Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2014; 10(7):733-745. doi:10.7150/ijbs.8097 This issue Cite
Research Paper
MicroRNA-135a Inhibits Cell Proliferation by Targeting Bmi1 in Pancreatic Ductal Adenocarcinoma
Zheng Dang1,2,*, Wei-Hua Xu1,*, Peng Lu1,3,*, Nan Wu1,*, Jie Liu1, Bai Ruan1, Liang Zhou4, Wen-Jie Song1, ![]() , Ke-Feng Dou1,
, Ke-Feng Dou1, ![]()
1. Department of Hepatobiliary Surgery, Xijing Hospital, The Fourth Military Medical University, Xi'an, Shaanxi 710032, P.R. China
2. Department of Hepatobiliary Surgery, Lanzhou General Hospital of PLA, Lanzhou, Gansu 730050, P.R. China
3. Department of General Surgery, The 518 Central Hospital of PLA, Xi'an, Shanxi 710043, P.R. China
4. Department of General Surgery, The 155 Central Hospital of PLA, Kaifeng, He'nan 471000 P.R. China
* Zheng Dang, Wei-Hua Xu, Peng Lu, and Nan Wu are co-first authors and contributed equally to this work.
Received 2013-11-11; Accepted 2014-5-31; Published 2014-6-24
Abstract
Pancreatic ductal adenocarcinoma (PDAC) is a highly lethal solid tumor due to the lack of reliable early detection markers and effective therapies. MicroRNAs (miRNAs), noncoding RNAs that regulate gene expression, are involved in tumorigenesis and have a remarkable potential for the diagnosis and treatment of malignancy. In this study, we investigated aberrantly expressed miRNAs involved in PDAC by comparing miRNA expression profiles in PDAC cell lines with a normal pancreas cell line and found that miR-135a was significantly down-regulated in the PDAC cell lines. The microarray results were validated by qRT-PCR in PDAC tissues, paired adjacent normal pancreatic tissues, PDAC cell lines, and a normal pancreas cell line. We then defined the tumor-suppressing significance and function of miR-135a by constructing a lentiviral vector to express miR-135a. The overexpression of miR-135a in PDAC cells decreased cell proliferation and clonogenicity and also induced G1 arrest and apoptosis. We predicted Bmi1 may be a target of miR-135a using bioinformatics tools and found that Bmi1 expression was markedly up-regulated in PDAC. Its expression was inversely correlated with miR-135a expression in PDAC. Furthermore, a luciferase activity assay revealed that miR-135a could directly target the 3'-untranslated region (3'-UTR) of Bmi1. Taken together, these results demonstrate that miR-135a targets Bmi1 in PDAC and functions as a tumor suppressor. miR-135a may offer a new perspective for the development of effective miRNA-based therapy for PDAC.
Keywords: miR-135a, pancreatic ductal adenocarcinoma, Bmi1, cell proliferation, apoptosis
Introduction
Pancreatic cancer (PC) is a highly malignant tumor type that is characterized by aggressive invasion and a high incidence of metastasis. As a result of late diagnosis and the lack of effective treatment measures, PC has an extremely poor prognosis, ranking 4th among the leading causes of cancer-related deaths in the United States [1]. Pancreatic ductal adenocarcinoma (PDAC) is the most common type of PC, involving more than 90% of PCs, and its 5-year survival rate does not exceed 5% [2]. Although some progress has been made with regard to many aspects of PC in recent years, the survival rate of PC has not increased significantly. As cancer is a complex disease involving numerous genetic and epigenetic changes, a better understanding of the molecular mechanism of PC is urgently needed to develop an effective diagnosis and treatment.
MicroRNAs (miRNAs) are a class of short, noncoding RNAs ranging approximately from 17-25 nucleotides, which contain a seed sequence for binding to the 3'-untranslated region of specific target mRNAs to regulate translational repression and/or mRNA degradation [3,4]. Since the abnormal expression of miR-15 and miR-16 was reported in chronic lymphocytic leukemia (CLL) [5-8], an increasing number of studies have shown that the aberrant expression of specific miRNAs is a common phenomenon in many human cancers. More and more evidences have shown that miRNA mutations or mis-expression correlate with various human cancers and indicate that miRNAs can act as either tumor suppressors or oncogenes [9,10]. MiRNAs directly or indirectly regulate the expression of many target mRNAs simultaneously, playing a critical role in the tumorigenesis, invasion, metastasis, and chemoresistance of cancer cells [11-13]. Indeed, the aberrant expression of various miRNAs has been identified in many aspects of PDAC [14-16] and may provide a new therapeutic opportunity, leading to a better clinical outcome for patients with PDAC.
In the present study, we found that the expression level of miR-135a was significantly down-regulated in PDAC cell lines compared to a normal pancreas cell line through a comprehensive analysis of miRNA expression profiles using microarrays. We then validated the results of the microarray experiments by qRT-PCR in PDAC tissues, paired adjacent normal pancreatic tissues, PDAC cell lines, and a normal pancreas cell line. Further experiments indicated that miR-135a overexpression blocked PDAC cell proliferation. We identified Bmi1 as a target gene of miR-135a using in silico prediction and the luciferase reporter assay. Together, our results suggest that miR-135a may play an important role in the pathogenesis of PDAC by targeting Bmi1.
Materials and Methods
Cell culture and clinical samples
Three human PDAC cell lines, PANC-1, BxPC-3, and ASPC-1, and a normal pancreatic ductal epithelial cell line, HPDE6c7, were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA). All of the cells were cultured in Dulbecco's Modified Eagle's Medium (GIBCO, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (HyClone, Logan, UT, USA) at 37°C in a humidified atmosphere containing 5% CO2.
After obtaining informed consent from all patients involved in the study, 11 specimens of PDAC tissues and their adjacent non-tumor tissues were collected immediately after surgical removal at the Department of Hepatobiliary Surgery, Xijing Hospital of the Fourth Military Medical University (Xi'an, China), from September 2012 to April 2013. None of the patients received chemotherapy prior to surgery. The tissues were snap frozen in liquid nitrogen and stored until further use. The study was approved by the Ethics Committee for Clinical Research of the Fourth Military Medical University.
MiRNA microarray analysis
The microRNA expression profile was determined using the Agilent Human microRNA array kit (V18.0) (Agilent Technologies, Santa Clara, CA, USA), which contains probes for 1523 human and 364 viral microRNAs from the Sanger database (v.18.0). Total RNA from 4 cell lines (PANC-1, BxPC-3, ASPC-1, and HPDE6c7) was extracted with the Trizol reagent (Invitrogen, Carlsbad, CA, USA) and then purified using the mirVanaTM miRNA Isolation Kit (Cat#AM1560, Ambion, Austin, TX, USA). The miRNA from each cell line was labeled with the miRNA Complete Labeling and Hyb Kit (Cat#5190-0456, Agilent Technologies, Santa Clara, CA, USA) according to the manufacturer's instructions; each array slide was hybridized with 100 ng Cy3-labeled miRNA for 20 hours at 55°C and 20 rpm. After hybridization, the slides were washed with the Gene Expression Wash Buffer Kit (Cat#5188-5327, Agilent Technologies, Santa Clara, CA, USA) in staining dishes (Cat#121, Thermo Shandon, Waltham, MA, USA). The slides were scanned using an Agilent Microarray Scanner and Feature Extraction software 10.7 (Agilent Technologies, Santa Clara, CA, USA) with the default settings. Lastly, the raw data were normalized by the Quantile algorithm, Gene Spring Software 11.0 (Agilent Technologies, Santa Clara, CA, USA).
