ISSN: 1449-2288International Journal of Biological Sciences

Int J Biol Sci 2014; 10(7):798-806. doi:10.7150/ijbs.9470 This issue Cite
Research Paper
Up-Regulated Lipocalin-2 in Pulmonary Hypertension Involving in Pulmonary Artery SMC Resistance to Apoptosis
Guoliang Wang1, Xiaoyan Liu2, Liukun Meng1, Shenghua Liu1, Li Wang1, Jun Li1, Chuanjue Cui1, Jian Meng1, Shengshou Hu1, Yingjie Wei1 ![]()
1. State Key Laboratory of Cardiovascular Disease, Fuwai Hospital, National Center for Cardiovascular Diseases, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, 100037, People's Republic of China.
2. Medical Research Center, Beijing Chao-Yang Hospital, Capital Medical University, Beijing, 100020, People's Republic of China.
Abstract
A key feature of pulmonary hypertension (PH) is the remodeling of small pulmonary arteries due to abnormal pulmonary artery smooth muscle cell (PASMC) proliferation and resistance to apoptosis. However, the cellular mechanisms underlying how PASMCs in the pathological condition of pulmonary hypertension become resistant to apoptosis remain unknown. It was recently reported that lipocalin 2 (Lcn2) is up-regulated in a wide array of malignant conditions, which facilitates tumorigenesis partly by inhibiting cell apoptosis. In this study, we observed that the expression levels of Lcn2 were significantly elevated in a rat PH model induced with monocrotaline and in patients with congenital heart disease-associated PH (CHD-PH) when compared with respective control. Therefore, we hypothesize that Lcn2 could regulate human PASMC (HPASMC) apoptosis through a mechanism. By the detection of DNA fragmentation using the TUNEL assay, the detection of Annexin V/PI-positive cells using flow cytometry, and the detection of cleaved caspase-3 and caspase-3 activity, we observed that Lcn2 significantly inhibited HPASMC apoptosis induced by serum withdrawal and H2O2 treatment. We also observed that Lcn2 down-regulated the proapoptotic protein Bax, decreased the levels of cellular ROS, and up-regulated the expression of superoxide dismutases (SOD1 and SOD2). In conclusion, Lcn2 significantly inhibits HPASMC apoptosis induced by oxidative stress via decreased intracellular ROS and elevated SODs. Up-regulation of Lcn2 in a rat PH model and CHD-PH patients may be involved in the pathological process of PH.
Keywords: apoptosis, oxidative stress, Lipocalin-2 (Lcn2), SOD, ROS, pulmonary hypertension (PH)
Introduction
Pulmonary hypertension (PH) is a severe disease with poor prognosis. It is characterized by persistent elevated pulmonary arterial pressure (PAP) and ultimately progresses to right ventricular failure and death. A key feature of vascular pathology in PH is the remodeling of small pulmonary arteries, which results in a persistent elevation of pulmonary arterial resistance [1]. Abnormal pulmonary artery smooth muscle cell (PASMC) proliferation and resistance to apoptosis have been proposed to be essential for vascular remodeling in PH [2,3]. In animal experiments, the induction of apoptosis or coupling with the inhibition of the proliferation of PASMCs prevents the progression of medial hypertrophy [4-6]. However, the cellular mechanisms underlying how normal PASMCs become resistant to apoptosis remain unknown.
Increased oxidative stress and elevated levels of reactive oxygen species (ROS) have been observed in animal models of PH and PH patients [7,8]. ROS play essential roles in the regulation of several physiological and pathological processes, and excessive amounts of ROS are lethal [9]. However, ROS have been demonstrated as important proliferative rather than apoptotic mediators in pulmonary vascular cells [10,11]. The mechanisms underlying how increased levels of ROS exert a proliferative effect rather than a proapoptotic effect in PH remain unclear.
Lipocalin 2 (Lcn2), also known as neutrophil gelatinase-associated lipocalin (NGAL), is a 25-KDa secretary glycoprotein that belongs to the lipocalin family. Lcn2 is an acute phase protein and proinflammatory marker [12-14]. Recent studies have shown that Lcn2 plays a role in cell survival in a wide array of benign and malignant conditions. It has been reported as a survival factor and can protect cells against apoptosis [15-18]. Lcn2 is over-expressed in cancers of diverse histological origin and it facilitates tumorigenesis by promoting survival, growth, and metastasis [18,19]. Paradoxically, Lcn2 induces apoptosis in other conditions. For example, Lcn2 can induce the apoptosis of H9c2 cardiomyocytes [20] and A549 human lung adenocarcinoma cells [21]. Thus, Lcn2 appears to be a double-edged sword in apoptosis and cell survival.
The up-regulation of Lcn2 in malignant conditions and its roles in cell survival motivated us to examine whether Lcn2 is over-expressed and its effect on HPASMC apoptosis, which is an important component of small pulmonary artery remodeling. Remarkably, prior to this paper, there has been no report on the role of Lcn2 in PH and in the survival of PASMCs. Because ectopically expressed Lcn2 has been reported to induce the expression of heme oxygenase-1,2 and SOD1,2 in CHO cells [17], we hypothesized that the up-regulation of antioxidant enzymes is an important mechanism in Lcn2-inhibited HPASMC apoptosis. Thus, this study aimed to determine whether Lcn2 expression is altered in a rat PH model induced with monocrotaline and in patients with congenital heart disease associated PH (a large subtype of PH) and to analyze the role of Lcn2 on HPASMC apoptosis at the cellular and molecular levels, which may help us better understand the pathophysiological role of Lcn2 in PH.
