Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Structure of c-Myc
Diverse Functions of c-Myc
c-Myc as a Therapeutic Target
Future Prospects
Abbreviations
Acknowledgements
References
Author biography

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2014; 10(10):1084-1096. doi:10.7150/ijbs.10190 This issue Cite
Review
Small Molecules Targeting c-Myc Oncogene: Promising Anti-Cancer Therapeutics
Bing-Jia Chen1, Yan-Ling Wu2, 1 ![]() , Yoshimasa Tanaka3, Wen Zhang1
, Yoshimasa Tanaka3, Wen Zhang1 ![]()
1. Lab of Chemical Biology and Molecular Drug Design, College of Pharmaceutical Science, Zhejiang University of Technology, 18 Chaowang Road, Hangzhou, 310014, China.
2. Lab of Molecular Immunology, Virus Inspection Department, Zhejiang Provincial Center for Disease Control and Prevention, 630 Xincheng Road, Hangzhou, 310051, China.
3. Center for Innovation in Immunoregulative Technology and Therapeutics, Graduate School of Medicine, Kyoto University, Kyoto, 606-8501, Japan.
Received 2014-7-25; Accepted 2014-8-25; Published 2014-9-13
Abstract
The nuclear transcription factor c-Myc is a member of the Myc gene family with multiple functions and located on band q24.1 of chromosome 8. The c-Myc gene is activated by chromosomal translocation, rearrangement, and amplification. Its encoded protein transduces intracellular signals to the nucleus, resulting in the regulation of cell proliferation, differentiation, and apoptosis, and has the ability to transform cells and bind chromosomal DNA. c-Myc also plays a critical role in malignant transformation. The abnormal over-expression of c-Myc is frequently observed in some tumors, including carcinomas of the breast, colon, and cervix, as well as small-cell lung cancer, osteosarcomas, glioblastomas, and myeloid leukemias, therefore making it a possible target for anticancer therapy. In this minireview, we summarize unique characteristics of c-Myc and therapeutic strategies against cancer using small molecules targeting the oncogene, and discuss the prospects in the development of agents targeting c-Myc, in particular G-quadruplexes formed in c-Myc promoter and c-Myc/Max dimerization. Such information will be of importance for the research and development of c-Myc-targeted drugs.
Keywords: c-Myc, small molecule, targeted therapy, tumor, G-quadruplex.
Introduction
As chronic non-infectious diseases, malignancy has become a serious threat to human health, and morbidity and mortality rates have been rising in recent years. At its most basal level, cancer is a genetic disease. It is challenging and interesting to develop effective therapy for cancer, because it comprises a heterogeneous group of diseases. Multiple factors are generally involved in its onset. As a result of synergistic or sequential damages of DNA, several proto-oncogenes such as c-Myc are activated and tumor suppressor genes are inactivated, leading to the alteration of DNA repair system and apoptosis regulation. Accumulation of the DNA damages may ultimately cause cell transformation.
Myc gene family comprises c-Myc, l-Myc, n-Myc, and so on. Based on the analysis so far, as many as 20% of human cancers can be associated with the overexpression of c-Myc. As a vital transcription regulator, c-Myc plays an essential role in the regulation of many physiological processes including cell cycle control, apoptosis, protein synthesis, and cell adhesion [1]. Aberrant expression of c-Myc is likely to ascribable to direct gene alteration, which associates with tumorigenesis and sustained tumor growth [2-4]. Thus, the inhibition of c-Myc has promise as a therapeutic strategy for human cancer [5, 6]. Here we review structural and functional features of c-Myc and highlight to discuss possible small molecule modulators of c-Myc as promising anti-cancer therapeutics.
Structure of c-Myc
Some 20 years ago, c-Myc was discovered in human Burkitt's lymphoma, as a celluar homologue of the viral oncogene v-Myc which was isolated from an avian retrovirus [7, 8]. On the basis of the study of v-Myc in chicken, the expression of human c-Myc was found to be altered in cancer. c-Myc is deregulated in a wide range of malignancies, such as mammary carcinoma, colon carcinoma, cervical carcinoma, myeloid leukaemia, melanoma osteosarcoma, glioblastoma, and small-cell lung carcinoma [8]. Human c-Myc (Fig. 1) is located on chromosome 8 q24.1 and consists of three exons and two introns [9].
A schematic diagram of human c-Myc gene structure. The diagram shows the location of exons (
 ) and promoters indicated by arrows. The region (
) and promoters indicated by arrows. The region (
 ) of a combination of transcription activators with DNA, the nuclease hypersensitivity element III1 (NHE III1), is located in the upstream of the P1 promoter.
) of a combination of transcription activators with DNA, the nuclease hypersensitivity element III1 (NHE III1), is located in the upstream of the P1 promoter.
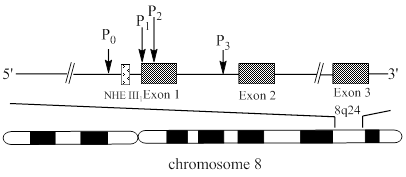
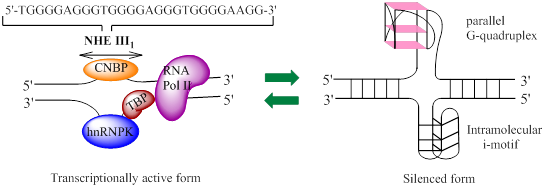
The c-Myc transcription is regulated by multiple promoters. The nuclease hypersensitivity element III1 (NHE III1), also known as Pu27 with 27bp, controls 80-90% of the transcriptional activity of this gene [10-12]. This guanine (G)-rich element is located at -142~-115 bp upstream of the P1 promoter and forms transcriptionally active double helix structure (Fig. 2). The G-rich strand can also form intramolecular G-quadruplex structure consisting of repeated sequences with three or four guanine residues to suppress c-Myc transcription in a silenced form [13]. Hence, the element is a potential target to down-regulate c-Myc overexpression in tumor cells.