Quantitative real-time reverse transcription PCR
To verify the microarray results, we performed qRT-PCR to assay screen the miR-135a expression in prepared tissues samples and cell lines. Total RNA was isolated with the Trizol reagent (Invitrogen, Carlsbad, CA, USA). The expression of miR-135a was analyzed with the miScript system (Qiagen, Hilden, Germany) in accordance with the manufacturer's protocol; this system contains the miScript Reverse Transcription kit, miScript Primer assays, and miScript SYBR Green PCR kit. Human U6 RNA was amplified for normalization. SYBR green real-time RT-PCR was adopted to detect Bmi1 mRNA, and the first-strand complementary DNA was synthesized using MMLV reverse transcriptase (Epicentre, Paris, France). Human glyceraldehyde 3-phosphatedehydrogenase (GAPDH) RNA was used as an internal control. The primers used were as follows: Bmi1 forward, 5'GCTTCAAGATGGCCGCTTG3', and reverse, 5'TTCTCGTTGTTCGATGCATTTC3'. All RT-PCR reactions were analyzed using the ABI Prism 7700 Sequence Detector (Applied Biosystems, Foster City, CA, USA) in triplicate for each sample. The fold change for the relative expression levels of each miRNA was calculated using the ΔΔCt method [17].
Cell transfection
Viruses were harvested at 48 h after co-transfection with the Lentiviral vector Lenti-miR-135a or Lenti-scramble and the Lentiviral packaging plasmid (Genechem, Shanghai, China) into HEK 293T cells using the LipoFectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA). PANC-1 and ASPC-1 cells were infected with 1×106 recombinant lentivirus-transducing units plus 6 mg/ml polybrene (Sigma-Aldrich, St. Louis, MO, USA). Stable clones were acquired at 2 weeks after antibiotic selection. The miR-135a mimics or inhibitor and corresponding negative control were designed and synthesized by Genechem (Shanghai, China). pcDNA-Bmi1 carrying a wild-type or mutant-type 3'-UTR for miR-135a was constructed to re-express Bmi1. Target cells were transfected with these oligonucleotide, plasmid vectors and negative control using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocol. Cells were collected at 48 h after transfection.
CCK-8 assay
The Cell Counting Kit-8 (CCK-8) assay kit (Dojindo, Kumamoto, Japan) was used to determine the biological effect of miR-135a on cell proliferation. Transfected cells were seeded in 96-well plates at a density of 1×103 cells per well; 10 µl CCK-8 solution was added to each well the next day. The cells were incubated for 2 h at 37°C, and the absorbance at 450 nm was determined using an enzyme-linked immunosorbent assay reader (Dasit, Milan, Italy). The experiments were repeated 3 times for 5 days, and the average of the results was analyzed.
Colony formation assay
Cells were trypsinized and placed in each well of a 6-well plate at a density of 1×103 cells per well; visible cell colonies appeared after 2 weeks in complete medium at 37°C. The cell colonies were then fixed with methanol for 15 min and stained with 0.1% crystal violet for 20 min. The number of colonies was counted per well, and each experiment was performed in triplicate.
EdU incorporation assay
The incorporation of 5-ethynyl-2'-deoxyuridine (EdU), a thymidine analog, can label cells undergoing DNA replication [18]. The Cell-LightTM EdU Apollo567 imaging kit (RiboBio, Guangzhou, China) was used to perform the EdU incorporation assay. Lentiviral-transfected PANC-1 and ASPC-1 cells were added 100 µl EdU (50 µM) per well in 96-well plates and cultured for 2 h. The EdU medium mixture was discarded, and 4% paraformaldehyde was added for 30 min at room temperature to fix the cells. The cells were washed with glycine (2 mg/ml) for 5 min in a decolorization shaker, 0.5% Trion X-100 was added for 10 min, and the cells were washed twice with PBS. Next, 100 µl Apollo 550 stain reaction buffer was added to each well for 30 min while shielding from light. The cells were washed three times with 0.5% Triton X-100 and stained with 100 µl Hoechst 33342 (5 mg/ml) for 30 min at room temperature; 100 µl PBS was then added. Images were captured using a fluorescence microscope (Olympus, Tokyo, Japan), and the number of EdU-positive cells was calculated using the formula EdU add-in cells/Hoechst-stained cells×100%.
Analysis of cell cycle and apoptosis
To analyze the cell cycle distribution, cells were harvested and fixed with 70% ethanol at 4°C overnight and washed twice with PBS. Staining Solution (50 µg/ml propidium iodide, 1 mg/mL RNase A, and 0.1% TritonX-100 in PBS) was added to the cells for 30 min at 37°C in the dark. For the apoptosis analysis, cells were incubated in a serum-free medium for 72 h and stained with fluorescein isothiocyanate (FITC)-conjugated Annexin V and propidium iodide (PI) using the Annexin V-FITC Apoptosis Detection kit (Jingmei, Shanghai, China) according to the manufacturer's protocol. All of the samples were analyzed using the FACS Caliber II sorter and Cell Quest FACS system (BD Bio-sciences, San Jose, CA, USA). The flow cytometry analysis was repeated three times.
Luciferase activity assay
The 3'-UTR of Bmi1 containing an intact miR-135a recognition sequence was cloned into the pGL3-control-mcs2 reporter vector (constructed by our laboratory). Oligonucleotides (59bp) harboring wild-type or mutated miR-135a binding sites from the human BMI-1 3'-UTR were annealed and ligated into the EcoR I and Pst I sites of the pGL3-control mcs2 reporter vector. The oligonucleotide sequences were as follows: Bmi1-wt (F: 5'AATTCGAATAACGATTTCTTGCAGCTATTTAGCCATTTTGATTGCTGTTTGATTCTGCA3' and R:5'GAATCAAACAGCAATCAAAATGGCTAAATAGCTGCAAGAAATCGTTATTCG3') and Bmi1-mut (F: 5'AATTCGAATAACGATTTCTTGCAGCTATTTTCGGTATTTGATTGCTGTTTGATTCTGCA3' and R: 5'GAATCAAACAGCAATCAAATACCGAAAATAGCTGCAAGAAATCGTTATTCG3'). For the luciferase assays, the cells were transfected with the appropriate plasmids in 24-well plates and harvested for luciferase activity assays using the dual-luciferase reporter assay system (Promega, Madison, WI, USA) at 48 h after transfection. The relative luciferase activity was normalized to that of firefly luciferase. The transfection experiments were performed in triplicate for each plasmid construct.
