International Journal of Biological Sciences
ISSN: 1449-2288
10
Impact Factor
ISSN: 1449-2288
- Current Issue
- Volume 22; 2026
- Volume 21; 2025
- Volume 20; 2024
- Volume 19; 2023
- Volume 18; 2022
- Archive
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Top
Introduction
CR-CaP: Dependence on AR...
CR-CaP: Increased Protein...
Activation of AR by Direct...
Tyrosine Kinase Antagonist...
Future Directions
References
Introduction
CR-CaP: Dependence on AR...
CR-CaP: Increased Protein...
Activation of AR by Direct...
Tyrosine Kinase Antagonist...
Future Directions
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2014; 10(6):620-626. doi:10.7150/ijbs.8264 This issue Cite
Review
Androgen Receptor Activation in Castration-Recurrent Prostate Cancer: The Role of Src-Family and Ack1 Tyrosine Kinases
Irwin H. Gelman ![]()
Department of Cancer Genetics, Roswell Park Cancer Institute, Elm and Carlton Streets, Buffalo, NY 14263, USA.
Received 2013-12-2; Accepted 2014-1-6; Published 2014-6-5
Citation:
Gelman IH. Androgen Receptor Activation in Castration-Recurrent Prostate Cancer: The Role of Src-Family and Ack1 Tyrosine Kinases. Int J Biol Sci 2014; 10(6):620-626. doi:10.7150/ijbs.8264. https://www.ijbs.com/v10p0620.htm
Other stylesAbstract
There is growing appreciation that castration-recurrent prostate cancer (CR-CaP) is driven by the continued expression of androgen receptor (AR). AR activation in CR-CaP through various mechanisms, including AR overexpression, expression of AR splice variants or mutants, increased expression of co-regulator proteins, and by post-translational modification, allows for the induction of AR-regulated genes in response to very low levels of tissue-expressed, so-called intracrine androgens, resulting in pathways that mediate CaP proliferation, anti-apoptosis and oncogenic aggressiveness. The current review focuses on the role played by Src-family (SFK) and Ack1 non-receptor tyrosine kinases in activating AR through direct phosphorylation, respectively, on tyrosines 534 or 267, and how these modifications facilitate progression to CR-CaP. The fact that SFK and Ack1 are central mediators for multiple growth factor receptor signaling pathways that become activated in CR-CaP, especially in the context of metastatic growth in the bone, has contributed to recent therapeutic trials using SFK/Ack1 inhibitors in monotherapy or in combination with antagonists of the AR activation axis.
Keywords: Src-family tyrosine kinases, Ack1, androgen receptor, prostate cancer, castration-recurrence.
Introduction
In 2013, 238,590 new cases of prostate cancer (CaP) are estimated to be identified, typically through increased levels of serum prostate specific antigen (PSA), digital rectal exams and pathologic analyses of biopsy samples. Roughly 12-13% of these cases will result in death (29,720 estimated in 2013), making it the second leading cause of cancer deaths in U.S. men (http://seer.cancer.gov/statfacts/html/prost.html). Since the initial description by Huggins and Hodges in 1941 [1] that CaP can be clinically reduced by castration, which causes deprivation of testicular androgen, most treatment-naïve CaP cases have been treated with some combination of androgen-deprivation therapy (ADT) plus surgical prostatectomy or radio-ablation. This has resulted in high initial cure rates of early, localized disease and palliation of metastatic disease. However, the so-called lethal clinical phenotype of CaP relates to disease recurrence following ADT, producing castration-recurrent (CR) metastatic disease that is found primarily in the bones and lymph nodes, and that responds poorly to standard chemotherapy and radiation [2].
CR-CaP: Dependence on AR signaling
Most CR-CaP cases continue to express the androgen receptor (AR) as well as significant androgen- and AR-driven genes such as PSA [3]. Indeed, CR-CaP cases typically express increased levels of AR protein [4-6], sometimes attributable to AR gene amplification [7], and as well, increased levels of androgen-responsive genes such as PSA and TMPRSS2 [8]. The notion that CR-CaP is AR-dependent is supported by the recent clinical success of AR antagonists such as enzalutamide (MDV3100), which increases survival rates in CR-CaP patients but only for several months [9]. The ultimate failure of AR antagonists to produce durable cures has been associated with the generation of AR splice variants lacking the ligand-binding domain (LBD) [10] or a F876L mutation in the LBD [11, 12]. Less understandable, however, are enzalutamide-resistant cases that continue to express WT-AR; these may reflect that AR overexpression may convert AR antagonists to agonists [13]. In addition to AR activation by direct tyrosine phosphorylation by SFK and Ack1 kinases- which will be the focus of the current review, other studies have identified additional AR-related changes that may contribute to CR-CaP: i) AR mutations (primarily in the LBD) that increase binding for non-androgen agonists [14], ii) AR stabilization [15], iii) induction of AR co-regulators and iv) post-translational modification [14]. These CR-associated changes are thought to facilitate AR-driven tumor progression in response to the post-castration expression of low levels tissue androgens [16].
The notion that the sustained expression of AR-driven genes is required for CR-CaP generation is supported by several studies using mouse models of human CaP. For example, Yuan et al. [17] showed that growth of a cell line (R3) derived from CR-growths of the human CWR22 xenograft in castrates was suppressed by the shRNA-mediated knockdown of AR, even though growth was not inhibited by the anti-androgen, bicalutimide. Campagno et al. [18] showed that intratumoral injections of AR-siRNA inhibited growth of LNCaP-C4-2 and CWR22Rv1 cells, two CR-CaP lines. Similar findings were demonstrated by Snoek et al. [19] using a tetracycline-regulated AR-shRNA in LNCaP-C4-2 cells and by Guo et al. [20] using AR-shRNA in CWR22Rv1 cells in castrates. An interesting twist on this concept comes from the study of Gan et al., [21], who showed that the ability of paclitaxel to inhibit the CR growth of CWR22Rv1 cells correlated with the downregulation of AR caused by the expression of the PTEN tumor suppressor and the nuclear accumulation of the AR suppressive transcription factor, FOXO1. Thus, siRNA-mediated FOXO1 knockdown attenuated the inhibitory effect of paclitaxel on AR-driven CR-promoting genes.