Methods
Animal model of pulmonary hypertension
All protocols and surgical procedures were approved by the local animal care committee. Adult male Sprague-Dawley rats (6 to 7 weeks of age; weight 250 to 300 g) were administered a single subcutaneous injection of 60 mg/kg monocrotaline (MCT) or saline. The animals were killed 7, 14, 21, 28 and 35 days after the subcutaneous injection of MCT or saline, the body weight was recorded, and right-heart catheterization was performed to measure the pulmonary arterial pressure. The rats were exsanguinated immediately after hemodynamic assessment, and the lungs were removed for further analysis. A part of the lungs from all of the groups were fixed in situ in the distended state by infusion of 10% neutral buffered formalin into the pulmonary artery (at 25 mm Hg pressure) and trachea for 1 minute and then placed in 10% neutral buffered formalin at 4°C for 24 h prior to embedment in paraffin. The remaining lungs were immediately frozen in liquid nitrogen for protein and RNA isolation. To assess the extent of RVH, the heart was removed, and the RV-free wall was dissected from the left ventricle plus septum (LV+S) and weighed separately; the degree of right ventricular hypertrophy (RVHI) was determined from the ratio RV/(LV+S), as previously described [22].
Patients and sampling
This study was approved by the ethics committee at Fuwai Hospital and conforms to the principles outlined in the Declaration of Helsinki. Informed consent was obtained from the parents of all of the patients. Patients with congenital heart disease-associated pulmonary hypertension (CHD-PH, measured through echocardiography) aged 6 to 24 months who underwent a surgical correction were included in the study. Patients with congenital heart diseases with unrestricted left-to-right shunt but no PH (CHD-nonPH) aged 6 to 24 months, who underwent surgical correction, were also included in the study as control subjects. Patients with any other diseases or clinical signs that were not associated with congenital heart diseases were excluded. Citrated plasma samples were obtained and stored in aliquots at -70 °C. The level of plasma Lcn2 was measured using the Human Lipocalin-2/NGAL Quantikine ELISA Kit (DLCN20, R&D, USA) according to the manufacturer's instructions.
Immunohistochemistry
Immunostaining for Lcn2 was performed in the lungs of rats after 3 weeks of exposure to monocrotaline or saline. The lung samples were sectioned at 5-μm thickness. Rabbit polyclonal anti-Lcn2 (Santa Cruz Biotechnology, USA) was detected using the Strept ABC peroxidase technique (ZSGB-BIO, CHN) as previously described [23].
Cell culture
Human pulmonary artery SMCs were purchased from ScienCell (USA) and used according to the manufacturer's instructions or maintained in DMEM supplemented with 10% fetal bovine serum (FBS), 1% penicillin / streptomycin at 5% CO2 and 37°C as previously described [23]. The cells were starved for 24 hours in 0.1% FBS-containing medium prior to treatment with recombinant human Lcn2 (RayBiotech, USA) or H2O2 (Beijing Chemical Works, CHN) at the indicated concentrations.
Measurements of Apoptosis
The apoptotic cells were quantified using the terminal deoxyribonucleotide transferase-mediated dUTP nick end-labeling (TUNEL) assay (In Situ Cell Death Detection Kit, KeyGEN BioTECH, CHN) according to the manufacturer's instructions. Fluorescent staining was visualized using a BX-61 microscope (Olympus). To further quantify the number of apoptotic cells, flow cytometry (ACCURY C6, BD, USA) using the FITC Annexin V Apoptosis Detection Kit (KeyGEN BioTECH, CHN) was performed according to the manufacturer's instructions, as previously described [23]. Caspase-3 activity was measured using a Caspase-3 Activity Assay Kit (Beyotime, CHN) according to the manufacturer's instructions.
Quantitative real-time RT-PCR
Frozen rat lung tissue was homogenized with the Trizol reagent (Invitrogen, USA), and the total RNA was extracted. Cells grown in 25-cm2 flasks or 6-well plates were washed with PBS and lysed directly in the culture dish with the Trizol reagent. The total RNA was extracted and reverse-transcribed using Oligo (dT) and Transcriptor Reverse Transcriptase (Roche, GA) according to the manufacturer's instructions. The gene transcripts were quantified by real-time quantitative PCR (qPCR) using an Applied Biosystems 7300 Fast Real-Time PCR System (ABI, USA) with SYBR Green PCR Master Mix, as previously described [23]. The expression of gene transcripts in the test samples was normalized against the internal standard glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The primers used are listed in Table 1.
Western blotting analyses
Frozen lung tissue was homogenized in RIPA lysis buffer (Beyotime, CHN) and centrifuged for 10 minutes at 15,000 × g and 4°C. The cells were trypsinized, washed twice with PBS, lysed with RIPA lysis buffer on ice for at least 30 min, and centrifuged for 10 minutes at 15,000 × g and 4°C. The protein concentration was determined using the BCA Protein Assay Kit (Beyotime, CHN) with BSA as the standard. An equal amount of protein (50 μg for rat lung samples, 20 μg for cell samples) from each sample was diluted with 5× sample loading buffer and boiled for 5 minutes. The protein suspensions were separated on 12% SDS-PAGE gel and transferred onto a nitrocellulose membrane, and the proteins were detected using the following antibodies: rabbit anti-Lcn2, rabbit anti-cleaved caspase-3, rabbit anti-SOD1, rabbit anti-SOD2, rabbit anti-Bax (from Santa Cruz Biotechnology, USA), rabbit anti-Bcl-2 (from RayBiotech, GA), mouse anti-GAPDH, goat anti-rabbit IgG, and goat anti-mouse IgG (from ZSGB-BIO, CHN).