Protein extraction and western blotting
Cells were solubilized on ice in RIPA lysis buffer (50 mM Tris-HCl [pH 7.4], 1% Triton X-100, 5 mM EDTA, 1 mM leupeptin, 1 mM phenylmethylsulfonyl fluoride, 10 mM NaF, and 1 mM Na3VO4) and centrifuged at 20,000×g for 30 min at 4°C to remove the cellular debris. The protein concentration was determined using a BCA Protein Assay Kit (KeyGen, Nanjing, China). The protein extracts were separated by 12% SDS-PAGE and electrophoretically transferred to polyvinylidene difluoride (PVDF) membranes (Amersham Biosciences, New York, NY, USA). Nonspecific binding sites were blocked with 5% nonfat dry milk in Tris-buffered saline with 0.5% Tween-20 at room temperature for 1 h, and the membranes were then incubated at 4°C overnight with primary antibodies directed against Bmi1 (Upstate Biotechnology Inc., Norwalk, CT, USA), p21, cyclin D1, Cdk2, Cdk4, Akt, pAkt, Bcl-2, Bax, and GAPDH (Santa Cruz Biotechnology, Santa Cruz, CA, USA). The antigen-antibody complexes were visualized using horseradish peroxidase-conjugated secondary antibodies and an enhanced chemiluminescence western blotting detection system (Amersham Life Science, Piscataway, NJ, USA). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a protein loading control.
Statistical analysis
All of the data are presented as the mean±standard deviation (SD). The statistical significance was evaluated using Student's t-test for unpaired comparison, and the analyses were performed using the SPSS software (SPSS, Chicago, IL, USA). P<0.05 was considered statistically significant.
Results
Identification of cancer-related miRNAs in PDAC using a miRNA array
To identify miRNAs expressed differentially between PDAC and normal pancreatic ductal epithelial cells, we first examined the miRNA expression profiles in three human PDAC cell lines (PANC-1, BxPC-3, and ASPC-1) and a normal pancreatic ductal epithelial cell line (HPDE6c7) with the Agilent Human miRNA array (v.18.0). The microarray results revealed that the expression of 66 miRNAs significantly differed between the PDAC and normal pancreatic ductal epithelial cells. Compared to the normal pancreatic ductal epithelial cells, 23 miRNAs were significantly overexpressed (fold change > 2), whereas 43 miRNAs were significantly underexpressed (fold change < 0.5). Of the dysregulated miRNAs, miR-205-3p, miR-203, miR-630, and miR-135a were the most up-regulated miRNAs in the normal pancreatic ductal epithelial cells, whereas miR-193a-5p, miR-10a-5p, miR-10b-5p, and miR-301a-3p were noted for the largest decrease in expression (Table 1).
Most dysregulated miRNAs in PDAC cell lines compared with normal pancreatic ductal epithelial cell line.
| miRNA | Expression in HPDE6c7 | Fold change | ||
|---|---|---|---|---|
| HPDE6c7 vs PANC-1 | HPDE6c7 vs BxPC-3 | HPDE6c7 vs ASPC-1 | ||
| hsa-miR-205-3p | up | 670.0528 | 628.206 | 659.2028 |
| hsa-miR-203 | up | 433.3644 | 406.2996 | 426.3471 |
| hsa-miR-135a | up | 270.5584 | 210.7637 | 250.8373 |
| hsa-miR-630 | up | 95.58312 | 89.61368 | 94.03536 |
| hsa-miR-10a-5p | down | 0.000401 | 0.003872 | 0.000248 |
| hsa-miR-10b-5p | down | 0.004478 | 0.004396 | 0.003212 |
| hsa-miR-193a-5p | down | 0.012944 | 0.003627 | 0.085107 |
| hsa-miR-301a-3p | down | 0.048843 | 0.031575 | 0.042265 |
MiR-135a is down-regulated in PDAC tissue and cell lines
MiR-135a has recently been reported in several types of tumors, such as renal cell carcinoma, gastric cancer, lung cancer, Hodgkin lymphoma, and colorectal cancer [19-22], suggesting that the dysregulation of miR-135a may play an essential role in tumorigenesis. However, no studies have been performed to assess the significance of miR-135a in PDAC. Therefore, we chose miR-135a for further investigation.
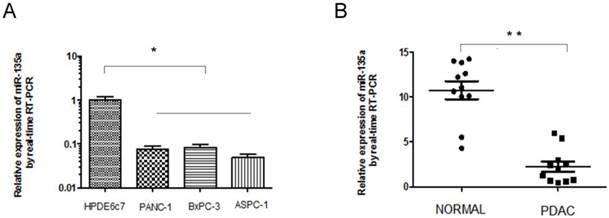
To further confirm the down-regulation of miR-135a in PDAC, we analyzed the expression of miR-135a in human PDAC cell lines and a normal pancreatic ductal epithelial cell line, 11 paired clinical PDACs, and the adjacent nontumorous pancreas tissues using quantitative real-time PCR; the results were normalized against an endogenous control (U6 RNA). Consistent with the microarray data, miR-135a expression was down-regulated in all three human PDAC cell lines (PANC-1, BxPC-3, and ASPC-1) compared to the normal pancreatic ductal epithelial cell line HPDE6c7 (Figure 1A). We also found that the miR-135a levels were significantly decreased in 9 of the 11 clinical PDAC tissues relative to the adjacent nontumorous pancreas tissues (Figure 1B). These results suggest that reduced miR-135a expression is a frequent event in human PDAC cell lines and PDAC tissues and this miRNA may be involved in pancreatic tumorigenesis.
Expression of miR-135a is decreased in PDAC cell lines and specimens. (A) The relative expression of miR-135a in PDAC cell lines and normal pancreas cell line was examined by qRT-PCR. (B) The relative expression of miR-135a in clinical PDAC patients was examined by qRT-PCR (*P < 0.05, **P < 0.01).