Several studies have demonstrated that androgen-independent or CR-CaP cell lines, or CR-CaP tumor samples, have AR-driven gene expression signatures that correlate with CR-CaP. Work from the Chinnaiyan and Brown labs [22, 23] used AR-ChIP-chip or -ChIP-seq combined with gene expression analyses to identify AR cistrome genes in an androgen-independent LNCaP variant cell line whose expression pattern correlated with CR disease in humans. For example, the study of Wang et al. [23] identified UBE2C, an AR-driven cell cycle regulated gene that overrides an M-phase checkpoint, as a marker of androgen-independent growth. Interestingly, by comparing the AR cistrome in CaP cells expressing full-length (FL) AR vs. a splice variant (V) of AR typically upregulated in abiraterone and enzalutamide-resistant CR-CaP cells and tumors, Hu et al. [24] also identified UBE2C expression as a marker of specific for CR-CaP. A more recent study by Sharma et al. [25] analyzed the AR cistrome in human clinical CR-CaP samples, identifying a 16-gene signature with stronger correlation to CR-CaP than the aforementioned Chinnaiyan and Brown lab studies. Importantly, these and other studies identified transcription factors such as GATA, OCT, PU1, TEF1 [26], FOXA1 [25], and the methyltransferase, EZH2 [27] as potential AR co-factors based on the proximity of their ChIP sites with those of AR. Some transcriptional co-factors, such as FOXA1, may only play a role in ligand-dependent AR function, since androgen-independent AR binding sites in CR-CaP, whose sequences increasingly diverge from the classic androgen responsive element (ARE: 5'-GGA/TACANNNTGTTCT-3'; [28]), form under conditions of FOXA1-independence [29].
CR-CaP: Increased Protein Tyrosine Phosphorylation, SFK and Ack1 activity
The regulation of AR transcriptional activity by post-translational modifications such as phosphorylation, acetylation, sumoylation, ubiquitination and methylation, have been well described [30]. CR-CaP clinical samples exhibit increased relative protein tyrosine phosphorylation levels compared to levels in AD-CaP [31]. This correlates with increased activation levels on non-receptor tyrosine kinases [32] as well as receptor tyrosine kinases such as Met [33], c-Kit [34] and the EGFR family [35, 36]. However, the role of AR tyrosine phosphorylation may have been underappreciated because it is not induced by androgens such as dihydrotestosterone (DHT), whereas it is induced by EGF, heregulin, IL-6 or serum [20, 37, 38]. In contrast, androgens induce IGF-1R activity in prostate cancer progression [39].
Although Src-family kinases are rarely mutated in human cancers [40, 41], there is growing appreciation that they play critical roles in cancer progression, especially associated with recurrent and metastatic disease [42-45], due to gene amplification [46], overexpression or activation by post-translational modification (reviewed in [47]). Indeed, SFK play central roles in mediating oncogenic signaling downstream of many receptor tyrosine kinases, which are themselves activated in cancer by multiple mechanisms [48]. FGR is the only SFK member gene that has been shown to be amplified in prostate cancer, specifically, in 37% of hormone-refractory disease [49]. In contrast, SFK members Src and Lyn are activated in CaP cell lines [50, 51] and tumor tissues [51], and Fyn is upregulated in primary prostate cancer vs. benign lesions [52], and even higher in metastases as evidenced by in silico analysis of Oncomine studies (Figure 1). Most SFK members share an autophosphorylation site (termed Y416 for chicken Src, Y419 for human Src) that is recognized by Src-poY416-specific antibodies and that is an appropriate surrogate marker for kinase activity in cells [53]. A limited set of studies has demonstrated a ~2.5-fold increase in relative Src-poY416 staining in CR- vs. AD-CaP samples [20, 54], and in metastases (bone and lymph node) vs. primary tumor sites (Figure 2, unpublished data, Gary Gallick, MD Anderson Cancer Center). Taken together, these data strongly support a role for increased tyrosine kinase and SFK activity for progression to CR- and metastatic CaP.
Figure 1
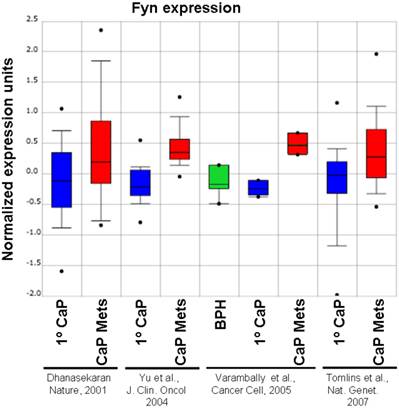
Relative expression level of Fyn in BPH, primary CaP (1º) and metastases (“Mets”) from Oncomine (http://www.oncomine.org) from the studies of Dhanasekaran et al. [116], Yu et al. [117], Varambally et al. [118] and Tomlins et al. [119].