Primers used for Real-Time RT-PCR
| Rat GAPDH forward | 5'-GGCACAGTCAAGGCTGAGAATG-3' |
|---|---|
| Rat GAPDH reverse | 5'-ATGGTGGTGAAGACGCCAGTA-3' |
| Rat Lcn2 forward | 5'-TTCCATCCTCGTCAGGGGCCA-3' |
| Rat Lcn2 reverse | 5'-CGGCCACTTGCACATCGTAGC-3' |
| Human GAPDH forward | 5'-GCACCGTCAAGGCTGAGAAC-3' |
| Human GAPDH reverse | 5'-TGGTGAAGACGCCAGTGGA-3' |
| Human SOD1 forward | 5'-TTTGCGTCGTAGTCTCCTGC-3' |
| Human SOD1 reverse | 5'-CCACACCTTCACTGGTCCAT-3' |
| Human SOD2 forward | 5'-GTTGGGGTTGGCTTGGTTTC-3' |
| Human SOD2 reverse | 5'-TGCTCCCACACATCAATCCC-3' |
| Human Bax forward | 5'-CCAAGAAGCTGAGCGAGTGT-3' |
| Human Bax reverse | 5'-GGAAAAAGACCTCTCGGGGG-3' |
Determination of reactive oxygen species [24]
The ROS levels were measured using the Reactive Oxygen Species Assay Kit (Beyotime, CHN) according to the manufacturer's instructions. A membrane-permeable fluorogenic probe, dichlorodihydrofluorescein diacetate (DCFDA), was used. The acetate and acetoxymethyl ester groups of this probe were enzymatically cleaved inside living cells. This probe can then be oxidized by intracellular oxidants (ROS) to obtain the product DCF, which emits a strong green fluorescence (λex=485 nm; λem=520 nm). The fluorescence intensity is proportional to the level of cellular ROS. After the indicated treatment, cells grown in a 6-well plate were loaded in PBS containing 10 μM DCFDA fluorescent probe at 37°C for 20 min and then washed 3 times with PBS and trypsinized. The cells were obtained after centrifugation at 200 × g for 5 min at 4°C, resuspended and incubated in ice-cold PBS. The fluorescence of the cells was monitored using flow cytometry (ACCURY C6, BD, USA). ROS production was calculated as the fold increase in the fluorescence compared with that observed in the control.
Statistical analyses
Each independent experiment was repeated at least three times, and at least 3 independent experiments were performed. The data are shown as the means ± SE. Student's t-test was used for comparisons between two groups, and p<0.05 was considered significant.
Results
Increased expression of Lcn2 in rat PH model and in patients with CHD-PH
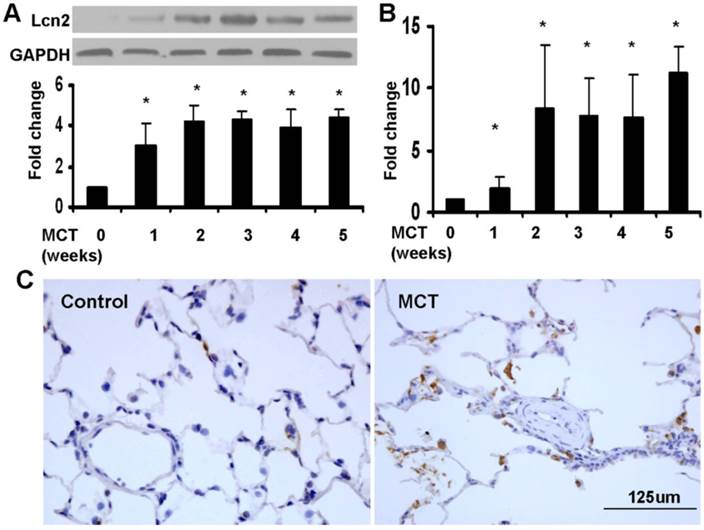
We used a monocrotaline-induced rat PH model to determine whether Lcn2 expression is altered in PH. Three weeks after the MCT injection, the hemodynamic assessment showed that MCT significantly enhanced the pulmonary arterial pressure (Supplementary Material: Fig. S1A) and resulted in significant right ventricular hypertrophy (Supplementary Material: Fig. S1B). The rat lung samples were collected to examine the expression level of Lcn2. Both western blotting analyses (Fig. 1A) and real time-PCR (Fig. 1B) revealed that the expression levels of Lcn2 were significantly increased after MCT injection. The protein levels increased significantly as early as 7 days after MCT injection (Fig. 1A). Immunostaining for Lcn2 also showed its up-regulation in the lungs of rats 3 weeks after MCT injection (Fig. 1C). Consistent with our cellular results obtained from HPASMCs (data not shown), the immunostaining analyses of Lcn2 in rat PASMCs showed undetectable expression.