MiR-135a could inhibit the proliferation of PDAC cells
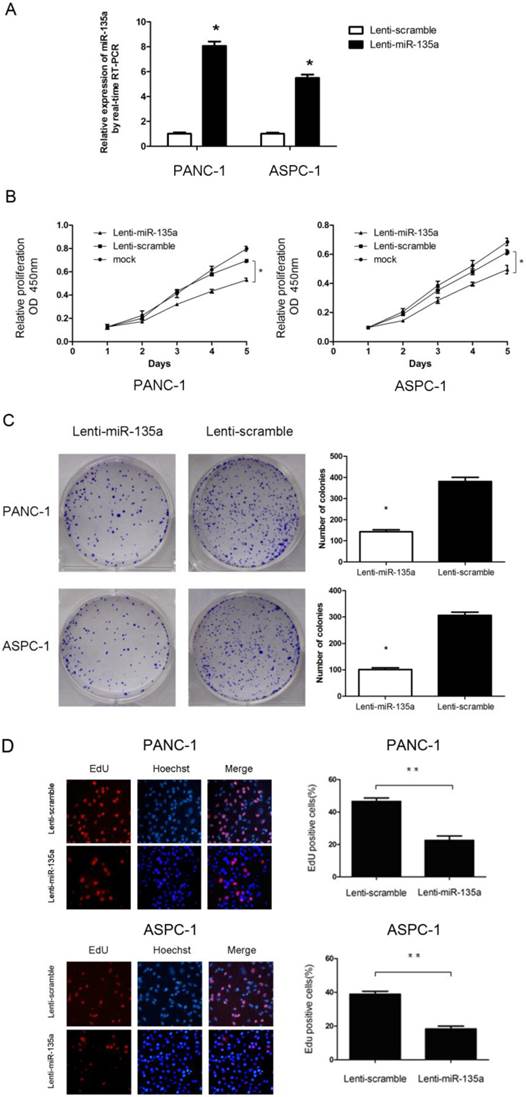
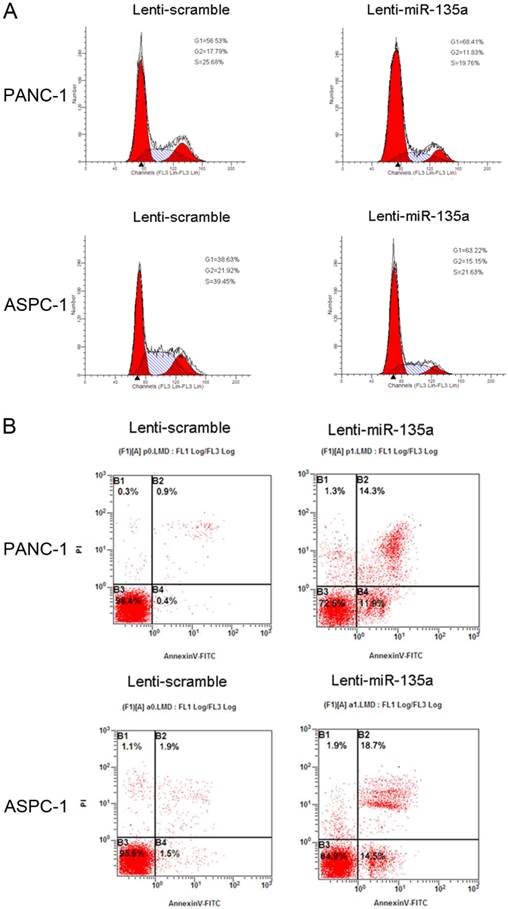
To assess the effect of miR-135a on PDAC cell proliferation, we established PANC-1 and ASPC-1 transfectants stably expressing miR-135a using lentiviral infection. A qRT-PCR analysis demonstrated that the miR-135a expression level in Lenti-miR-135a-PANC-1 and Lenti-miR-135a-ASPC-1 cells was significantly higher than in the control stable cell lines (Figure 2A). CCK-8 assays and colony formation analyses showed that the enforced expression of miR-135a resulted in a decrease in cell growth of the PANC-1 and ASPC-1 transfectants compared to cells transfected with the scrambled precursor (Figure 2B, C). Moreover, EdU cell proliferation assays also indicated that the growth of Lenti-miR-135a-PANC-1 and Lenti-miR-135a-ASPC-1 cells was significantly inhibited relative to the scrambled precursor- and negative control-transfected cells (Figure 2D). Previous reports have shown that the expression of miR-135a can induce cells in G0⁄ G1 arrest in RCC cell lines [22]. Our cell cycle assay showed that miR-135a transfection increased the percentage of cells in the G0⁄ G1 phase compared to the negative control, and the proportion of S and G2/M phases was lower in both the miR-135a-transfected cell lines compared to the negative control (Figure 3A). Furthermore, cell apoptosis analyses demonstrated that miR-135a overexpression increased the number of apoptotic cells in both cell lines compared to the negative control when these cells were maintained in serum-free media for 72h (Figure 3B). Taken together, these results suggest that miR-135a is able to inhibit proliferation and enhance the apoptosis of PDAC cells.
Overexpression of miR-135a inhibited the growth of PANC-1 and ASPC-1 cells. (A) Lenti-miR-135a transfection significantly increased the miR-135a level compared to Lenti-scramble transfection. (B) The CCK-8 assays results showed that the cell growth abilities of PANC-1 and ASPC-1 cells were inhibited after transfection with Lenti-miR-135a. (C) The effect of Lenti-miR-135a or Lenti-scramble on the growth of PANC-1 and ASPC-1 cells was examined by colony formation assay. Data are representative of three independent experiments. (D) The effect of transfection of Lenti-miR-135a or Lenti-scramble on the growth of PANC-1 and ASPC-1 cells was examined by EdU incorporation assay. Magnification: ×400; (*P < 0.05, **P < 0.01 versus Lenti-scramble).
(A) Flow cytometry analysis showed that transfection of Lenti-miR-135a arrested the accumulation of PANC-1 and ASPC-1 cells in the G0-G1 phase. The proportion of S and G2/M phase was lower in both Lenti-miR-135a transfected cell lines compared with the negative control. (B) Following 72h of culturing in serum-free medium, cell apoptosis indicated that PANC-1 and ASPC-1 cells transfection of Lenti-miR-135a increased the number of apoptotic cells versus the negative control.
Bmi1 is a direct target of miR-135a
To investigate the mechanisms by which miR-135a suppresses tumor growth, we further explored the putative targets of miR-135a using several prediction tools: miRanda, TargetScan, Pita, miTarget and RNAhybrid. Among the hundreds of different targets predicted, Bmi1 was notable because it is considered to be an oncogene that can promote cell proliferation in such cancers as lung cancer, breast cancer, and myelogenous leukemia [23-25]. Furthermore, our previous study demonstrated that Bmi1 expression was markedly up-regulated in pancreatic cancer cell lines and clinical cancer specimens [26].
We measured the levels of Bmi1 protein in PDAC cell lines. Bmi1 protein levels were relatively higher in the PANC-1, BxPC-3, and ASPC-1 cell lines, whereas the miR-135a levels were relatively lower in these cell lines. By contrast, relatively lower Bmi1 expression and higher miR-135a expression were validated in the normal pancreatic ductal epithelial cell line, HPDE6c7 (Figure 4A, 1B). The results indicated that the endogenous level of Bmi1 expression was inversely associated with that of miR-135a in PDAC cell lines. Bmi1 expression in 11 pairs of clinical PDAC and adjacent non-tumor samples were analyzed by western blotting. We found that the Bmi1 protein levels in a large part of PDAC samples were significantly higher than that in adjacent non-tumor samples (Figure 4B). Importantly, as shown in Figure 4B and Figure 4C, Bmi1 expression was upregulated in miR-135a-downregulated PDAC samples. Thus, the inverse expression levels of Bmi1 and miR-135a in PDAC indicated that miR-135a may participate in the regulation of Bmi1.