Figure 2
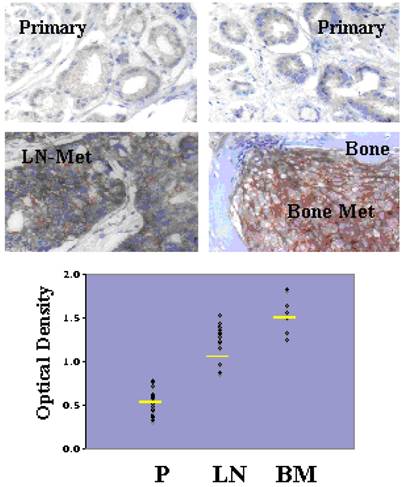
Increased levels of activated Src in lymph node (LN) and bone metastases (BM) compared to primary-site (P) CaP. De-identified patient formalin-fixed paraffin sections from pathology archives (MD Anderson Cancer Center, under IACUC approval) were stained using Src-poY416-specific antibody (Cell Signaling Technologies, Inc., cat. # 201, 1:100, as described in [58]). Top images- Immunohistochemistry staining analysis for Src-poY416 in typical P, LN and BM clinical samples. Bottom panel- Scoring for relative Src-poY416 staining using the 0 to 3+ system, with the mean values shown as yellow lines. Published with permission from Gary E. Gallick, M.D. Anderson Cancer Center.
In addition to the role of SFK in prostate cancer progression, several groups have reported that the non-receptor tyrosine kinase, Ack1 (Activated Cdc42-associated Kinase 1) may facilitate CaP progression through the direct activation of AR. Several mechanisms for Ack1 activation in prostate cancer have been identified, including gene amplification [55] or kinase hyperactivation [38] that occurs downstream of multiple receptor tyrosine kinases [56]. As shown in the study of Taylor et al. [57] (Figure 3), increasing levels of Ack1 (TNK2) message are found in primary site CaP compared to normal or benign prostate hyperplasias (BPH), and even higher levels are found in lymph node metastases.
Several lines of evidence indicate that the expression of specific SFK or Ack1 can drive the formation of CaP or progression to CR-CaP. Although not the main focus of this review, there is a large body of evidence showing that SFK play key roles in facilitating proliferation of CaP induced by various growth factors and in promoting oncogenic migration parameters such as invasiveness [58] (reviewed in [32, 59]). Indeed, Src is required for the lymph node metastasis of a metastatic variant of PC-3 CaP cells although its knockdown has no effect on primary tumor growth [58]. Gelman et al. [60] recently demonstrated that TRAMP mice, whose prostate cancer progression is induced by the prostate-specific transgenic expression of the SV40 Tag [61], had greatly diminished prostatic adenocarcinoma and metastasis formation rates when crossed into Src-null, and to a lesser extent, Lyn-null backgrounds, but no change in the rate or extent of conversion to neuroendocrine cancer in the prostate. The loss of Fyn had no effect on primary tumor or metastasis formation in TRAMP mice exhibiting CaP. However, an interesting finding, and one that merits further investigation in the context of human disease, is that in rare cases where primary CaP failed to form within the typical onset period (<20 weeks of age), the loss of Src, Lyn or Fyn resulted in highly aggressive metastatic disease exhibiting markers of adenocarcinoma. This might suggest that SFK suppress the growth of metastases in the absence of paracrine factors secreted by primary tumors, a phenomenon described in the TRAMP model [62] and in human cancers [63].
Figure 3
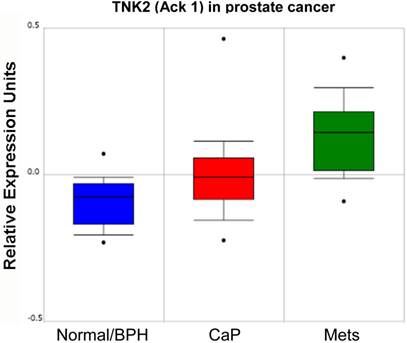
Relative expression level of Ack1 (TNK2) in normal/BPH, primary CaP and lymph node metastases (mets) from Oncomine (http://www.oncomine.org) from the study of Taylor et al. [57].
The notion that activated Src is sufficient to drive CaP initiation comes from the study of Cai et al. [64] who used a tissue recombination model to show that Src, and to lesser extents, Fyn and Lyn, can induce prostatic basal epithelial cells to form CaP tumors when mixed with urogenital sinus mesenchymal cells. Subsequent phosphoproteome analyses of mouse tumors induced by activated AKT plus AR, ERG, or activated K-Ras, as well as of metastatic CR-CaP patient tumors, showed evidence of Src-driven pathways [31, 65]. Interestingly, even the overexpression of non-mutated c-Src was able to induce CaP initiation in the context of AR overexpression [66], an important finding given that Src kinase-activating mutations are not readily found in primary or CR-CaP [67-69]. A recent paper by Su et al. [70] demonstrates that the frequency and time-to-onset of spontaneously generated CR-CaP in the CWR22 xenograft model are decreased by the siRNA-mediated knockdown of Src.
Activation of AR by Direct Phosphorylation: Role of SFK and Ack1
The landmark study by Guo et al. [20] demonstrated that AR activation could be induced by direct phosphorylation by Src on Y534, as identified by mass spectrometry. Kraus et al. [71] confirmed that the Src-mediated tyrosine phosphorylation of AR was positively regulated by the PKC scaffolding protein, RACK1. The concept that AR might serve as a Src substrate is strengthened by evidence that a Src-AR complex is required for androgen-induced CaP cell proliferation in vitro [72] and that Src kinase inhibition blocks AR-dependent transactivation of known androgen-inducible genes [20, 73]. Moreover, expression of activated Src and the subsequent phosphorylation of AR on Y534 are sufficient to induce androgen-independent growth in vitro in LNCaP and LAPC-4 cells [38] and CR growth in vivo [20]. Activated Ack1 could also facilitate androgen-independent CaP cell proliferation in vitro through the direct phosphorylation of AR on Y267 [38, 74]. EGF treatment of LNCaP or LAPC-4 cells could induce AR phosphorylation on both Y267 and Y534, whereas other pro-proliferative stimulants could only activate AR through either Ack1 or Src pathways: AR-poY267 could be induced by heregulin or the Mer receptor ligand, Gas6, whereas AR-poY534 could be induced by IL-6 or bombesin [37]. Importantly, increased AR-poY267 and -poY534 staining levels correlate with CR-CaP disease progression and worse survival prognosis [20, 75], strongly suggesting that the increased activation of SFK and Ack1, and subsequent AR activation through direct phosphorylation by these kinases, facilitates CaP malignancy, especially CR progression.
