10765-79
52. Zhang C, Wang L, Wu D, Chen H, Chen Z, Thomas-Ahner JM. et al. Definition of a FoxA1 Cistrome that is crucial for G1 to S-phase cell-cycle transit in castration-resistant prostate cancer. Cancer Res. 2011;71:6738-48
53. Dong XY, Chen C, Sun X, Guo P, Vessella RL, Wang RX. et al. FOXO1A is a candidate for the 13q14 tumor suppressor gene inhibiting androgen receptor signaling in prostate cancer. Cancer Res. 2006;66:6998-7006
54. Fan W, Yanase T, Morinaga H, Okabe T, Nomura M, Daitoku H. et al. Insulin-like growth factor 1/insulin signaling activates androgen signaling through direct interactions of Foxo1 with androgen receptor. J Biol Chem. 2007;282:7329-38
55. Burkhardt L, Fuchs S, Krohn A, Masser S, Mader M, Kluth M. et al. CHD1 is a 5q21 tumor suppressor required for ERG rearrangement in prostate cancer. Cancer Res. 2013;73:2795-805
56. Ma Q, Fu W, Li P, Nicosia SV, Jenster G, Zhang X. et al. FoxO1 mediates PTEN suppression of androgen receptor N- and C-terminal interactions and coactivator recruitment. Mol Endocrinol. 2009;23:213-25
57. Liu P, Li S, Gan L, Kao TP, Huang H. A transcription-independent function of FOXO1 in inhibition of androgen-independent activation of the androgen receptor in prostate cancer cells. Cancer Res. 2008;68:10290-9
58. Mediwala SN, Sun H, Szafran AT, Hartig SM, Sonpavde G, Hayes TG. et al. The activity of the androgen receptor variant AR-V7 is regulated by FOXO1 in a PTEN-PI3K-AKT-dependent way. Prostate. 2013;73:267-77
59. Callewaert L, Van Tilborgh N, Claessens F. Interplay between two hormone-independent activation domains in the androgen receptor. Cancer Res. 2006;66:543-53
60. Dehm SM, Regan KM, Schmidt LJ, Tindall DJ. Selective role of an NH2-terminal WxxLF motif for aberrant androgen receptor activation in androgen depletion independent prostate cancer cells. Cancer Res. 2007;67:10067-77
61. Bohrer LR, Liu P, Zhong J, Pan Y, Angstman J, Brand LJ. et al. FOXO1 binds to the TAU5 motif and inhibits constitutively active androgen receptor splice variants. Prostate. 2013;73:1017-27
62. Gan L, Chen S, Wang Y, Watahiki A, Bohrer L, Sun Z. et al. Inhibition of the androgen receptor as a novel mechanism of taxol chemotherapy in prostate cancer. Cancer Res. 2009;69:8386-94
63. Sunters A, Fernandez de Mattos S, Stahl M, Brosens JJ, Zoumpoulidou G, Saunders CA. et al. FoxO3a transcriptional regulation of Bim controls apoptosis in paclitaxel-treated breast cancer cell lines. J Biol Chem. 2003;278:49795-805
64. Sunters A, Madureira PA, Pomeranz KM, Aubert M, Brosens JJ, Cook SJ. et al. Paclitaxel-induced nuclear translocation of FOXO3a in breast cancer cells is mediated by c-Jun NH2-terminal kinase and Akt. Cancer Res. 2006;66:212-20
65. Goto T, Takano M, Hirata J, Tsuda H. The involvement of FOXO1 in cytotoxic stress and drug-resistance induced by paclitaxel in ovarian cancers. Br J Cancer. 2008;98:1068-75
66. Thadani-Mulero M, Nanus DM, Giannakakou P. Androgen receptor on the move: boarding the microtubule expressway to the nucleus. Cancer Res. 2012;72:4611-5
67. Li Y, Wang Z, Kong D, Li R, Sarkar SH, Sarkar FH. Regulation of Akt/FOXO3a/GSK-3beta/AR signaling network by isoflavone in prostate cancer cells. J Biol Chem. 2008;283:27707-16
68. Cornforth AN, Davis JS, Khanifar E, Nastiuk KL, Krolewski JJ. FOXO3a mediates the androgen-dependent regulation of FLIP and contributes to TRAIL-induced apoptosis of LNCaP cells. Oncogene. 2008;27:4422-33
69. Yang L, Xie S, Jamaluddin MS, Altuwaijri S, Ni J, Kim E. et al. Induction of androgen receptor expression by phosphatidylinositol 3-kinase/Akt downstream substrate, FOXO3a, and their roles in apoptosis of LNCaP prostate cancer cells. J Biol Chem. 2005;280:33558-65
Author contact
![]() Corresponding author: Donald J. Tindall, Ph.D. (tindall.donaldedu).
Corresponding author: Donald J. Tindall, Ph.D. (tindall.donaldedu).