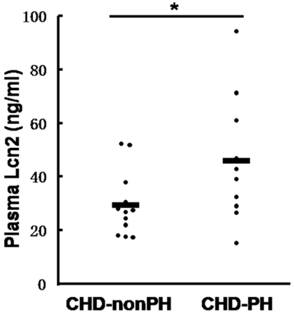
We then examined the Lcn2 levels in PH patients. Twenty-two patients with congenital heart disease were included in the study and divided into 2 groups: 10 CHD-PH patients (age 13.0 ± 3.7 months, 60% males) and 12 CHD-nonPH patients (age 12.8 ± 4.4 months, 50% males). The plasma samples from patients with CHD-PH and CHD-nonPH were subjected to immunoblotting. As shown in Fig. 2, the CHD-PH patients exhibited significantly higher plasma Lcn2 concentrations compared with the CHD-nonPH patients.
The level of Lcn2 in rat PH model induced with MCT. 7, 14, 21, 28 and 35 days after the subcutaneous injection of MCT or saline, the lung samples of rats were collected. A, Total protein was extracted and subjected to Western blotting analyses. Representative western blot and quantitative analysis are shown (n = 6; *, p < 0.05 compared with control). B, Total RNA was extracted and Real-time PCR for Lcn2 was performed (n = 6; *, p < 0.05 compared with control). C, Immunostaining for LCN2 in lungs of rat after 3 weeks of MCT (or saline) injection (400×).
The levels of Lcn2 in patients with CHD-PH and CHD-nonPH. Plasma samples from patients with CHD-PH (n = 10, age 13.0 ± 3.7 months, 60% males) and CHD-nonPH (n = 12, age 12.8 ± 4.4 months, 50% males) were collected. The level of Lcn2 was measured using the Human Lipocalin-2/NGAL Quantikine ELISA Kit. Heavy lines represent mean value of each group (*, p < 0.05).
Lcn2 inhibits HPASMC apoptosis
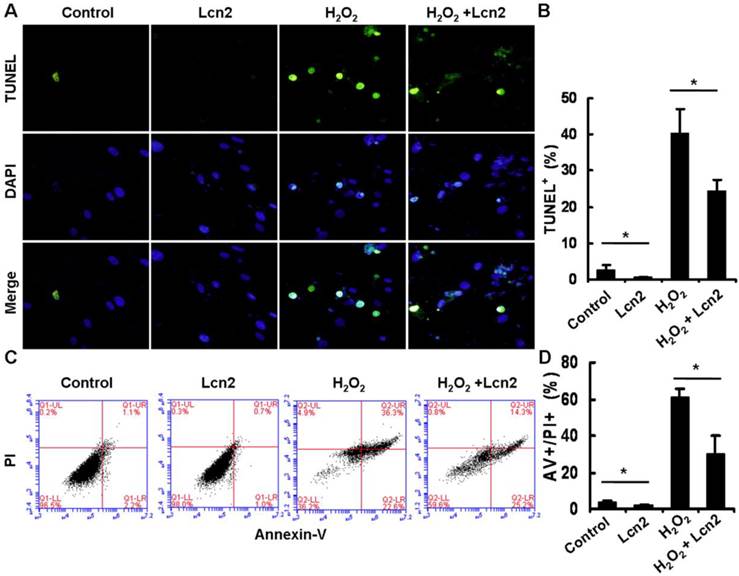
Under conditions of serum deprivation, a few cells will undergo apoptosis due to a lack of survival factors [25]. We used the TUNEL assay to detect DNA fragmentation and found that less apoptotic cells were observed after treatment of HPASMCs with 10 ng/ml recombinant human Lcn2 for 24 h compared with the serum-free medium alone (control). Furthermore, 100 μM H2O2 induced nearly half of the HPASMCs to undergo apoptosis. In the presence of 10 ng/ml Lcn2, the sensitivity of HPASMCs to H2O2 was decreased (Fig. 3A). The quantitative analysis of TUNEL-positive cells demonstrated a significant decrease in apoptotic cells in the Lcn2-treated groups compared with the no-Lcn2-treated groups (Fig. 3B). The role of Lcn2 in HPASMC apoptosis was analyzed further using Annexin V (AV) and propidium iodide (PI) double staining followed by flow cytometry (Fig. 3C). A significant decrease in the population of AV+/PI+ cells was observed in the Lcn2-treated groups (Fig. 3D).
Determination of apoptotic cells using TUNEL assay and flow cytometry. HPASMCs were treated with serum free medium or 100 μM H2O2, in the presence or absence of 10 ng/ml Lcn2 for 24 h. A, Apoptotic cells were examined using TUNEL assay for DNA fragmentation. TUNEL-positive nuclei are shown in green, total nuclei stained with DAPI in blue. HPASMCs were photographed by fluorescence microscopy (magnification, ×400). B, Quantitative analysis (% of apoptotic cells versus total, at lease 50 cells per group per experiment) from five randomly selected fields in each individual experiment are shown in histogram (n = 3; *, p < 0.05). C, After double staining of HPASMCs, the AV+ and PI+ cells were analyzed by flow cytometry (total 20, 000 events per group). D, Quantitative determination of data from FACS analysis showing treatment of Lcn2 (10 ng/ml) for 24h produced a significant decrease in the number of cells undergoing apoptosis (AV+/PI+) in both groups (n = 3; *, p < 0.05).