Several non-genotropic AR functions have been described and are thought to occur at the plasma membrane, i.e.- not through AR's transactivation function [76]. Interestingly, Src may controls these functions, which are poorly understood but which may regulate cell survival pathways, by direct binding to AR [77, 78] although data are lacking as to whether this control axis is dependent on Src phosphorylation of AR.
Tyrosine Kinase Antagonist Treatment in CR-CaP
Studies corroborating the involvement of activated Src in progression to androgen-independence or castration-recurrence [70, 73, 79, 80], increased invasiveness [66], or metastatic growth in bones [81], as well as preclinical studies demonstrating critical roles for SFK in prostate cancer metastasis [51, 58, 82-85] have spawned clinical trials using SFK-specific or pan-tyrosine kinase inhibitors (reviewed in [2, 36]). Indeed, a large set of review papers have addressed the rationale of targeting of SFK and Ack1 in CR-CaP, especially in its main manifestation as bone metastatic growths [36, 42, 43, 58, 86-90].
The effects of kinase inhibitors on CaP biology in vitro and in vivo depends, in most cases, on the specificity of the drugs studied. For example, Dasatinib (BMS-354825), originally described as a Src/Abl-specific inhibitor [91], likely inhibits a wide range of receptor- and non-receptor tyrosine kinases [92]. Thus, whereas initial reports demonstrated that Dasatinib could inhibit Src/FAK-mediated signaling pathways that control prostate cancer cell adhesion, motility and invasiveness [50, 93], subsequent reports showed that it could also inhibit CaP growth as boney metastases [94], androgen-independent growth [54] associated with the site-specific tyrosine phosphorylation of AR by Src or Ack1 [37], or spontaneous formation of CR (CWR22) tumors [70]. Although early clinical trials showed some efficacy of using Dasatinib as a monotherapy or in combination with docetaxel [95-99], the recent READY Phase III trial showed that adding Dasatinib had no greater effect on survival in cases of chemotherapy-naïve metastatic CR-CaP [100]. Importantly, serum markers, such as insulin-like growth factor-1 (IGF-1), have been identified that correlate with efficacy by Dasatinib in metastatic CR-CaP cases [101], prompting a study by Dayyani et al. [102] showing superior inhibition of CR-CaP growths in mouse models when combining the IGF-1receptor/insulin-receptor inhibitor, BMS-754807, with Dasatinib.
The argument that Dasatinib's broad tyrosine kinase specificity undercuts its clinical efficacy [103] has resulted in the increased focus on more specific SFK inhibitors in CR-CaP. For example, Saracatinib (AZD0530), a Src/Abl inhibitor which showed anti-CaP efficacy in preclinical CaP models [104, 105] and in PC-3 bone metastases [106], showed efficacy as a monotherapy in CR-CaP patients [107]. Bosutinib (SKI-606) is another Src/Abl inhibitor [108] that has shown efficacy against prostate cancer in preclinical models [109], has passed Phase I testing [110] and was FDA-approved for use in Gleevec-resistant chronic myelogenous leukemia, yet is not part of any current Phase II trials for prostate cancer. Cabozantinib (XL184), an inhibitor of the Met receptor tyrosine kinase, which manifests its oncogenic signaling in prostate cancer through Src activation [83], has shown efficacy in Phase II trials against CR-CaP [111]. KXO1 (KX2-391) is a novel non-ATP-competitive Src/tubulin polymerization inhibitor which, in recent Phase II studies [112], failed to reduce tumor burden in bone-metastasis CR-CaP cases, although a caveat noted was that the dosing was insufficient to achieve the drug's tubulin polymerization inhibition activity. Lastly, several new Ack1-specific inhibitors have been identified [113], including AIM-100, which has shown efficacy against CR-CaP cell lines in vitro and in vivo [75, 114].
Future Directions
The growing corpus of evidence showing that SFK and Ack1 are potent drivers of CR-CaP and metastasis via AR-dependent mechanisms has led to a renewed focus on developing specific tyrosine kinase inhibitors as mainline or adjunct therapies. In addition to solving issues of increased toxicities when combining such antagonists with newer generation inhibitors of AR or androgen synthesis, such as enzalutamide or abiraterone, respectively, there is no consensus a priori as to whether SFK-specific drugs might work better than those with broad specificity for tyrosine kinases. As the use of SFK/Ack1 inhibitors increases in CR-CaP populations, one area needing improvement is the identification and development of bio- or genetic markers of drug efficacy. One such example might be the development of immunohistochemistry-grade antibodies specific for AR-poY267 and -poY534. Indeed, newer pharmacogenetic paradigms are being developed to identify individualized response and toxicity signatures that will help stratify patient-specific treatments [115] with the aim of improving patient survival outcomes.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Huggins C, Hodges CV. Studies on Prostatic Cancer: I. The Effect of Castration, of Estrogen and of Androgen Injection on Serum Phosphatases in Metastatic Carcinoma of the Prostate. 1941;1:298-307