The diagram of equilibrium between two forms of NHE III1 (nuclease hypersensitivity element III1). The left part represents a transcriptionally active form, which can regulate 80-90% of c-Myc transcription, and the right one a silenced form, with both G-quadruplex and i-motif structures being shown, that represses the transcription of c-Myc. CNBP: cellular nucleic acid binding protein; hnRNP: heterogeneous nuclear ribonucleoprotein; TBP: TATA-box-binding protein; RNA Pol II: RNA polymerase II.
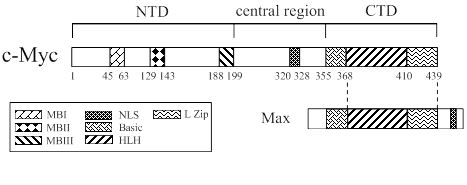
As shown in Fig. 3, c-Myc protein is a 65 kDa nuclear phosphoro-protein belonging to a basic helix-loop-helix leucine zipper (b/HLH/LZ) protein family [6, 13]. It is considered as a master regulatory factor of cell proliferation, metabolism, differentiation, and apoptosis. In humans, c-Myc dysregulation is one of the most common abnormalities found in cancer [14-19]. The c-Myc protein is composed of 439 amino acids (aa), which consists of an N-terminal transactivation domain (NTD), a C-termianl domain (CTD) and a central region. The N-terminal domain contains transcription activation domain (TAD) and three ~20 aa segments termed MYC box-I, II, and III (MBI, MBII, and MBIII), which are essential for the biological functions of this protein [19, 20]. MBI and MBII are located at aa 45-63 and 129-143, respectively, and are responsible for the regulation of transcription and transformation. The C-terminal domain extends aa 360-437, which is essential for the association with b/HLH/LZ-interacting proteins such as the Myc-associated factor X (MAX). The heterodimerization of c-Myc with MAX plays a cardinal role in the proliferation, transformation, and apoptosis [21, 22].
Structure of human c-Myc protein. The C-terminal domain (CTD) of c-Myc is pivotal for the association with MAX, a basic-helix-loop-helix (HLH)-leucine zipper (L Zip)-interacting protein. The N-terminal transactivation domain (NAD) consists of three elements, Myc box-I, II, III (MBI, MBII and MBIII), which are the regulatory motifs necessary for c-Myc functions.
Diverse Functions of c-Myc
The c-Myc oncoprotein has been shown to regulate the expression of about 15% of all human genes and to be involved in many physiological functions, such as cell cycle control, metabolism, protein biosynthesis, and microRNA regulation. In addition, c-Myc is involved in cell apoptosis and senescence, and DNA damage responses. c-Myc overexpression induces DNA damage response through the generation of reactive oxygen species and formation of aberrant DNA-replication intermediates. DNA damage response showed a dual role in tumor progression, including tumor suppression and tumor maintenance [1, 12, 23, 24]. The endogenous c-Myc thus has inherently contradictory features [24]. Herein, the current trends and perspectives in main c-Myc functions are summed up.
Regulation of cell cycle
The primary functions of c-Myc are to promote cell proliferation and to arrest cell differentiation [25-28]. The effect of c-Myc is mainly on the regulation of G1 phase transition in the cell cycle [33]. c-Myc has been shown to be involved in the activation and repression of cyclins D1 and D2, cyclin E, CDK4 (cyclin-dependent kinase 4) and cyclin B1 [22, 29-32]. By inducing the cyclin D-CDK2 (cyclin-dependent kinase 2) activity in the G1 phase of the cell cycle, c-Myc regulates the cell proliferation [33, 34]. The activation of CDK4 and cyclin D2 can lead to segregating p27Kip1 (cyclin-dependent kinase inhibitor 1B) from the cyclin D2-CDK4 complexes [35]. Under the conditions where KIPI is dissociated with the cyclin E-CDK2 complexes, the complexes can be phosphorylated by the CAK (cyclin activating kinase) [22]. Other studies demonstrated that at the core promoter, c-Myc serves as a repressor of CDK inhibitors P15 and P21 through the interaction of c-Myc-Max heterodimer with transcription factors such as MIZ-1 [36-40]. But, the control of cellular proliferation and differentiation is a complex process requiring the coordination of growth factor induced signaling pathways, such as NF-κB (nuclear factor κB), HIFs (hypoxia inducible factors). NF-κB is an important DNA-binding transcription factors, which can activate of c-Myc and cyclin D1 to promote cell proliferation. Meanwhile, c-Myc also can repress NF-κB transactivation and induce sensitivity to TNF (tumor necrosis factor)-induced apoptosis [41]. And HIFs can alter cell-cycle progression through a putative transcription target such as cyclin D1 and indirect modulation of p21 and p27. The two α subunits of HIF-α, HIF-1α and HIF-2α respectively inhibits cell cycle progression and increases proliferation by promoting c-Myc and opposing c-Myc [42]. c-Myc is thus considered as a direct regulator of cell cycle regulation.