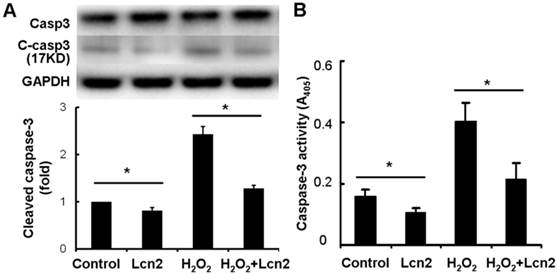
Next, we investigated whether caspase-3 cleavage was inhibited by Lcn2 in HPASMCs. The cells were cultured in serum-free conditions (control) or treated with 100 μM H2O2, in the presence or absence of 10 ng/ml recombinant human Lcn2 for 24 h. Lcn2-inhibited caspase-3 cleavage was confirmed in both conditions (with or without H2O2) using western blotting analyses (Fig. 4A). We observed that the levels of the cleaved form of caspase-3 (17 kDa) were significantly decreased in the Lcn2-treated groups (Fig. 4A). Next, we directly measured the activity of caspase-3 using an assay based on the cleavage of a fluorogenic substrate and found that the activity of caspase-3 was significantly decreased in cells exposed to Lcn2 (Fig. 4B).
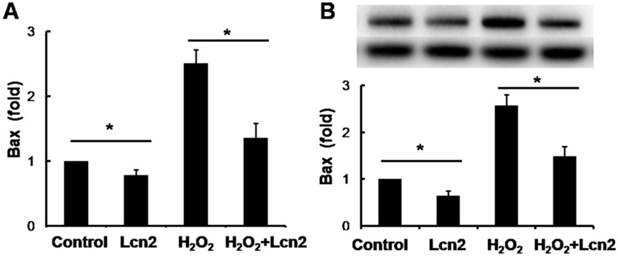
Lcn2 decreases the expression of Bax in HPASMCs
Bax is an important proapoptotic factor involved in the intrinsic pathway of apoptosis. Bax increases mitochondrial membrane permeability by translocating and integrating into the membrane, thereby promoting mitochondria-dependent apoptosis. The effect of Lcn2 on the expression of this protein was examined. RT-PCR (Fig. 5A) and western blotting analyses (Fig. 5B) revealed that the expression levels of Bax in HPASMCs were significantly decreased in the Lcn2-treated groups. We also examined mitochondria membrane-integrated Bax using an antibody against an epitope in the N-terminal domain of Bax, which is normally obscured and is only exposed when Bax changes its conformation as it integrates into the mitochondrial membrane [26]. A decrease in the endogenous active form of Bax in the Lcn2-treated group was observed compared with the H2O2-treated group (Supplementary Material: Fig. S2).
Detection of cleaved caspase-3 and its activity. HPASMCs were treated with serum free medium or 100 μM H2O2, in the presence or absence of 10 ng/ml Lcn2 for 24 h, total protein was extracted and subjected to Western blotting analyses. A, A representative blot and n = 3 independent experiments are quantitated (*, p < 0.05). B, Cell lysates were assayed for caspase-3 activity and normalized to total protein concentration, the value of absorbance at 405 nm is shown (n = 3; *, p < 0.05).
Lcn2 decreases Bax expression. HPASMCs were treated with serum free medium or 100 μM H2O2, in the presence or absence of 10 ng/ml Lcn2 for 24 h. A, Total RNA was extracted and reverse transcripted to cDNA, quantitative determination of Bax expression using real-time PCR, and normalized against the internal standard GAPDH. B, Total protein was extracted and subjected to Western blotting analyses. A representative blot and quantitative analysis are shown. (n = 3; *, p < 0.05).
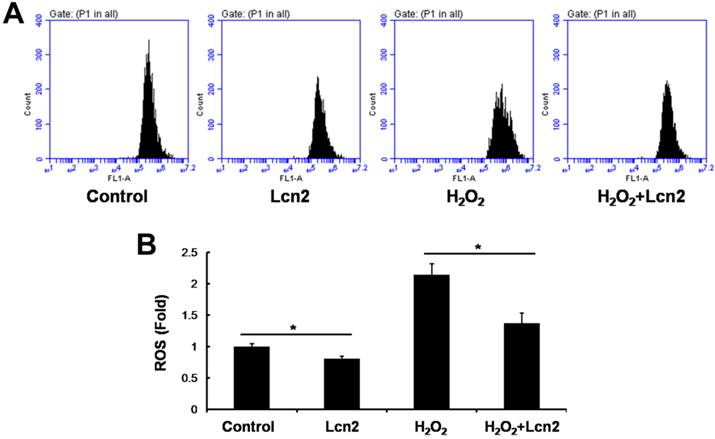
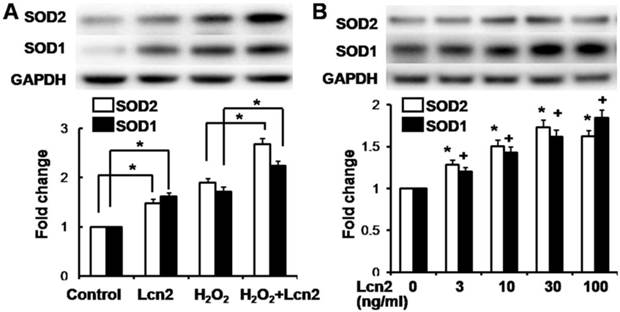
Lcn2 decreases intracellular ROS and increases SOD1 and SOD2 expression
ROS are important effectors in oxidative stress-induced apoptosis, and excessive amounts of ROS are lethal [9]. Thus, we used the general ROS indicator DCFDA to determine the level of intracellular ROS. The cells were cultured in serum-free conditions (control) or treated with 100 μM H2O2, in the presence or absence of 10 ng/ml recombinant human Lcn2 for 12 h. Lcn2 significantly decreased the observed fluorescence in both conditions (with or without H2O2) compared with the no-Lcn2-treated groups (Fig. 6). Superoxide dismutases (SOD1 and SOD2) are major antioxidant enzymes that act against ROS-mediated damage in cells [17]. The effects of Lcn2 on SOD1 and SOD2 expression were also examined using RT-PCR and western blotting analyses. Lcn2 significantly up-regulated the expression of SOD1 and SOD2 in both conditions (with or without H2O2) compared with the no-Lcn2-treated groups (Fig. 7A). The promoting effects of Lcn2 on SOD1 and SOD2 expression were dose-dependent (Fig. 7B).