2. George D, Moul JW. Emerging treatment options for patients with castration-resistant prostate cancer. Prostate. 2012;72:338-349
3. Azzouni F, Mohler J. Biology of castration-recurrent prostate cancer. Urol Clin North Am. 2012;39:435-452
4. Sharifi N. Mechanisms of androgen receptor activation in castration-resistant prostate cancer. Endocrinology. 2013;154:4010-4017
5. Mohler JL. A role for the androgen-receptor in clinically localized and advanced prostate cancer. Best Pract Res Clin Endocrinol Metab. 2008;22:357-372
6. Schmidt LJ, Tindall DJ. Androgen receptor: past, present and future. Curr Drug Targets. 2013;14:401-407
7. El Gedaily A, Bubendorf L, Willi N. et al. Discovery of new DNA amplification loci in prostate cancer by comparative genomic hybridization. Prostate. 2001;46:184-190
8. Ryan CJ, Tindall DJ. Androgen receptor rediscovered: the new biology and targeting the androgen receptor therapeutically. J Clin Oncol. 2011;29:3651-3658
9. Scher HI, Fizazi K, Saad F. et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012;367:1187-1197
10. Li Y, Chan SC, Brand LJ. et al. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res. 2013;73:483-489
11. Joseph JD, Lu N, Qian J. et al. A Clinically Relevant Androgen Receptor Mutation Confers Resistance to Second-Generation Antiandrogens Enzalutamide and ARN-509. Cancer Discov. 2013;3:1020-1029
12. Korpal M, Korn JM, Gao X. et al. An F876L Mutation in Androgen Receptor Confers Genetic and Phenotypic Resistance to MDV3100 (Enzalutamide). Cancer Discov. 2013;3:1030-1043
13. Chen CD, Welsbie DS, Tran C. et al. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10:33-39
14. Lamont KR, Tindall DJ. Minireview: Alternative activation pathways for the androgen receptor in prostate cancer. Mol Endocrinol. 2011;25:897-907
15. Gregory CW, Johnson RT Jr, Mohler JL. et al. Androgen receptor stabilization in recurrent prostate cancer is associated with hypersensitivity to low androgen. Cancer Res. 2001;61:2892-2898
16. Mohler JL. Castration-recurrent prostate cancer is not androgen-independent. Adv Exp Med Biol. 2008;617:223-34
17. Yuan X, Li T, Wang H. et al. Androgen receptor remains critical for cell-cycle progression in androgen-independent CWR22 prostate cancer cells. Am J Pathol. 2006;169:682-696
18. Compagno D, Merle C, Morin A. et al. SIRNA-directed in vivo silencing of androgen receptor inhibits the growth of castration-resistant prostate carcinomas. PLoS One. 2007;2(e):1006
19. Snoek R, Cheng H, Margiotti K. et al. In vivo knockdown of the androgen receptor results in growth inhibition and regression of well-established, castration-resistant prostate tumors. Clin Cancer Res. 2009;15:39-47
20. Guo Z, Dai B, Jiang T. et al. Regulation of androgen receptor activity by tyrosine phosphorylation. Cancer Cell. 2006;10:309-319
21. Gan L, Chen S, Wang Y. et al. Inhibition of the androgen receptor as a novel mechanism of taxol chemotherapy in prostate cancer. Cancer Res. 2009;69:8386-8394
22. Wang Q, Li W, Liu XS. et al. A hierarchical network of transcription factors governs androgen receptor-dependent prostate cancer growth. Mol Cell. 2007;27:380-392
23. Wang Q, Li W, Zhang Y. et al. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell. 2009;138:245-256
24. Hu R, Lu C, Mostaghel EA. et al. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res. 2012;72:3457-3462
25. Sharma NL, Massie CE, Ramos-Montoya A. et al. The androgen receptor induces a distinct transcriptional program in castration-resistant prostate cancer in man. Cancer Cell. 2013;23:35-47
26. Lin B, Wang J, Hong X. et al. Integrated expression profiling and ChIP-seq analyses of the growth inhibition response program of the androgen receptor. PLoS One. 2009;4(e):6589
27. Chng KR, Chang CW, Tan SK. et al. A transcriptional repressor co-regulatory network governing androgen response in prostate cancers. EMBO J. 2012;31:2810-2823
28. Roche PJ, Hoare SA, Parker MG. A consensus DNA-binding site for the androgen receptor. Mol Endocrinol. 1992;6:2229-2235
29. Decker KF, Zheng D, He Y. et al. Persistent androgen receptor-mediated transcription in castration-resistant prostate cancer under androgen-deprived conditions. Nucleic Acids Res. 2012;40:10765-10779
30. Coffey K, Robson CN. Regulation of the androgen receptor by post-translational modifications. J Endocrinol. 2012;215:221-237
31. Drake JM, Graham NA, Stoyanova T. et al. Oncogene-specific activation of tyrosine kinase networks during prostate cancer progression. Proc Natl Acad Sci U S A. 2012;109:1643-1648
32. Chang YM, Kung HJ, Evans CP. Nonreceptor tyrosine kinases in prostate cancer. Neoplasia. 2007;9:90-100
33. Knudsen BS, Edlund M. Prostate cancer and the met hepatocyte growth factor receptor. Adv Cancer Res. 2004;91:31-67
34. Paronetto MP, Farini D, Sammarco I. et al. Expression of a truncated form of the c-Kit tyrosine kinase receptor and activation of Src kinase in human prostatic cancer. Am J Pathol. 2004;164:1243-1251
35. Lorenzo GD, Bianco R, Tortora G. et al. Involvement of growth factor receptors of the epidermal growth factor receptor family in prostate cancer development and progression to androgen independence. Clin Prostate Cancer. 2003;2:50-57
36. Gallick GE, Corn PG, Zurita AJ. et al. Small-molecule protein tyrosine kinase inhibitors for the treatment of metastatic prostate cancer. Future Med Chem. 2012;4:107-119