Ribosome biogenesis and protein synthesis
Besides as a cell cycle regulator, c-Myc has been also shown to coordinate protein synthesis. Many observations suggested that protein synthesis is regulated by c-Myc in multiple ways, in which the transcription of various RNA, iRNA, and ribosomes is controlled by the oncogene product [43]. In vivo, the rate of protein synthesis in c-Myc-overexpressed fibroblasts is three times higher than that in their parent cell lines [44]. The mechanisms underlying the regulation of protein synthesis by c-Myc are attributed to the regulation of transcription and ribosome biogenesis. Ribosome biogenesis has its own role in many coordination steps, such as synthesis and processing of ribosome RNA and ribosome proteins [43]. Until now, many genes of ribosomal proteins have been shown to be controlled by c-Myc [29, 45, 46]. Notably, c-Myc has a coordination action with nuclear RNA polymerases (RNA pol I and III) required in ribosome biogenesis and translation [33, 47-49]. Promotion of pol III transcription by c-Myc that directly binds TFIIIB (transcription factor ШB), a pol III-specific transcription factor, and pol III-transcribed tRNA and 5SrRNA genes plays an essential role in the regulation of cell cycle progression [33]. In addition, c-Myc also stimulates the transcription of rRNA (ribosomal RNA), resulting in enhanced protein synthesis [47, 48, 50]. And recently research showed that c-Myc enhances protein synthesis during tumorigenesis not only through transcriptional control but also by activating mTOR (the mammalian target of rapamycin) -dependent phosphorylation of 4EBP1 (eukaryotic translation initiation factor 4E binding protein 1). 4EBP1 is a master regulator of protein synthesis control and essential for cancer cells survival in c-Myc-dependent tumor development [51].
Regulation of stem cell functions
Many studies have proven that c-Myc plays a role in the regulation of stem cell function. With the cooperation of n-Myc, c-Myc inhibits the differentiation of stem cells, such as embryonic stem cells [52] and neural stem cells [52-54], and maintains their pluripotency and self-renewal. In addition, the functions of hematopoietic stem cells are regulated by c-Myc and n-Myc [55]. The ability of c-Myc to regulate the expression and function of stem cells is thus inextricably linked to its oncogenic acitivity.
Cell apoptosis
c-Myc is not only an inducer of cell proliferation, but also has the ability to regulate cell apoptosis via various signaling pathways [56, 57]. The mechanism by which c-Myc induces apoptosis has not been fully elucidated. Two major pathways have been, however, proposed [57]. One signaling pathway is that c-Myc induces expression of ARF, a tumor suppression protein which is transcribed from an alternate reading frame of the INK4a/ARF locus. When P 19Arf binds Mdm2 (mouse double minute 2), P53 (tumor protein p53) is activated and promotes apoptosis via activation of proapoptotic genes and cell cycle mediators [56, 58-60]. The other one is that c-Myc regulates apoptosis by repressing the expression of anti-apoptotic proteins, such as Bcl-2 (B-cell lymphoma 2), which blocks mitochondrial outer membrane permeabilization and inhibits cytochrome c release from mitochondria [61-63].
miRNA expression
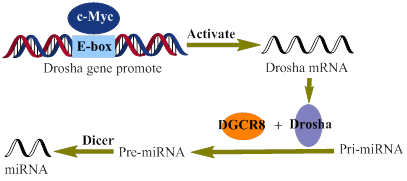
Recently, evidence has been accumulating that c-Myc also regulates the expression of miRNAs, which are a set of small, non-protein-coding RNAs and regulate gene expression at the post-transcriptional level [64-69]. Little is, however, known about how c-Myc regulates the expression of miRNAs [70]. So far, many factors, such as SMAD (drosophila mothers against decapentaplegic) [71, 72], P53 (tumor protein p53) [73], ATM (ataxia telangiectasia mutated) [74], MutLα (MLH1-PMS2 heterodimer) [75], and BRCAl (breast cancer 1) [76], have been shown to interact with microprocessor complexes and modulate the expression of miRNAs. Through enhancing the expression of Drosha (Fig. 4), a c-Myc target gene, c-Myc indirectly promotes the processing of miRNAs [70]. Proper manipulation of the c-Myc-niRNA may lead to a novel therapy for malignancies.
A schematic representation of the c-Myc regulation of miRNAs biogenesis. Directly interacting with the E-box of the drosha gene promoter, c-Myc activates the transcription of Drosha, which in turn promotes the biogenesis of miRNAs.
c-Myc as a Therapeutic Target
c-Myc is expressed in most human hepatic cells, and is closely involved in cell cycle, differentiation, protein synthesis, and apoptosis in normal as well as neoplastic cells. Hence, c-Myc is one of the most important targets in the development of cancer treatment [77]. Here, we particularly review current progress in main small-molecule ligands that can induce and stabilize G-quadruplexes formed in c-Myc promoter sequences and inhibit c-Myc/Max dimerization, leading to the inhibition of cancer cell proliferation. Whereas until now people have not succeeded in developing any drug targeting c-Myc, which can be utilized in clinics [78]. Therefore, it is worth challenging to create novel small molecule compounds that can specifically inhibit the functions of c-Myc and be used to alleviate the suffering of cancer patients.