Lcn2 decreases the level of intracellular ROS. HPASMCs were treated with serum free medium or 100 μM H2O2, in the presence or absence of 10 ng/ml Lcn2 for 24 h, then loaded in PBS containing 10 μM DCFDA fluorescent probe at 37°C for 20 min. Wash and incubate the cells in ice-cold PBS after tripsinized, the fluorescence of the cells was monitored using flow cytometry. Representative dot plots (A) and quantitative analysis of mean fluorescent intensity (B) are shown (n = 3; *, p < 0.05).
Lcn2 increases the expression of SOD1 and SOD2. A, HPASMCs were treated with serum free medium or 100 μM H2O2, in the presence or absence of 10 ng/ml Lcn2 for 24 h. Total protein was extracted and subjected to Western blotting analyses. A representative blot and quantitative analysis is shown (n = 3; *, p < 0.05). B, HPASMCs were treated with 0, 3, 10, 30 and 100 ng/ml Lcn2 in serum free medium for 24 h, total protein was extracted and subjected to Western blotting analyses. A representative blot and quantitative analysis are shown. (n = 3; *, p < 0.05 compared with SOD2 control; +, p < 0.05 compared with SOD1 control).
Discussion
Lcn2 was initially shown to combat bacterial infection via the impedance of bacterial iron sequestration [14]. Its small size and secreted nature motivated its investigation as a diagnostic and prognostic biomarker in numerous diseases, including inflammation and cancer [27]. Recent studies have shown that Lcn2 is over-expressed in cancers of diverse histological origin and it facilitates tumorigenesis by promoting survival, growth, and metastasis [18,19], indicating a pivotal role of this molecule in several physiological and pathological conditions. Conflicting results have been reported regarding the role of Lcn2 in cell apoptosis. On the one hand, Lcn2 can protect a cell from apoptosis induced by cellular stress, such as H2O2 [15-18]. On the other hand, Lcn2 can induce cell apoptosis under specific conditions [20,21]. This motivated us to examine the effect of Lcn2 on HPASMC apoptosis, an important component of small pulmonary arteries remodeling, which results in PH. In this study, we examined the up-regulated expression of Lcn2 in the lungs of rats treated with MCT (Fig. 1). The plasma levels of Lcn2 in CHD-PH patients were also significantly elevated compared with the CHD-nonPH subjects (Fig. 2). These results enhanced the probability that Lcn2 may play a role in PH.
Using TUNEL staining and flow cytometry analysis, we demonstrated that recombinant Lcn2 can protect HPASMCs from apoptosis induced by serum withdrawal and H2O2 (Fig. 3). This novel observation was confirmed using measurements of cleaved caspase-3 and its activity (Fig. 4). Although Lcn2 can protect cells against apoptosis, little is known regarding its intracellular mechanisms, particularly in HPASMCs. Thus, we investigated the expression and translocation of Bax, which is involved in the promotion of mitochondria-dependent apoptosis [26]. The expression of Bax was inhibited by Lcn2 at both the protein and mRNA levels (Fig. 5). In particular, Lcn2 inhibited Bax translocation and integration into the mitochondrial membrane, an important step in the activation of mitochondria-dependent apoptosis, as assessed using an antibody against the conformationally changed form of Bax (Supplementary Material: Fig. S2).
ROS are important effectors in oxidative stress-induced apoptosis, and oxidative damage to macromolecules is mediated by intracellular ROS. Our results confirmed decreased levels of intracellular ROS in the Lcn2-treated groups (Fig. 6). Antioxidant enzymes play a critical role in the regulation of intracellular oxidative stress [17]. Superoxide dismutases (SODs) are important antioxidant enzymes, and an increased amount of SODs reduces the intracellular ROS levels. In this study, we found that Lcn2 up-regulates the expression of SOD1 and SOD2 at both the transcriptional and translational levels (Fig. 7). These findings were consistent with the report of Parisa et al. [17], who found that ectopically expressed Lcn2 is a protective factor against oxidative stress via the induction of heme oxygenase-1,2 and SOD1,2 expression in CHO cells. However, the induction of Lcn2 on the expression of heme oxygenase was undetectable in this study (data not shown). Roudkenar et al. [15] reported that Lcn2 exerts its cytoprotective effect independent of HO-1. These reports and our results suggest that SOD1,2 play important roles, at least partially, in Lcn2-inhibited HPASMC apoptosis.
ROS play an essential role in the regulation of several physiological and pathological processes. Moderate increases in ROS can promote cell proliferation [28], whereas extremely excessive amounts of ROS are lethal [9]. Patients with PH are characterized by increased oxidative stress [7]. Elevated levels of ROS are also observed in animal models of PH [8]. ROS have been demonstrated as important proliferative mediators in pulmonary vascular cells. However, whether elevated Lcn2 levels in PH protect pulmonary vascular cells from apoptosis caused by potentially excessive amounts of ROS and maintains the proliferative effect of ROS by up-regulating SODs deserves further study. Indeed, inducing the apoptosis of hypertrophied PASMCs in animal studies prevents the progression of PH [18].