37. Liu Y, Karaca M, Zhang Z. et al. Dasatinib inhibits site-specific tyrosine phosphorylation of androgen receptor by Ack1 and Src kinases. Oncogene. 2010;29:3908-3916
38. Mahajan NP, Liu Y, Majumder S. et al. Activated Cdc42-associated kinase Ack1 promotes prostate cancer progression via androgen receptor tyrosine phosphorylation. Proc Natl Acad Sci U S A. 2007;104:8438-8443
39. 39. Pandini G, Mineo R, Frasca F, et al. Androgens up-regulate the insulin-like growth factor-I receptor in prostate cancer cells. Cancer Res. 2005;65:1849-1857
40. Frame MC. Newest findings on the oldest oncogene; how activated src does it. J Cell Sci. 2004;117:989-998
41. Irby RB, Mao WG, Coppola D. et al. Activating SRC mutation in a subset of advanced human colon cancers. Nature Genet. 1999;21:187-190
42. Boyce B, Xing L. Src inhibitors in the treatment of metastatic bone disease: rationale and clinical data. Clin Investig (Lond). 2011;1:1695-1706
43. Edwards J. Src kinase inhibitors: an emerging therapeutic treatment option for prostate cancer. Expert Opin Investig Drugs. 2010;19:605-614
44. Kim MP, Park SI, Kopetz S. et al. Src family kinases as mediators of endothelial permeability: effects on inflammation and metastasis. Cell Tissue Res. 2009;335:249-259
45. Summy JM, Gallick GE. Src family kinases in tumor progression and metastasis. Cancer Metastasis Rev. 2003;22:337-358
46. Kiyose S, Nagura K, Tao H. et al. Detection of kinase amplifications in gastric cancer archives using fluorescence in situ hybridization. Pathol Int. 2012;62:477-484
47. Gelman IH. Src-family tyrosine kinases as therapeutic targets in advanced cancer. Front Biosci (Elite Ed). 2011;3:801-807
48. Wheeler DL, Iida M, Dunn EF. The role of Src in solid tumors. Oncologist. 2009;14:667-678
49. Edwards J, Krishna NS, Witton CJ. et al. Gene amplifications associated with the development of hormone-resistant prostate cancer. Clin Cancer Res. 2003;9:5271-5281
50. Nam S, Kim D, Cheng JQ. et al. Action of the Src family kinase inhibitor, dasatinib (BMS-354825), on human prostate cancer cells. Cancer Res. 2005;65:9185-9189
51. Goldenberg-Furmanov M, Stein I, Pikarsky E. et al. Lyn is a target gene for prostate cancer: sequence-based inhibition induces regression of human tumor xenografts. Cancer Res. 2004;64:1058-1066
52. Posadas EM, Al-Ahmadie H, Robinson VL. et al. FYN is overexpressed in human prostate cancer. BJU Int. 2009;103:171-177
53. Frame MC. Src in cancer: deregulation and consequences for cell behaviour. Biochimica Et Biophysica Acta-Reviews On Cancer. 2002;1602:114-130
54. Tatarov O, Mitchell TJ, Seywright M. et al. SRC family kinase activity is up-regulated in hormone-refractory prostate cancer. Clin Cancer Res. 2009;15:3540-3549
55. van der Horst EH, Degenhardt YY, Strelow A. et al. Metastatic properties and genomic amplification of the tyrosine kinase gene ACK1. Proc Natl Acad Sci U S A. 2005;102:15901-15906
56. Mahajan K, Mahajan NP. ACK1 tyrosine kinase: targeted inhibition to block cancer cell proliferation. Cancer Lett. 2013;338:185-192
57. Taylor BS, Schultz N, Hieronymus H. et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11-22
58. Park SI, Zhang J, Phillips KA. et al. Targeting SRC family kinases inhibits growth and lymph node metastases of prostate cancer in an orthotopic nude mouse model. Cancer Res. 2008;68:3323-3333
59. Fizazi K. The role of Src in prostate cancer. Ann Oncol. 2007 doi: 10.1093/annonc/mdm086
60. Gelman IH, Peresie J, Eng KH. et al. Differential requirement for Src-family tyrosine kinases for prostate cancer, initiation progression and metastasis in the TRAMP mouse model. Molecular Cancer Research. 2013 in press
61. Kaplan-Lefko PJ, Chen TM, Ittmann MM. et al. Pathobiology of autochthonous prostate cancer in a pre-clinical transgenic mouse model. Prostate. 2003;55:219-237
62. Kwon ED, Foster BA, Hurwitz AA. et al. Elimination of residual metastatic prostate cancer after surgery and adjunctive cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) blockade immunotherapy. Proc Natl Acad Sci U S A. 1999;96:15074-15079
63. Peeters CF, de Waal RM, Wobbes T. et al. Metastatic dormancy imposed by the primary tumor: does it exist in humans? Ann Surg Oncol. 2008;15:3308-3315
64. Cai H, Smith DA, Memarzadeh S. et al. Differential transformation capacity of Src family kinases during the initiation of prostate cancer. Proc Natl Acad Sci U S A. 2011;108:6579-6584
65. Drake JM, Graham NA, Lee JK. et al. Metastatic castration-resistant prostate cancer reveals intrapatient similarity and interpatient heterogeneity of therapeutic kinase targets. Proc Natl Acad Sci U S A. 2013
66. Cai H, Babic I, Wei X. et al. Invasive prostate carcinoma driven by c-Src and androgen receptor synergy. Cancer Res. 2011;71:862-872
67. Beltran H, Yelensky R, Frampton GM. et al. Targeted next-generation sequencing of advanced prostate cancer identifies potential therapeutic targets and disease heterogeneity. Eur Urol. 2013;63:920-926
68. Kumar A, White TA, Mackenzie AP. et al. Exome sequencing identifies a spectrum of mutation frequencies in advanced and lethal prostate cancers. Proc Natl Acad Sci U S A. 2011;108:17087-17092