Small-molecule ligands targeting the c-Myc promoter G-quadruplexes
Advances in the development of anti-tumor drugs targeting the c-Myc promoter G-quadruplexes. G-quadruplexes have been shown to be a promising target for anti-cancer therapy, based on their functions in regulating c-Myc transcription and suppressing tumorigenicity. As shown in Fig. 1, nuclease hypersensitive element (NHE) III1, a G-rich sequence of c-Myc promoter, has two different forms, transcriptionally active and silenced forms; When the G-rich sequence exists in the form of double helix, RNA polymerases in cooperation with various other factors can transcribe c-Myc gene, and in contrast, the silenced form, containing G-quadruplexes, prevents the various transcription factors from interacting with the element, leading to a down-regulation of c-Myc transcription [77]. Under physiological conditions, however, very few G-quadruplexes exist in the c-Myc promoter region [7880]. Since G-quadruplexes play important roles in the repression of c-Myc, small-molecule ligands that can specifically induce the formation of and stabilize the G-quadruplex in vivo may be developed as promising anti-cancer drugs [77, 81]. Until now, only one small-molecule compound CX-3543 (Quarfloxin) (Fig. 5) has entered phase II clinical trials for neuro-endocrine carcinomas (NCT00780663), which targets selectively the Myc G-quadruplexes by interacting with the site between planes in the π-π patterns in vitro based on the determination through NMR (nuclear magnetic resonance), PCR-stop, and MSi (molecular simulation studies). Although CX-3543 was selected as a binder of Myc G-quadruplex, it was preferred to disrupt nucleolin/G-quadruplex complexes in the nucleolus to interact with the biosynthesis of ribosomal RNA in cancer cells and induce apoptosis in cancer cells [82, 83].
Some small-molecules that bind c-Myc promoter G-quadruplexs.
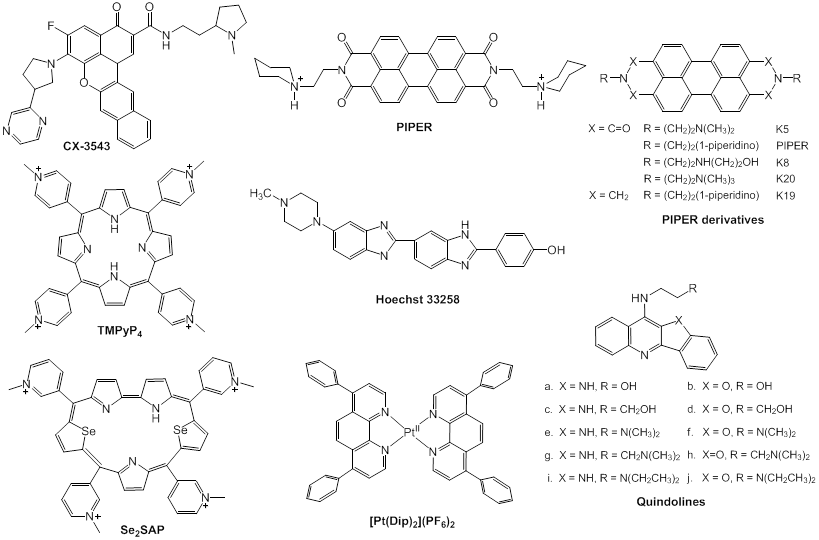
Perylene derivatives: Perylene compounds have been reported to strongly interact with G-quadruplexes [86]. As shown in Fig. 5, a representative of these derivatives, N,N'-bis(2-(1-piperidino)ethyl)-3,4,9,10-perylenetetracarboxylic acid diimide (PIPER), induces the formation of G-quadruplex from duplex of the c-Myc Pu27-mer sequence through end stacking interaction with G-tetrads [84, 85]. In addition, Pivetta and colleagues [86] synthesized a number of perylene derivatives [Fig. 5] with linear or cyclic amines in the side chains. The binding affinity and selectivity of perylenes to G-quadruplexes depends on the structure of the side chains. To develop novel compounds with higher affinity on the basis of the structure-affinity relationship, further synthetic studies should be carried out.
Cationic porphyrins: As a G-quadruplex ligand and a representative of cationic porphyrins, TMPyP4 (Fig. 5) [87] is of special concern. Based on microcalorimetric (ITC, isothermal titration calorimitry, and DSC, differential scanning calorimetry), spectrometric (UV-vis and CD, circular dichroism), and molecular simulation (MSi) studies, TMPyP4 has been proven to down-regulate the expression level of c-Myc through combining with G-quadruplexs of Pu27 [77]. Upon incubation with TMPyP4, G-quadruplex structure undergoes transformation from a parallel type to hybrid with parallel and anti-parallel types, with the ratio of 1:4 at a saturated condition [89]. In addition, TMPyP4 can also interact with some G-rich sequences and stabilize i-motif structures that promote the formation of G-quadruplexes [89, 90. TMPyP4 is thus a promising agent as a lead compound for a novel anti-cancer therapy targeting G-quadruplexes [91]. Owing to the poor selectivity of TMPyP4, however, TMPyP4 itself cannot be used as a therapeutic. Hurley and colleagues designed and synthesized a novel derivative, Se2SAP (Fig. 5), with a porphyrin ring core [92]. Compared to TMPyP4, Se2SAP has higher affinity to G-quadruplexes of c-Myc in comparison with double-stranded DNA and other G-quadruplexes. Se2SAP can convert the parallel G-quadruplexes of the c-Myc Pu27 into a hybrid G-quadruplex with parallel and anti-parallel structures. Based on these studies, Se2SAP is hopefully to become the first compound that can identify different conformations of G-quadruplexes because of its lower photosensitiveness and less toxicity [12].
Quindolines: Gu and colleagues designed and synthesized a series of quindolines [93], which can induce the sequence of the c-Myc promoter Pu27 to form G-quadruplex structure and stabilize it. By repressing the transcription of c-Myc in HepG2 cells, these compounds inhibit the growth of tumour cells. These derivatives have higher affinity to G-quadruplexes than to other DNA structures. The side chains are shown to determine the binding affinity and selectivity. Quindoline derivatives with tertiary amine in the side chains (Fig. 5), such as SYUIQ-05, are more likely to interact with c-Myc G-quadruplexes than telomere G-quadruplexes [94]. The π-π docking is considered to be the primary determinant in the interaction.