Lcn2 has been investigated as a diagnostic and prognostic biomarker in numerous diseases [27]. It is expressed at very low concentrations in healthy children and is markedly elevated in the case of epithelial damage [29,30]. Fadel et al. [31] reported that plasma Lcn2 can be used as an early biomarker for the diagnosis of acute kidney injury following cardio-pulmonary bypass in pediatric cardiac surgery. In this study, the plasma Lcn2 levels in CHD-PH children were significantly elevated compared with those found in CHD-nonPH subjects (Fig. 1). Recent studies have reported that pulmonary vascular remodeling caused by an abnormal hemodynamic state plays an important role in the development of congenital heart disease-associated PH [32,33]. Left-to-right shunt is an important characteristic of congenital heart disease-associated PH. Under higher oxygen saturation and the shear stress of increased blood flow, pulmonary vascular endothelial cells may be damaged and induced to release inflammatory cytokines, which results in inflammatory cell infiltration [34]. This may be the reason why the plasma Lcn2 level was elevated in CHD-PH, which suggests a more serious hemodynamic state than CHD-nonPH. From this perspective, it is possible that the plasma Lcn2 level may be a new biomarker of PH, at least for CHD-PH. Further experiments are required to understand the in vivo roles of Lcn2 in PH.
Supplementary Material
Figures S1 and S2.
Acknowledgements
This work was supported by research grants from the National 973 Program of China (grant 2010CB529505 to SS.H.). We thank Dr. Yu Nie for his valuable suggestions in preparation of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Runo JR, Loyd JE. Primary pulmonary hypertension. Lancet. 2003;361:1533-1544
2. Burg ED, Remillard CV, Yuan JX. Potassium channels in the regulation of pulmonary artery smooth muscle cell proliferation and apoptosis: Pharmacotherapeutic implications. Br J Pharmacol. 2008;153(Suppl 1):S99-S111
3. Perros F, Montani D, Dorfmuller P, Durand-Gasselin I, Tcherakian C, Le Pavec J. et al. Platelet-derived growth factor expression and function in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2008;178(1):81-88
4. Rajagopalan N, Simon MA, Suffoletto MS, Shah H, Edelman K, Ma thier MA. et al. Noninvasive estimation of pulmonary vascular resistance in pulmonary hypertension. Echocardiography. 2009;26:489-494
5. Abe K, Toba M, Alzoubi A, Ito M, Fagan KA, Cool CD. et al. Formation of plexiform lesions in experimental severe pulmonary arterial hypertension. Circulation. 2010;121:2747-2754
6. Peter Dromparis, Roxane Paulin, Trevor H Stenson, Alois Haromy, Gopinath Sutendra, Evangelos D Michelakis. Attenuating endoplasmic reticulum stress as a novel therapeutic strategy in pulmonary hypertension. Circulation. 2013;127:115-125
7. Wong CM, Bansal G, Pavlickova L, Marcocci L, Suzuki YJ. Reactive oxygen species and antioxidants in pulmonary hypertension. Antioxid Redox Signal. 2013;18(14):1789-1796
8. Aggarwal S, Gross CM, Sharma S, Fineman JR, Black SM. Reactive oxygen species in pulmonary vascular remodeling. Compr Physiol. 2013;3(3):1011-1034
9. Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach?. Nat Rev Drug Discov. 2009;8:579-591
10. Hu HL, Zhang ZX, Chen CS, Cai C, Zhao JP, Wang X. Effects of mitochondrial potassium channel and membrane potential on hypoxic human pulmonary artery smooth muscle cells. Am J Respir Cell Mol Biol. 2010;42:661-666
11. Wedgwood S, Dettman RW, Black SM. ET-1 stimulates pulmonary arterial smooth muscle cell proliferation via induction of reactive oxygen species. Am J Physiol. 2001;281:L1058-L1067
12. Nilsen-Hamilton M, Liu Q, Ryon J, Bendickson L, Lepont P, Chang Q. Tissue involution and the acute phase response. Ann NY Acad Sci. 2003;995:94-108
13. Wang Y, Lam KS, Kraegen EW, Sweeney G, Zhang J, Tso AW. et al. Lipocalin-2 is an inflammatory marker closely associated with obesity, insulin resistance, and hyperglycemia in humans. Clin Chem. 2007;53:34-41
14. DH Goetz, MA Holmes, N Borregaard, ME Bluhm, KN Raymond, RK Strong. The neutrophil lipocalin NGAL is a bacteriostatic agent that interferes with siderophore-mediated iron acquisition. Mol Cell. 2002;10:1033-1043
15. Roudkenar MH, Halabian R, Bahmani P, Roushandeh AM, Kuwahara Y, Fukumoto M. Neutrophil gelatinase-associated lipocalin: a new antioxidant that exerts its cytoprotective effect independent on Heme Oxygenase-1. Free Radic Res. 2011;45(7):810-819