69. Grasso CS, Wu YM, Robinson DR. et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239-243
70. Su B, Gillard B, Gao L. et al. Src controls castration recurrence of CWR22 prostate cancer xenografts. Cancer Med. 2013;2(6):784-92
71. Kraus S, Gioeli D, Vomastek T. et al. Receptor for activated C kinase 1 (RACK1) and Src regulate the tyrosine phosphorylation and function of the androgen receptor. Cancer Res. 2006;66:11047-11054
72. Migliaccio A, Varricchio L, De FA. et al. Inhibition of the SH3 domain-mediated binding of Src to the androgen receptor and its effect on tumor growth. Oncogene. 2007;26:6619-6629
73. Asim M, Siddiqui IA, Hafeez BB. et al. Src kinase potentiates androgen receptor transactivation function and invasion of androgen-independent prostate cancer C4-2 cells. Oncogene. 2008;27:3596-3604
74. Mahajan NP, Whang YE, Mohler JL. et al. Activated tyrosine kinase Ack1 promotes prostate tumorigenesis: role of Ack1 in polyubiquitination of tumor suppressor Wwox. Cancer Res. 2005;65:10514-10523
75. Mahajan K, Challa S, Coppola D. et al. Effect of Ack1 tyrosine kinase inhibitor on ligand-independent androgen receptor activity. Prostate. 2010;70:1274-1285
76. Yu J, Akishita M, Eto M. et al. Src kinase-mediates androgen receptor-dependent non-genomic activation of signaling cascade leading to endothelial nitric oxide synthase. Biochem Biophys Res Commun. 2012;424:538-543
77. Kousteni S, Bellido T, Plotkin LI. et al. Nongenotropic, sex-nonspecific signaling through the estrogen or androgen receptors: dissociation from transcriptional activity. Cell. 2001;104:719-730
78. Migliaccio A, Castoria G, Di Domenico M. et al. Steroid-induced androgen receptor-oestradiol receptor beta-Src complex triggers prostate cancer cell proliferation. EMBO J. 2000;19:5406-5417
79. Kung HJ, Evans CP. Oncogenic activation of androgen receptor. Urol Oncol. 2009;27:48-52
80. Asim M, Hafeez BB, Siddiqui IA. et al. Ligand-dependent corepressor acts as a novel androgen receptor corepressor, inhibits prostate cancer growth, and is functionally inactivated by the Src protein kinase. J Biol Chem. 2011;286:37108-37117
81. Putzke AP, Ventura AP, Bailey AM. et al. Metastatic progression of prostate cancer and e-cadherin regulation by zeb1 and SRC family kinases. Am J Pathol. 2011;179:400-410
82. Inoue S, Branch CD, Gallick GE. et al. Inhibition of Src kinase activity by Ad-mda7 suppresses vascular endothelial growth factor expression in prostate carcinoma cells. Mol Ther. 2005;12:707-715
83. Kim SJ, Johnson M, Koterba K. et al. Reduced c-Met expression by an adenovirus expressing a c-Met ribozyme inhibits tumorigenic growth and lymph node metastases of PC3-LN4 prostate tumor cells in an orthotopic nude mouse model. Clin Cancer Res. 2003;9:5161-5170
84. Putzke AP, Ventura AP, Bailey AM. et al. Metastatic progression of prostate cancer and e-cadherin regulation by zeb1 and SRC family kinases. Am J Pathol. 2011;179:400-410
85. Yano A, Tsutsumi S, Soga S. et al. Inhibition of Hsp90 activates osteoclast c-Src signaling and promotes growth of prostate carcinoma cells in bone. Proc Natl Acad Sci U S A. 2008;105:15541-15546
86. Agarwal N, Sonpavde G, Sternberg CN. Novel Molecular Targets for the Therapy of Castration-Resistant Prostate Cancer. Eur Urol. 2012;61(5):950-60
87. Rove KO, Crawford ED. Evolution of treatment options for patients with CRPC and bone metastases: bone-targeted agents that go beyond palliation of symptoms to improve overall survival. Oncology (Williston Park). 2011;25:1362-81 1387
88. Saad F. Src as a therapeutic target in men with prostate cancer and bone metastases. BJU Int. 2009;103:434-440
89. Saad F, Lipton A. SRC kinase inhibition: targeting bone metastases and tumor growth in prostate and breast cancer. Cancer Treat Rev. 2010;36:177-184
90. Sturge J, Caley MP, Waxman J. Bone metastasis in prostate cancer: emerging therapeutic strategies. Nat Rev Clin Oncol. 2011;8:357-368
91. Lombardo LJ, Lee FY, Chen P. et al. Discovery of N-(2-chloro-6-methyl- phenyl)-2-(6-(4-(2-hydroxyethyl)- piperazin-1-yl)-2-methylpyrimidin-4- ylamino)thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J Med Chem. 2004;47:6658-6661
92. Li J, Rix U, Fang B. et al. A chemical and phosphoproteomic characterization of dasatinib action in lung cancer. Nat Chem Biol. 2010;6:291-299
93. Rice L, Lepler S, Pampo C. et al. Impact of the SRC inhibitor dasatinib on the metastatic phenotype of human prostate cancer cells. Clin Exp Metastasis. 2012;29:133-142
94. Koreckij T, Nguyen H, Brown LG. et al. Dasatinib inhibits the growth of prostate cancer in bone and provides additional protection from osteolysis. Br J Cancer. 2009;101:263-268
95. Araujo JC, Mathew P, Armstrong AJ. et al. Dasatinib combined with docetaxel for castration-resistant prostate cancer: results from a phase 1-2 study. Cancer. 2012;118:63-71
96. Araujo JC, Trudel GC, Paliwal P. Long-term use of dasatinib in patients with metastatic castration-resistant prostate cancer after receiving the combination of dasatinib and docetaxel. Cancer Manag Res. 2013;6:25-30 doi: 10.2147/CMAR.S41667. Epub;%2013 Mar 9.:25-30
97. Twardowski PW, Beumer JH, Chen CS. et al. A phase II trial of dasatinib in patients with metastatic castration-resistant prostate cancer treated previously with chemotherapy. Anticancer Drugs. 2013;24:743-753