Hoechst 33258: It has been demonstrated that the synthetic dye Hoechst 33258 (Fig. 5) can convert Pu27 to G-quadruplexes [84]. The compound interacts with AAGT loop of G-quadruplexes of Pu27.
Alkaloids: From a long-term perspective, natural products have been a prodigious source of compounds with therapeutic activity and low toxicity in the development of tumor-selective therapies [95, 96], of which telomestatin has been shown to be one of the most potent G-quadruplex ligands [97]. Thus, natural products are a pre-existing small molecule library for screening new anti-cancer targeted drugs [98]. Natural alkaloids, a series of compounds of traditional Chinese medicine (TCM), can interact with DNA to form complexes [99]. Ji and coworkers examined the interaction of a number of natural alkaloids [Fig. 6] with G-quadruplexes formed by c-Myc Pu27.
Structures of some natural alkaloids. (A) Structures of four natural alkaloids with similar structures: sanguinarine (San), palmatine (Pal), berberine (Beb), tetrahydropalmatine (Tep) and several Beb derivatives; (B) structures of 7 alkaloids with a backbone of bis-benzltetrahydroisoquinoline.
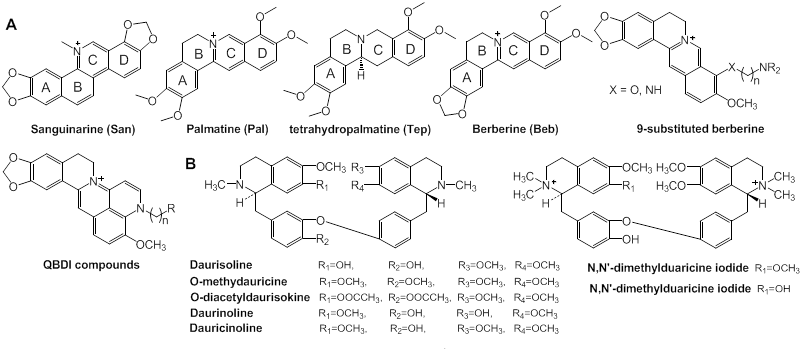
Among them, sanguinarine (San), beberine (Beb), palmatine (Pal), and tetrahydropalmatine (Tep) (Fig. 6) can induce the formation of and stabilize G-quadruplexes. Except for Tep, the examined compounds contain unsaturated ring C and positively charged centers N+ that enhance the interaction with G-quadruplexes through expanding the conjugate system. Especially, the study by Ji et al showed that San had the highest ability to stabilize the structure of G-quadruplexes and inhibit cell growth and that the interaction is mediated by external stacking or intercalating [100]. Other alkaloids with similar structures in Fig. 6 also have comparable ability to stabilize G-quadruplexes. A 9-substituted derivative of Beb with an alkyl side chain carried with a terminal amino group synthesized by Huang and colleagues had higher binding affinity to G-quadruplexes than the original compound [101, 102]. Furthermore, quinolino-benzo dihydroisoquindolium (QBDI) was synthesized by modifying the 9-substituted Beb. All of the Beb and QBDI derivatives can interact with the c-Myc G-quadruplex with high selectivity [103].
Metal complexes: Varieties of metal-containing small molecules have been demonstrated to interact with G-quadruplexes [104, 105]. As clinical drugs used in chemotherapy, cisplatin and its analogs have conceivable adverse effects, such as renal toxicity and treatment-induced resistance. It is thus indispensable to design novel, well-tolerated metal complexes for anti-cancer therapies [106, 107]. In recent years, a number of metal complexes have been reported to interact with G-quadruplexes and stabilize them [108]. Platinum (II) complexes (Pt (II)) can serve as stabilizers of G-quaduplexes [108, 109], since they can strongly interact with G-quadruplexes. It is worthy of note that these studied complexes show only weak binding affinity to duplex DNA. Wang and coworkers designed and synthesized a Pt (II) (Fig. 5) which improved the selectivity of Pt (II) binding to G-quadruplexes over duplex DNA. Based on the experimental data, [Pt(Dip)2](PF6)2 shows greater binding affinity to c-Myc parallel G-quadruplexes than to duplex DNA [110]. In addition, many other complexes containing Zn+, Ni+, Cu+, Mn2+, Ru2+, V4+ and so on, have been evaluated for binding to G-quadruplexes [109]. As well as traditional organic heteroaromatic compounds, various metal complexes have emerged as an increasing important type of compounds in search of novel G-quadruplex binders with strong and selective affinity and potential anticancer activity.
Many small-molecule ligands discussed above, such as quindolines [86], cationic porphyrins [93], platinum complexes [111], can repress the transcription of c-Myc by inducing the formation of c-Myc G-quadruplexes and stabilizing them. Most G-quadruplex ligands containing aromatic planes can stack on the end of G-quadruplexes by a π-π interaction [105]. Only a few ligands, such as Hoechst 33258, have been reported to bind to G-quadruplex grooves and loops [112], which have high selectivity in interacting with different topologies of G-quadruplexes [113]. Also, Chen et al. predicted that 3, 3'-diethyloxadicarbocyanine (DODC) can bind to G-quadruplex grooves [114], and it was proved in their subsequent studies [115, 116].
Carbamide and its analogues: Different from most above-discussed compounds with a few large conjugated aromatic planes, Carbamide 1 (Fig. 7) identified by Ma and colleagues, a natural product, can act as a stabilizer of c-Myc G-quadruplexes through binding with G-quadruplex grooves. NMR and MSi revealed that carbamide 1 could control c-Myc gene transcription. The interaction of carbamide analogues 1-5 with G-quadruplexes was examined and it was demonstrated that carbamide 1 with variable diphenyl ether units had the highest activity in binding to G-quadruplexes [117].