16. Mehryar Habibi Roudkenar, Raheleh Halabian, Zahra Ghasemipour, Amaneh Mohammadi Roushandeh, Mahdi Rouhbakhsh, Mahin Nekogoftar. et al. Neutrophil gelatinase-associated lipocalin acts as a protective factor against H2O2 toxicity. Archives of medical research. 2008;39:560-566
17. Parisa Bahmani, Raheleh Halabian, Mehdi Rouhbakhsh, Amaneh Mohammadi Roushandeh, Nasser Masroori, Majid Ebrahimi. et al. Neutrophil gelatinase-associated lipocalin induces the expression of hemeoxygenase-1 and superoxide dismutase. Cell stress and chaperones. 2010;15:395-403
18. Lin HH, Liao CJ, Lee YC, Hu KH, Meng HW, Chu ST. Lipocalin-2-induced cytokine production enhances endometrial carcinoma cell survival and migration. Int J Biol Sci. 2011;7(1):74-86
19. Rodvold J, Mahadevan N, Zanetti M. Lipocalin 2 in cancer: when good immunity goes bad. Cancer Lett. 2012;316(2):132-8
20. Guoxiong Xu, JinHee Ahn, SoYoung Chang, Megumi Eguchi, Arnaud Ogier, SungJun Han. et al. Lipocalin-2 induces cardiomyocyte apoptosis by increasing intracellular iron accumulation. J Biol Chem. 2012;287:4808-4817
21. I-Lun Hsin, Yueh-Chieh Hsiao, Ming-Fang Wu, Ming-Shiou Jang, Sheau-Chung Tang, Yu-Wen Lin. et al. Lipocalin 2, a new GADD153 target gene, as an apoptosis inducer of endoplasmic reticulum stress in lung cancer cells. Toxicology and applied pharmacology. 2012;263:330-337
22. Meng L, Liu X, Zheng Z, Li J, Meng J, Wei Y, Hu S. Original rat model of high kinetic unilateral pulmonary hypertension surgically induced by combined surgery. J Thorac Cardiovasc Surg. 2013;146(5):1220-1226
23. Liu X, Meng L, Shi Q, Liu S, Cui C, Hu S, Wei Y. Dermatopontin promotes adhesion, spreading and migration of cardiac fibroblasts in vitro. Matrix Biol. 2013;32(1):23-31
24. Mari C Asensio-Lo´ pez, Jesu´ sSa´ nchez-Ma´ s, Domingo A Pascual-Figal, Sergio Abenza. et al. Involvement of ferritin heavy chain in the preventive effect of metformin against doxorubicin-induced cardiotoxicity. Free Radic Biol Med. 2013;57(10):188-200
25. Chen J, Baydoun AR, Xu R, Deng L, Liu X, Zhu W. et al. Lysophosphatidic acid protects mesenchymal stem cells against hypoxia and serum deprivation-induced apoptosis. Stem Cells. 2008;26(1):135-45
26. Shin EJ, Schram K, Zheng XL, Sweeney G. Leptin attenuates hypoxia/reoxygenation-induced activation of the intrinsic pathway of apoptosis in rat H9c2 cells. J Cell Physiol. 2009;221:490-497
27. Chakraborty S, Kaur S, Guha S, Batra SK. The multifaceted roles of neutrophil gelatinase associated lipocalin (NGAL) in inflammation and cancer. Biochim Biophys Acta. 2012;1826(1):129-169
28. Barrera C. Oxidative stress and lipid peroxidation products in cancer progression and therapy. ISRN Oncology. 2012:137289
29. Schmidt-Ott KM, Mori K, Kalandadze A, Li JY, Paragas N, Nicholas T. et al. Neutrophil gelatinase-associated lipocalin-mediated iron traffic in kidney epithelia. Curr Opin Nephrol Hypertens. 2006;15:442-449
30. Zap pitelli M, Washburn KK, Arikan AA, Loftis L, Ma Q, Devarajan P. et al. Urine neutrophil gelatinase-associated lipocalin is an early marker of acute kidney injury in critically ill children: a prospective cohort study. Crit Care. 2007;11:R84
31. Fadel FI, Abdel Rahman AM, Mohamed MF, Habib SA, Ibrahim MH, Sleem ZS. et al. Plasma neutrophil gelatinase-associated lipocalin as an early biomarker for prediction of acute kidney injury after cardio-pulmonary bypass in pediatric cardiac surgery. Arch Med Sci. 2012;8(2):250-255
32. Jeffery TK, Morrell NW. Molecular and cellular basis of pulmonary vascular remodeling in pulmonary hypertension. Prog Cardiol Dis. 2002;45:173-202
33. Mandegar M, Fung YC, Huang W, Remillard CV, Rubin LJ, Yuan JX. Cellular and molecular mechanisms of pulmonary vascular remodeling: role in the development of pulmonary hypertension. Microvasc Res. 2004;68:75-103
34. Dorfmüller P, Perros F, Balabanian K, Humbert M. Inflammation in pulmonary arterial hypertension. Eur Respir J. 2003;22:358-363
Author contact
![]() Corresponding author: Yingjie Wei, State Key Laboratory of Cardiovascular Disease, Fuwai Hospital, National Center for Cardiovascular Diseases, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, 100037, People's Republic of China. Tel: 86-10-60866393. E-mail: weiyingjieorg.
Corresponding author: Yingjie Wei, State Key Laboratory of Cardiovascular Disease, Fuwai Hospital, National Center for Cardiovascular Diseases, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, 100037, People's Republic of China. Tel: 86-10-60866393. E-mail: weiyingjieorg.
Received 2014-4-23
Accepted 2014-6-16
Published 2014-7-17