The protein expression of Bmi1 in PDAC and miR-135a regulated endogenous Bmi1 protein levels. (A) The expression of Bmi1 in PDAC cell lines was examined by western blotting. (B) The expression of Bmi1 in PDAC tissues and adjacent non-tumor samples were examined by western blotting. (C) miR-135a expression was inversely related to Bmi1 expression in PDAC tissues. Bmi1 was decreased in adjacent non-tumor samples that showed increased miR-135a expression, whereas Bmi1 was increased in PDAC tissues that showed decreased miR-135a expression. (D) PANC-1 and ASPC-1 cells were transfected with miR-135a mimics or inhibitor. The levels of Bmi1 protein were analyzed by western blotting 72h later. (E) The expression of Bmi1 mRNA in PANC-1 and ASPC-1 cells treated as described above were measured by qRT-PCR. N, adjacent non-tumor tissues; T, tumor tissues.
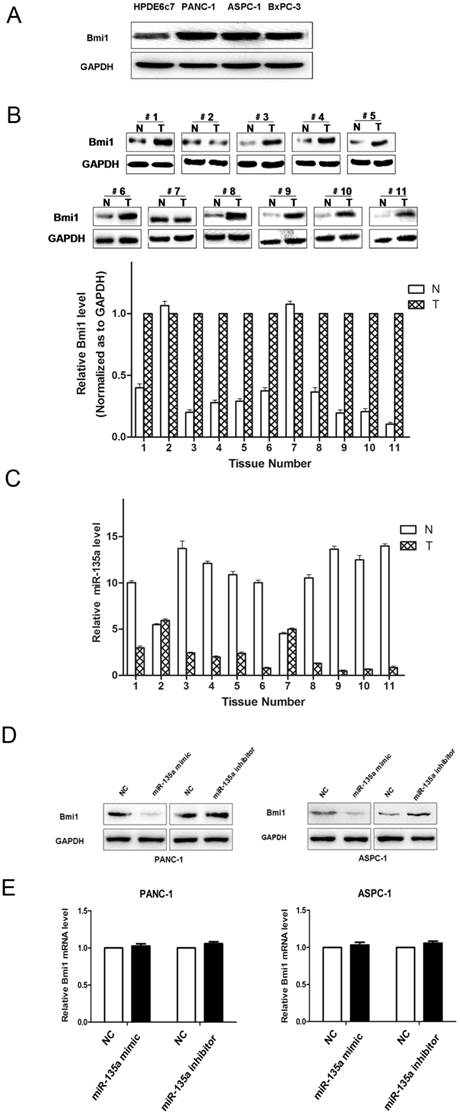
Based on these observations, we addressed whether the tumor suppressor function of miR-135a in PDAC was attributable to its suppressive effect on Bmi1 expression. Thus, we examined the effect of miR-135a on the endogenous Bmi1 expression level by qRT-PCR and western blotting in PANC-1 and ASPC-1 cells. Transfection with the miR-135a mimic led to an obvious down-regulation of Bmi1 protein in both PANC-1 and ASPC-1 cells, whereas the miR-135a inhibitor was able to up-regulate the expression of Bmi1 in these cells (Figure 4D). However, the Bmi1 mRNA expression levels in the miR-135 mimic-transfected cells were not significantly affected compared to the empty vector-transfected cells (Figure 4E). These results suggest that miR-135a may suppress Bmi1 expression via inhibiting the translation of Bmi1 but do not have any impact on mRNA stability.
As shown in Figure 5A, an in silico analysis revealed that the human Bmi1 mRNA 3'-UTR contains a conserved putative target site for miR-135a. Thus, to verify whether Bmi1 is a direct target of miR-135a, a dual-luciferase reporter system was used with the co-transfection of miR-135a or miR-NC and a luciferase reporter plasmid containing a region of the 3'-UTR of human Bmi1 complementary to miR-135a. Correspondingly, we also generated a Bmi1 3'-UTR mutant in the miR-135a target region to disrupt binding (Figure 5A). In PANC-1 and ASPC-1 cells, miR-135a remarkably suppressed the expression of luciferase containing a wild-type construct of Bmi1 3'-UTR in comparison to NC. Conversely, this effect was not observed in either cell line carrying the mutant-type construct of the Bmi1 3'-UTR (Figure 5B).
Bmi1 is a target of miR-135a. (A) Predicted duplex formation between human Bmi1 3'-UTR and miR-135a. Schematic representation of firefly luciferase reporter construct containing Bmi1 3'UTR with either wild-type (WT) or mutant (MUT) miR-135a target site. (B) Analysis of the luciferase activity of the luciferase reporter plasmid containing either wild-type (WT) or mutant (MUT) Bmi1 3'-UTR in PANC-1 and ASPC-1 cells (*P < 0.05). (C) The inhibition of Bmi1 by miR-135a was partially recovered after co-transfection with pcDNA-Bmi1 (mutant 3'-UTR for miR-135a) and miR-135a mimics. (D) The CCK-8 assays results showed that the cell growth abilities of PANC-1 and ASPC-1 cells were recovered after co-transfection with pcDNA-Bmi1 (mutant 3'-UTR for miR-135a) and miR-135a mimics (*P < 0.05).
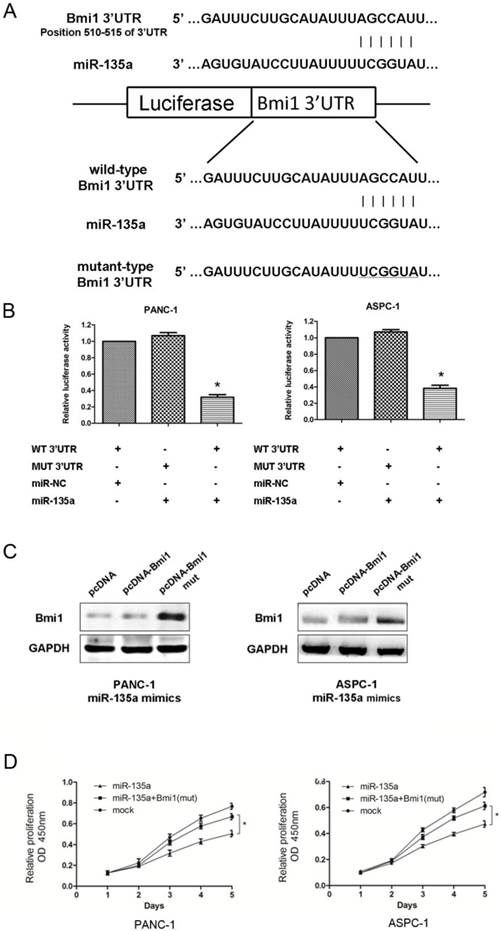
To further elucidate whether the growth-suppressive effect of miR-135a was mediated by repression of Bmi1 in PDAC cells, we introduced pcDNA-Bmi1 with wild or mutant 3'-UTR for miR-135a and miR-135a mimics into PDAC cells. As shown in Figure 5C, the expression levels of Bmi1 was recovered after exogenous expression of pcDNA-Bmi1 (mutant 3'-UTR for miR-135a). Figure 5D showed that PANC-1 and ASPC-1 cell proliferation was promoted when Bmi1 was re-expressed in both cells treated with miR-135a. Taken together, these data imply that miR-135a may attenuate the expression of Bmi1 by directly targeting its 3'-UTR.