Chemical structures of a variety of G-quadruplex ligands. (A) Structures of carbamide and its analogues; (B) structures of low-molecular weight ligands of telomerase promoter with affinity to the c-Myc gene promoter G-quadruplexes
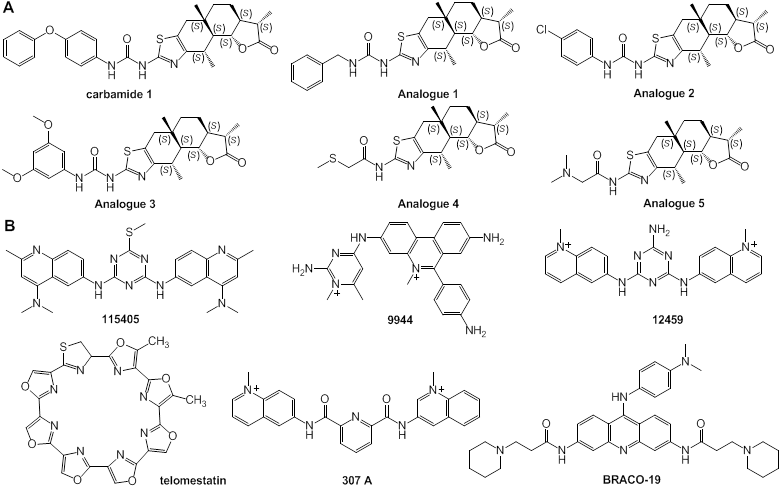
Other ligands: In process of exploring small-molecule ligands, the effect of telomerase inhibitors (Fig. 7) on the stabilization of c-Myc promoter G-quadruplexes was examined. It was demonstrated that a variety of telomerase inhibitors effectively stabilized G-quadruplexes of the c-Myc promoter, and that the affinity of compounds TMPyP4 and 12459 to the c-Myc promoter G-quadruplexes was two-fold higher than that to the G-quadruplexes of telomere [118].
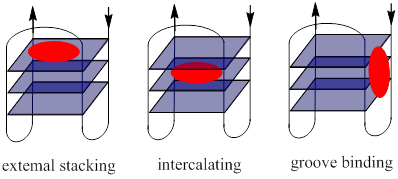
Binding modes of ligands to the c-Myc gene promoter G-quadruplexes. Through targeting G-quadruplexes, small-molecule ligands play pivotal roles in the regulation of c-Myc gene transcription. There are three basic binding modes of the ligands with G-quadruplexes (Fig. 8) [104, 119]: (1) external stacking: ligands with a π-delocalized system stack on the end of G-quadruplexes via π-π stacking; (2) intercalating: ligands insert into the interspace of two G-tetrad planes; and (3) groove or loop nonspecific binding: ligands bind to the grooves or loops of the G-quadruplexes. In the absence of the interaction between internal cation and electrical body of G-quadruplexes, the binding of the molecules at the end of the G-tetrad is easier and more stable than intercalating in lower energy [120]. Therefore, the small-molecule ligands are prone to interact with the c-Myc G-quadruplexes in an external stacking-manner.
Three modes of interaction of small ligands with G-quadruplexes.
Small molecule modulators of c-Myc/Max dimerization
c-Myc, a DNA-binding transcriptional regulator, functions through heterodimerization with another basic-helix-loop-helix leucine zipper (BHLH-LZ) transcription factor, MAX [1, 121]. The c-Myc/Max complex recognizes DNA response elements, such as a specific sequence E-box or other target genes to activate c-Myc-mediated transcription [121]. The interaction and heterodimerization between c-Myc and Max are required in the regulation of all known functions of c-Myc, including regulation of cell proliferation, apoptosis, and target gene transcription [1, 2, 121]. There are two ways to modulate the c-Myc/Max functions (Fig. 9): to inhibit c-Myc/Max dimerization and to disrupt their DNA recognition [122]. Some small molecules can downregulate c-Myc functions through not only inducing the formation of and stabilizing G-quadruplexes, but also targeting c-Myc in a different manner, such as inhibiting c-Myc/Max dimerization or their binding to E-box motifs, and disturbing the interaction of c-Myc with other factors. Thus, small molecules that can inhibit the c-Myc/Max dimerization or binding to E-box motifs may serve as potential tumor therapeutics.
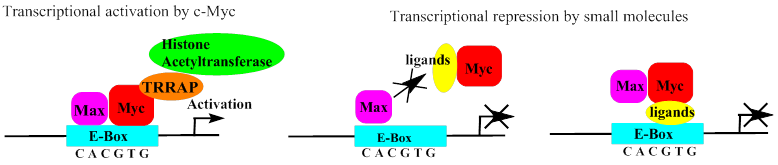
Transcriptional regulation by c-Myc and inhibition of c-Myc functions by small molecules. Left: The binding of c-Myc/Max dimers to E-box elements (CACGTG) activates the transcription of c-Myc-regulated genes. Right: The inhibition of the c-Myc/Max dimerization or c-Myc/Max DNA binding represses the transcription of genes regulated by c-Myc/Max.