98. Yu EY, Wilding G, Posadas E. et al. Phase II study of dasatinib in patients with metastatic castration-resistant prostate cancer. Clin Cancer Res. 2009;15:7421-7428
99. Yu EY, Massard C, Gross ME. et al. Once-daily dasatinib: expansion of phase II study evaluating safety and efficacy of dasatinib in patients with metastatic castration-resistant prostate cancer. Urology. 2011;77:1166-1171
100. Araujo JC, Trudel GC, Saad F. et al. Docetaxel and dasatinib or placebo in men with metastatic castration-resistant prostate cancer (READY): a randomised, double-blind phase 3 trial. Lancet Oncol. 2013
101. Dayyani F, Varkaris A, Araujo JC. et al. Increased serum insulin-like growth factor-1 levels are associated with prolonged response to dasatinib-based regimens in metastatic prostate cancer. Prostate. 2013;73:979-985
102. Dayyani F, Parikh NU, Varkaris AS. et al. Combined Inhibition of IGF-1R/IR and Src family kinases enhances antitumor effects in prostate cancer by decreasing activated survival pathways. PLoS ONE. 2012;7(e):51189
103. Lara PN, Evans CP. Dasatinib and docetaxel in advanced prostate cancer. Lancet Oncol. 2013
104. Evans CP, Lara PN Jr, Kung H. et al. Activity of the Src-kinase inhibitor AZD0530 in androgen-independent prostate cancer (AIPC): Pre-clinical rationale for a phase II trial. J Clin Oncol (ASCO Annual Meeting Proceedings). 2006;24:14542
105. Chang YM, Bai L, Liu S. et al. Src family kinase oncogenic potential and pathways in prostate cancer as revealed by AZD0530. Oncogene. 2008
106. Yang JC, Bai L, Yap S. et al. Effect of the specific Src family kinase inhibitor saracatinib on osteolytic lesions using the PC-3 bone model. Mol Cancer Ther. 2010;9:1629-1637
107. Lara PN Jr, Longmate J, Evans CP. et al. A phase II trial of the Src-kinase inhibitor AZD0530 in patients with advanced castration-resistant prostate cancer: a California Cancer Consortium study. Anticancer Drugs. 2009;20:179-184
108. Boschelli DH, Wu B, Barrios Sosa AC. et al. Identification of 7-phenylaminothieno- [3,2-b]pyridine-6-carbonitriles as a new class of Src kinase inhibitors. J Med Chem. 2004;47:6666-6668
109. Rabbani SA, Valentino ML, Arakelian A. et al. SKI-606 (Bosutinib) blocks prostate cancer invasion, growth, and metastasis in vitro and in vivo through regulation of genes involved in cancer growth and skeletal metastasis. Mol Cancer Ther. 2010;9:1147-1157
110. Daud AI, Krishnamurthi SS, Saleh MN. et al. Phase I study of bosutinib, a src/abl tyrosine kinase inhibitor, administered to patients with advanced solid tumors. Clin Cancer Res. 2012;18:1092-1100
111. Smith DC, Smith MR, Sweeney C. et al. Cabozantinib in patients with advanced prostate cancer: results of a phase II randomized discontinuation trial. J Clin Oncol. 2013;31:412-419
112. Antonarakis ES, Heath EI, Posadas EM. et al. A phase 2 study of KX2-391, an oral inhibitor of Src kinase and tubulin polymerization, in men with bone-metastatic castration-resistant prostate cancer. Cancer Chemother Pharmacol. 2013;71:883-892
113. Phatak SS, Zhang S. A novel multi-modal drug repurposing approach for identification of potent ACK1 inhibitors. Pac Symp Biocomput. 2013:29-40
114. Mahajan K, Coppola D, Rawal B. et al. Ack1-mediated androgen receptor phosphorylation modulates radiation resistance in castration-resistant prostate cancer. J Biol Chem. 2012;287:22112-22122
115. Hertz DL, McLeod HL. Use of pharmacogenetics for predicting cancer prognosis and treatment exposure, response and toxicity. J Hum Genet. 2013;58:346-352
116. Dhanasekaran SM, Barrette TR, Ghosh D. et al. Delineation of prognostic biomarkers in prostate cancer. Nature JID - 0410462. 2001;412:822-826
117. Yu YP, Landsittel D, Jing L. et al. Gene expression alterations in prostate cancer predicting tumor aggression and preceding development of malignancy. J Clin Oncol. 2004;22:2790-2799
118. Varambally S, Yu J, Laxman B. et al. Integrative genomic and proteomic analysis of prostate cancer reveals signatures of metastatic progression. Cancer Cell. 2005;8:393-406
119. Tomlins SA, Mehra R, Rhodes DR. et al. Integrative molecular concept modeling of prostate cancer progression. Nat Genet. 2007;39:41-51
Author contact
![]() Corresponding author: Tel: 716-845-7681 Fax: 716-845-1698 Irwin.gelmanorg.
Corresponding author: Tel: 716-845-7681 Fax: 716-845-1698 Irwin.gelmanorg.
Citation styles
APA
Gelman, I.H. (2014). Androgen Receptor Activation in Castration-Recurrent Prostate Cancer: The Role of Src-Family and Ack1 Tyrosine Kinases. International Journal of Biological Sciences, 10(6), 620-626. https://doi.org/10.7150/ijbs.8264.
ACS
Gelman, I.H. Androgen Receptor Activation in Castration-Recurrent Prostate Cancer: The Role of Src-Family and Ack1 Tyrosine Kinases. Int. J. Biol. Sci. 2014, 10 (6), 620-626. DOI: 10.7150/ijbs.8264.
NLM
Gelman IH. Androgen Receptor Activation in Castration-Recurrent Prostate Cancer: The Role of Src-Family and Ack1 Tyrosine Kinases. Int J Biol Sci 2014; 10(6):620-626. doi:10.7150/ijbs.8264. https://www.ijbs.com/v10p0620.htm
CSE
Gelman IH. 2014. Androgen Receptor Activation in Castration-Recurrent Prostate Cancer: The Role of Src-Family and Ack1 Tyrosine Kinases. Int J Biol Sci. 10(6):620-626.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.