MiR-135a regulates the expression of several cell cycle- and survival-related proteins via Bmi1
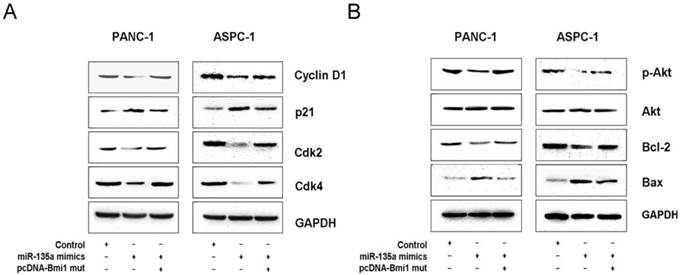
Bmi1 has been shown to be overexpressed in many cancer cells and is known to regulate cell growth, survival, and invasion through some important signal pathways [27-29]. To further investigate the mechanism by which miR-135a inhibits PDAC growth, we examined several related molecules downstream of Bmi1. Our western blotting data demonstrate that the overexpression of miR-135a significantly reduced the expression of cyclin D1, cyclin-dependent kinase (Cdk) 2, and Cdk4 in PANC-1 and ASPC-1 cells; in contrast, the expression of cyclin-dependent kinase inhibitor (p21) was up-regulated. Moreover, co-transfection with the miR-135a mimics and pcDNA-Bmi1 (mutant 3'-UTR for miR-135a) increased the expression of cyclin D1, Ckd2, Cdk4 and suppressed that of p21 at protein levels (Figure 6A). In addition, we also examined the expression of various apoptotic components by a western blotting analysis. Following the restoration of miR-135a in PANC-1 and ASPC-1 cells, the expression of phospho-Akt and Bcl-2 was inhibited, whereas the level of Bax was elevated. But the expression levels of phospho-Akt and Bcl-2 proteins were increased and that of Bax was suppressed after co-transfection with miR-135a mimics and pcDNA-Bmi1 (mutant 3'-UTR for miR-135a) (Figure 6B). Together, our data suggest that miR-135a regulates several important molecules through Bmi1 down-regulation in PDAC cells.
MiR-135a regulates several cell cycle and survival related proteins expression via Bmi1. (A) The expression of several cell cycle-related regulatory factors was detected by western blotting in PANC-1 and ASPC-1 cells infected with negative control, miR-135a mimics, miR-135a mimics and pcDNA-Bmi1 (mutant 3'-UTR for miR-135a). (B) The expression of several survival-related regulatory factors was detected by western blotting in PANC-1 and ASPC-1 cells infected with negative control, miR-135a mimics, miR-135a mimics and pcDNA-Bmi1 (mutant 3'-UTR for miR-135a). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as the loading control.
Discussion
In recent years, a significant number of miRNAs functioning as oncogenes or tumor suppressors have been identified and studied. With regard to PDAC, for example, miR-21 was found to be associated with tumor progression and an adverse clinical course through targeting PDCD4 and TIMP3 [30]. Conversely, Zhao et al. found that the frequently downregulated miR-217 can regulate KRAS and function as a tumor suppressor in PDAC [31]. However, the results from different research teams are lack of consistency [32]. PDAC specimens from different patients typically have different cell types and proportions, which may result in inaccurate miRNA profiles. Thus, we utilized cell lines for the microarray analysis to avoid the disadvantage of using PDAC specimens. The results indicated that several miRNAs, such as miR-21 and miR-10b, are overexpressed in PDAC cell lines and that miR-203 expression is significantly decreased in PDAC cell lines compared to the normal pancreas cell line [30,33,34]. These data were consistent with previous reports, indicating that the use of cell lines was helpful and reliable. Among these dysregulated miRNAs, we chose miR-135a for further investigation. Its function and molecular mechanism in PDAC have not been characterized.
To verify the results of the microarray analysis, we examined the expression of miR-135a in human PDAC cell lines and clinical PDAC specimens using qRT-PCR and found that the expression of miR-135a was down-regulated in the PADC cell lines and tissues. Such evidence indicated a possible tumor-suppressive role of miR-135a in PDAC, and the results of CCK-8 assays and colony formation analyses suggested that miR-135a overexpression inhibited the growth of PDAC cell lines. EdU cell proliferation assays further indicated that the restoration of miR-135a expression decreased cell proliferation, and we also found that miR-135a overexpression could induce cells in G0⁄ G1 arrest and lead to the dramatic induction of apoptosis in PDAC cell lines under serum-starved conditions. Taken together, our study showed that miR-135a plays a tumor-suppressive role in PDAC.
Navarro A et al. identified miR-135a as a novel prognostic marker of relapse in cHL and showed that miR-135a could modulate apoptosis and decreases cell growth by targeting JAK2 [21], and Wu H et al. found that miR-135a could target JAK2 to inhibit gastric cancer cell proliferation [20]. JAK2 is the predominant member of the JAK family. The JAK/STAT signaling pathway is regulated at various steps via a number of distinct mechanisms, and deregulation of this pathway is associated with several aspects of tumor pathogenesis [35]. Moreover, several studies also have indicated that the overexpression of miR-135a could inhibit cell proliferation and promote apoptosis via target c-MYC, STAT6, SMAD5, BMPR2 and HOXA10 in various cancers [22,36,37]. These target genes of miR-135a were shown to be involved in several cellular processes, including cell proliferation, cell cycle and/or apoptosis in many types of human cancers [36,38,39]. However, miR-135a was reported to promote the migration and invasion of breast cancer cells by targeting HOXA10 [40]. In colorectal cancer, miR-135a was shown to promote colorectal cancer cell proliferation, migration, and invasion by targeting the tumor suppressor gene MTSS1 [19]. Liu et al. found that miR-135a was overexpressed in portal vein tumor thrombosis and in vivo assays indicated that blockade of miR-135a could reduce the incidence of portal vein tumor thrombosis. MTSS1 also was identified as the direct and functional target of miR-135a in their study [41]. MTSS1 gene was reported in some cancers including gastric cancer, breast cancer and hepatocellular carcinoma, but its exact roles and biochemical mechanisms in oncogenesis remain largely unknown [42-44]. Besides oncology researches, miR-135a was reported in other fields such as megakaryocytopoiesis, bone and muscle development, hypertension. The different roles of miR-135a may reflect the ability of a single miRNA to perform different functions in different cellular environments. Therefore, these studies along with our findings indicate cancer and/or cell type-specific functions for miR-135a.