Small-molecule inhibitors of c-Myc/Max dimerization. As well as small nonpeptide molecules, which interfere with the dimerization of c-Myc/Max, a number of small-molecule inhibitors of c-Myc/Max dimerization have been discovered in the last decade [123-125]. However, the design of c-Myc inhibitors is still challenging due to a lack of obvious binding sites in the c-Myc/Max interface [1]. Most of the inhibitors thus far obtained were screened from chemical libraries [126]. The peptide mimetic compound IIA6B17 (Fig. 10) was identified as the first small- molecule inhibitor of c-Myc/Max by Vogt and colleagues [127]. Recently, using a reporter system, it was shown that IIA6B17 inhibited c-Myc transcription and interfered with c-Myc/Max dimerization [128]. These high throughput screening studies pave the way of the discovery of new inhibitors of c-Myc/Max heterodimer or c-Myc/Max/DNA complex formation [123-125, 129-135]. Likewise, a compound NY2267 (Fig. 10) and its structurally related derivatives were shown to strongly inhibit c-Myc/Max binding to DNA by groups of Janda and Vogt [124]. NY2267, however, indiscriminately inhibits transcription mediated by c-Jun and c-Myc, since they are similar in their dimer structures [123]. Because all of these small molecules so far identified require high concentrations for the inhibition of c-Myc/Max/DNA complex, it has been difficult to develop c-Myc-targeted anti-cancer therapeutics [124]. Prochwnik and co-workers designed and reported seven small-molecule inhibitors of c-Myc/Max including 10058-F4 and 10074-G5 (Fig. 10) [136]. They first synthesized derivatives based on the structure of 10058-F4 (IC50 = 49 μM on HL60 cells), of which 28RH-NCN-1 exhibited more potent activity in inhibiting the binding of c-Myc/Max to E-box motifs than the parent compound 10058-F4 [124, 129]. On the basis of the structure-activity relationship of the derivatives, pharmacophore analysis demonstrated that 31122-64-2 and 307545-04-6 inhibited the c-Myc/Max/DNA complex formation through inhibiting DNA binding of c-Myc [137]. Then, it was reported that JY-3-094 (Fig. 10), an analog of 10074-G5, could interfere with the association between recombinant c-Myc and Max five-times more efficiently than the parent compound, whereas the analog has poor cell permeability. Thus, a p-carboxylic acid moiety of the analog was esterified to optimize the cellular uptake and retention. The prodrug compound JY-3-094 [138] and its derivatives would warrant further optimization of small-molecule c-Myc/Max inhibitors.
Small molecules capable of inhibiting transcriptional regulation by c-Myc. (A) Small-molecule inhibitors of c-Myc/Max dimerization; (B) small-molecule inhibitors of c-Myc/Max/DNA complex formation.
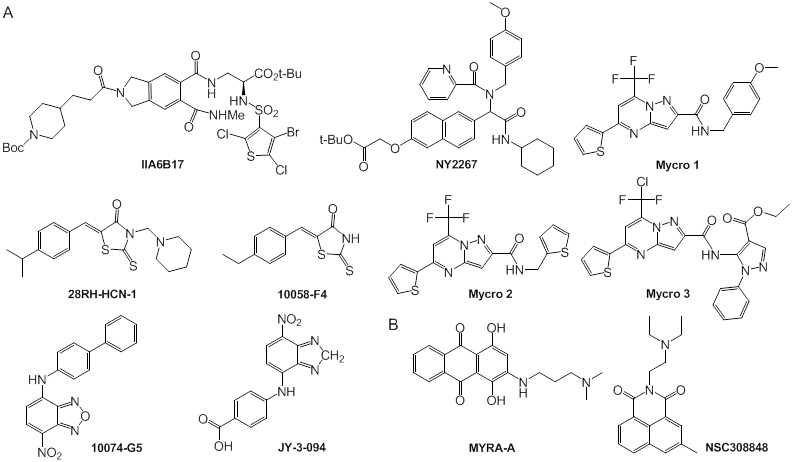
Berg and co-workers designed and synthesized two other small molecules, the pyrazolo [1,5-a] pyrimidine Mycro l and its derivative Mycro 2 (Fig. 10), which inhibit the interaction between c-Myc and Max [125]. Based on the structure of the compounds, pyrazolo [1,5-a] pyrimidine Mycro 3 was also synthesized, which had high selectivity and inhibited c-Myc/Max dimerization and conjugation with DNA [131]. Because Mycro 3 is potent and selective for c-Myc in whole cell assays, with weak inhibitory activity against AP-1, this compound has a superior specificity profile to its predecessors. Mycros are the first inhibitors of c-Myc/Max dimerization, and also inhibit c-Myc/Max/DNA binding, in which the compounds preferentially interact with c-Myc/Max over other related dimeric transcription factors [125].
The mechanisms underlying the inhibition of c-Myc/Max heterodimerization by these small molecules have been unclear until recently. Small-molecule inhibitors were shown to bind one of the three discrete binding sites on the c-Myc bHLH-Zip domain [132, 139]. Using a fluorescence polarization (FP) assay, Metallo and colleagues demonstrated that 10058-F4 and 10074-G5 interacted with c-Myc402-412 and c-Myc363-381, respectively. These two compounds could bind to their specific amino acid residues simultaneously and independently. Besides these two distinct binding sites, a third one in the c-Myc bHLH-Zip domain was identified, with which the compound 10074-A4 could interact. It is worthy of note that the three binding sites are functionally independent and mutually exclusive [132]. When the molecules bind to their distinct binding sites of c-Myc, the oncoprotein may form a more rigid and defined conformation than c-Myc/Max heterodimer, resulting in the dissociation between c-Myc and Max [139].
Small-molecule inhibitors of c-Myc/Max DNA binding. Inhibitors of c-Myc/Max/DNA complexes block c-Myc induced transcription. It is worth noting that two compounds, MYRA-A and NSC308848 (Fig. 10), have high selectivity in targeting the DNA binding domain of c-Myc/Max. The inhibitors thus can suppress the expression of gene products regulated by c-Myc and the induction of apoptosis [136, 140].