MicroRNAs play an important role in many biological processes of cancer cells by interacting with specific mRNAs and inhibiting the expression of target genes. It is noteworthy that one miRNA can regulate many target genes simultaneously; for example, miR-203 regulates a cohort of pro-metastatic genes, such as ZEB2, Bmi1, and survivin, in prostate cancer [45]. The in silico analysis showed that miR-135a may potentially interact with the 3'-UTR of a large number of genes; of these target genes, Bmi1 was reported in our earlier studies to be up-regulated in PDAC. We found that overexpressed Bmi1 in PDAC cell lines and clinical cancer specimens was associated with an unfavorable prognosis for patients with PDAC [26]. Bmi1, a member of the polycomb gene family, has been reported to be associated with the initiation, progression, and prognosis of various cancers, acting as an important regulator of several cellular processes, including cell cycle regulation, cell immortalization, and cell senescence [18-23,45]. Moreover, many studies have shown that Bmi1 is involved in the self-renewal and differentiation of stem cells through multiple pathways [46-50]. However, the molecular mechanism underlying the process of Bmi1 overexpression in PDAC has not been elucidated. In the present study, our results showed that Bmi1 is overexpressed and inversely associated with miR-135a expression in both PDAC cell lines and primary PDAC tissues. Transfection of miR-135a mimics or inhibitors was also found to alter the Bmi1 protein level without affecting its mRNA level. Then, we co-transfected miR-135a and Bmi1 and showed that the down-regulation of Bmi1 by miR-135a relies on the Bmi1 3'-UTR. A luciferase activity assay also revealed that miR-135a binds to the 3'-UTR of Bmi1 mRNA and represses its translation. This is the first evidence to explain that Bmi1 overexpression in PADC may result from a lack of miR-135a.
Furthermore, it has also been reported that miR-135a regulates several pathways, including the cell cycle, apoptosis, and cancer-related pathways [22]. Thus, we tested the expression of several known related molecules in miR-135a-overexpressing and control cells. Cyclin D1, Cdk2, and Cdk4 are important proteins regulating the cell cycle and are related to the development of many cancers, including PDAC. p21 is perceived mainly as a tumor suppressor gene belonging to the Cip/Kip families of cyclin-dependent kinase inhibitors because of its antiproliferative effects [51,52]. Our results showed that the expression of cyclin D1, Cdk2, and Cdk4 was reduced when miR-135a was overexpressed, though the expression of p21 was up-regulated. Furthermore, the expression of phospho-Akt and Bcl-2 was inhibited, whereas the level of Bax was elevated following miR-135a overexpression. Importantly, Bmi1 re-expression antagonized the effects of miR-135a on these proteins. Taken together, our data suggest that miR-135a exerts its tumor-suppressive action in PDAC through the modulation of Bmi1 expression and its several downstream proteins and signaling, including some cyclins (cyclin D1, Cdk2, and Cdk4) and a CDK inhibitor (p21), Bcl-2 family proteins, and the PI3K/AKT pathway. We plan to conduct a further investigation with in vivo studies and clinical specimens to confirm the importance of miR-135a in PDAC.
In conclusion, we determined that miR-135a is frequently down-regulated in PDAC and is a candidate tumor-suppressing miRNA in PDAC cells. The overexpression of miR-135a inhibits the proliferation of PDAC cells, at least in part via the regulation of Bmi1. Therefore, the identification of miR-135a as a regulator of BMI1 may provide insight for the establishment of new strategies and useful miRNA-based therapies for the treatment of PDAC.
Abbreviations
PDAC: pancreatic ductal adenocarcinoma; miRNAs: microRNAs; PC: pancreatic cancer; qRT-PCR: quantitative real-time reverse transcription PCR; 3'-UTR: 3'-untranslated region; CCK-8: Cell Counting Kit-8; EdU: 5-ethynyl-2'-deoxyuridine; Bmi1: B-cell-specific Moloney murine leukemia virus insertion site 1; NC: negative control.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (Grants No. 81101820).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel R, Ward E, Brawley O. et al. Cancer statistics, 2011: the impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J Clin. 2011;61:212-236
2. Jemal A, Bray F, Center MM. et al. Global cancer statistics. CA Cancer J Clin. 2011;61:69-90
3. Garzon R, Calin GA, Croce CM. MicroRNAs in Cancer. Annu Rev Med. 2009;60:167-179
4. Iorio MV, Croce CM. MicroRNAs in cancer: small molecules with a huge impact. J Clin Oncol. 2009;27:5848-5856
5. Calin GA, Dumitru CD, Shimizu M. et al. Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci USA. 2002;99:15524-15529
6. Volinia S, Calin GA, Liu CG. et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci USA. 2006;103:2257-2261
7. Rosenfeld N, Aharonov R, Meiri E. et al. MicroRNAs accurately identify cancer tissue origin. Nat Biotechnol. 2008;26:462-469
8. Mitchell PS, Parkin RK, Kroh EM. et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc Natl Acad Sci USA. 2008;105:10513-10518
9. Lu J, Getz G, Miska EA. et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834-838
10. Engels BM, Hutvagner G. Principles and effects of microRNA-mediated post-transcriptional gene regulation. Oncogene. 2006;25:6163-6169
11. Budhu A, Jia HL, Forgues M. et al. Identification of metastasis-related microRNAs in hepatocellular carcinoma. Hepatology. 2008;47:897-907
12. Cheng AM, Byrom MW, Shelton J. et al. Antisense inhibition of human miRNAs and indications for an involvement of miRNA in cell growth and apoptosis. Nucleic Acids Res. 2005;33:1290-1297
13. He L, He X, Lim LP. et al. A microRNA component of the p53 tumour suppressor network. Nature. 2007;447:1130-1134
14. Szafranska AE, Davison TS, John J. et al. MicroRNA expression alterations are linked to tumorigenesis and non-neoplastic processes in pancreatic ductal adenocarcinoma. Oncogene. 2007;26:4442-4452
15. Wang J, Chen J, Chang P. et al. MicroRNAs in plasma of pancreatic ductal adenocarcinoma patients as novel blood-based biomarkers of disease. Cancer Prev Res (Phila). 2009;2:807-813
16. Lee EJ, Gusev Y, Jiang J. et al. Expression profiling identifies microRNA signature in pancreatic cancer. Int J Cancer. 2007;120:1046-1054
17. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402-408
18. Salic A, Mitchison TJ. A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc Natl Acad Sci USA. 2008;105:2415-2420
19. Zhou W, Li X, Liu F. et al. MiR-135a promotes growth and invasion of colorectal cancer via metastasis suppressor 1 in vitro. Acta Biochim Biophys Sin (Shanghai). 2012;44:838-846
20. Wu H, Huang M, Cao P. et al. MiR-135a targets JAK2 and inhibits gastric cancer cell proliferation. Cancer Biol Ther. 2012;13:281-288
21. Navarro A, Diaz T, Martinez A. et al. Regulation of JAK2 by miR-135a: prognostic impact in classic hodgkin lymphoma. Blood. 2009;114:2945-2951
22. Yamada Y, Hidaka H, Seki N. et al. Tumor-suppressive microRNA-135a inhibits cancer cell proliferation by targeting the c-MYC oncogene in renal cell carcinoma. Cancer Sci. 2013;104:304-312
23. Yu Q, Su B, Liu D. et al. Antisense RNA-mediated suppression of Bmi-1 gene expression inhibits the proliferation of lung cancer cell line A549. Oligonucleotides. 2007;17:327-335
24. Guo WJ, Zeng MS, Yadav A. et al. Mel-18 acts as a tumor suppressor by repressing Bmi-1 expression and down-regulating Akt activity in breast cancer cells. Cancer Res. 2007;67:5083-5089
25. Meng XX, Liu WH, Liu DD. et al. Construction of antisense Bmi-1 expression plasmid and its inhibitory effect on k562 cells proliferation. Chin Med J (Engl). 2005;118:1346-1350
26. Song W, Tao K, Li H. et al. Bmi-1 is related to proliferation, survival and poor prognosis in pancreatic cancer. Cancer Sci. 2010;101:1754-1760
27. Xiao J, Deng C. Knockdown of Bmi-1 impairs growth and invasiveness of human gastric carcinoma cells. Oncol Res. 2009;17:613-620
28. Qin L, Zhang X, Zhang L. et al. Downregulation of BMI-1 enhances 5-fluorouracil-induced apoptosis in nasopharyngeal carcinoma cells. Biochem Biophys Res Commun. 2008;371:531-535
29. Guo BH, Feng Y, Zhang R. et al. Bmi-1 promotes invasion and metastasis, and its elevated expression is correlated with an advanced stage of breast cancer. Mol Cancer. 2011;10:10
30. Nagao Y, Hisaoka M, Matsuyama A. et al. Association of microRNA-21 expression with its targets, PDCD4 and TIMP3, in pancreatic ductal adenocarcinoma. Mod Pathol. 2012;25:112-121
31. Zhao WG, Yu SN, Lu ZH. et al. The miR-217 microRNA functions as a potential tumor suppressor in pancreatic ductal adenocarcinoma by targeting KRAS. Carcinogenesis. 2010;31:1726-1733
32. Gramantieri L, Fornari F, Callegari E. et al. MicroRNA involvement in hepatocellular carcinoma. J Cell Mol Med. 2008;12:2189-2204
33. Nakata K, Ohuchida K, Mizumoto K. et al. MicroRNA-10b is overexpressed in pancreatic cancer, promotes its invasiveness, and correlates with a poor prognosis. Surgery. 2011;150:916-922
34. Wellner U, Schubert J, Burk UC. et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol. 2009;11:1487-1495
35. Shuai K, Liu B. Regulation of JAK-STAT signalling in the immune system. Nat Rev Immunol. 2003;3:900-911
36. Wu S, Lin Y, Xu D. et al. MiR-135a functions as a selective killer of malignant glioma. Oncogene. 2012;31:3866-3874
37. Tang WW, Wan GP, Wan YC. et al. Effects of miR-135a on HOXA10 expression, proliferation and apoptosis of ovarian cancer cells. Zhonghua Fu Chan Ke Za Zhi. 2013;48:364-369
38. Dang CV, O'Donnell KA, Zeller KI. et al. The c-Myc target gene network. Semin Cancer Biol. 2006;16:253-264
39. Chariot A, Gielen J, Merville MP. et al. The homeodomain-containing proteins: an update on their interacting partners. Biochem Pharmacol. 1999;58:1851-1857
40. Chen Y, Zhang J, Wang H. et al. miRNA-135a promotes breast cancer cell migration and invasion by targeting HOXA10. BMC Cancer. 2012;12:111
41. Liu S, Guo W, Shi J. et al. MicroRNA-135a contributes to the development of portal vein tumor thrombus by promoting metastasis in hepatocellular carcinoma. J Hepatol. 2012;56:389-396
42. Liu K, Wang G, Ding H. et al. Downregulation of metastasis suppressor 1 (MTSS1) is associated with nodal metastasis and poor outcome in Chinese patients with gastric cancer. BMC Cancer. 2010;10:428
43. Parr C, Jiang WG. Metastasis suppressor 1 (MTSS1) demonstrates prognostic value and anti-metastatic properties in breast cancer. Eur J Cancer. 2009;45:1673-1683
44. Ma S, Guan XY, Lee TK. et al. Clinicopathological significance of missing in metastasis B expression in hepatocellular carcinoma. Hum Pathol. 2007;38:1201-1206
45. Saini S, Majid S, Yamamura S. et al. Regulatory role of mir-203 in prostate cancer progression and metastasis. Clin Cancer Res. 2011;17:5287-5298
46. Lessard J, Sauvageau G. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature. 2003;423:255-260
47. Park IK, Qian D, Kiel M. et al. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature. 2003;423:302-305
48. Sawa M, Yamamoto K, Yokozawa T. et al. BMI-1 is highly expressed in M0-subtype acute myeloid leukemia. Int J Hematol. 2005;82:42-47
49. Yang J, Chai L, Liu F. et al. Bmi-1 is a target gene for SALL4 in hematopoietic and leukemic cells. Proc Natl Acad Sci USA. 2007;104:10494-10499
50. Raaphorst FM. Self-renewal of hematopoietic and leukemic stem cells: a central role for the polycomb-group gene Bmi-1. Trends Immunol. 2003;24:522-524
51. Aprelikova O, Xiong Y, Liu ET. Both p16 and p21 families of cyclin-dependent kinase (CDK) inhibitors block the phosphorylation of cyclin-dependent kinases by the CDK-activating kinase. J Biol Chem. 1995;270:18195-18197
52. Zupkovitz G, Grausenburger R, Brunmeir R. et al. The cyclin-dependent kinase inhibitor p21 is a crucial target for histone deacetylase 1 as a regulator of cellular proliferation. Mol Cell Biol. 2010;30:1171-1181
Author contact
![]() Corresponding author: Wen-jie Song and Ke-Feng Dou, Department of Hepatobiliary Surgery, Xijing Hospital, Fourth Military Medical University, No. 127 West Changle Road, Xi'an, 710032, Shaanxi, P.R. China. Phone: 86-29-84775255; Fax: 86-29-84775255; E-mail: wjsongedu.cn (Wen-jie Song); gdwkdkfcom (Ke-Feng Dou)
Corresponding author: Wen-jie Song and Ke-Feng Dou, Department of Hepatobiliary Surgery, Xijing Hospital, Fourth Military Medical University, No. 127 West Changle Road, Xi'an, 710032, Shaanxi, P.R. China. Phone: 86-29-84775255; Fax: 86-29-84775255; E-mail: wjsongedu.cn (Wen-jie Song); gdwkdkfcom (Ke-Feng Dou)