Future Prospects
Because of its oncogenic properties in neoplastic cells, c-Myc has become an interesting and feasible target for novel therapies of a variety of human malignancies related to c-Myc. It has been shown that several small molecules target the transcription of c-Myc gene directly or the c-Myc downstream pathway. Especially, G-rich region of c-Myc promoter has become a promising target. Many reports demonstrate that low-molecular weight compounds have a potential to be developed into therapeutic drugs in indivisualized cancer therapy. Even with the advances in the field of drug design and in the mechanisms underlying the c-Myc overexpression in tumor cells, it is still difficult to obtain highly specific and active anti-cancer drugs. Through the identification of various small molecular compounds that interfere with c-Myc, it may be possible to develop novel and effective therapeutic agents to treat cancer.
Abbreviations
CNBP: cellular nucleic acid binding protein; hnRNP: heterogeneous nuclear ribonucleoprotein; TBP: TATA-box-binding protein; RNA Pol II: RNA polymerase II; CDK4: cyclin-dependent kinase4; CDK2: cyclin-dependent kinase 2; p27Kip1: cyclin-dependent kinase inhibitor 1B; CAK: cyclin activating kinase; NF-κB: nuclear factor κB; HIFs: hypoxia inducible factors; TNF: tumor necrosis factor; TFIIIB: transcription factor ШB; rRNA: ribosomal RNA; mTOR: the mammalian target of rapamycin; 4EBP1: eukaryotic translation initiation factor 4E binding protein 1; Mdm2: mouse double minute 2; P53: tumor protein p53; Bcl-2: B-cell lymphoma 2; SMAD: drosophila mothers against decapentaplegic; ATM: ataxia telangiectasia mutated; MutLα: MLH1-PMS2 heterodimer; BRCAl: breast cancer 1; NMR: nuclear magnetic resonance; MSi: molecular simulation; ITC: isothermal titration calorimitry; DSC: differential scanning calorimetry; CD: circular dichroism.
Acknowledgements
We gratefully acknowledge the financial support from Zhejiang Provincial Natural Science Foundation of China (LY12B02019), the Qianjiang Talents Program of Zhejiang Province (2009R10002) and the Program for Zhejiang Leading Team of Science and Technology Innovation (2011R50021).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Dang C V. MYC on the Path to Cancer. Cell. 2012;149(1):22-35
2. Fernandez PC, Frank S R, Wang L. et al. Genomic targets of the human c-Myc protein. Genes & development. 2003;17(9):1115-1129
3. Li Z, Van Calcar S, Qu C. et al. A global transcriptional regulatory role for c-Myc in Burkitt's lymphoma cell. PNAS. 2003;100(14):8164-8169
4. Lin CY, Lovén J, Rahl PB. et al. Transcriptional amplification in tumor cells with elevated c-Myc. Cell. 2012;151(1):56-67
5. Zhang X, Zhao X, Fiskus W. et al. Coordinated silencing of MYC-mediated miR-29 by HDAC3 and EZH2 as a therapeutic target of histone modification in aggressive B-cell lymphomas. Cancer cell. 2012;22(4):506-523
6. Berg T. Small-molecule modulators of c-Myc/Max and Max/Max interaction. Curr Top Microbio Immunol. 2011;348:139-149
7. Lin CP, Liu CR, Lee CN. et al. Targeting c-Myc as a novel approach for hepatocellular carcinoma. World J Hepatol. 2010;2(1):16-20
8. Zhang W, Kater AP, Widhopf GF. et al. B-cell activating factor and v-Myc myelocytomatosis viral oncogene homolog (c-Myc) influence progression of chronic lymphocytic leukemia. PNAS. 2010;107(44):18956-18960
9. Watt R, Nishikura K, Sorrentino J. et al. The structure and nucleotide sequence of the 5'end of the human c-myc oncogene. PNAS. 1983;80(20):6307-6311
10. Islam MA, Thomas SD, Murty VV. et al. c-Myc quadruplex-forming sequence Pu-27 induces extensive damage in both telomeric and non-telomeric regions of DNA. J Biol Chem. 2014: jbc.M113.505073.
11. Simonsson T, Pribylova M, Vorlickova M. A Nuclease Hypersensitive Element in the human c-myc promoter adopts several distinct i-tetraplex structures. Biochem Biophys Res Commun. 2000;278(1):158-166
12. Seenisamy J, Bashyam S, Gokhale V. et al. Design and synthesis of an expanded porphyrin that has selectivity for the c-MYC G-quadruplex structure. J Am Chem Soc. 2005;127(9):2944-2959
13. Cashman DJ, Buscaglia R, Freyer MW. et al. Molecular modeling and biophysical analysis of the c-MYC NHE-III1 silencer element. J Mol Model. 2008;14(2):93-101
14. Soucek L, Evan G. Myc-Is this the oncogene from Hell? Cancer Cell. 2002;1(5):406-408
15. Vafa O, Wade M, Kern S. et al. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: a mechanism for oncogene-induced genetic instability. Mol Cell. 2002;9(5):1031-1044
16. Zhang Q, Spears E, Boone DN. et al. Domain-specific c-Myc ubiquitylation controls c-Myc transcriptional and apoptotic activity. PNAS. 2013;110(3):978-983
17. Song G, Li Y, Zhang Z. et al. c-myc but not Hif-1α-dependent downregulation of VEGF influences the proliferation and differentiation of HL-60 cells induced by ATRA. Oncol Rep. 2013;29(6):2378-2384
18. Alblihn A, Johnsen JI, Henriksson MA. MYC in oncogenesis and as a target for cancer therapies. Adv Cancer Res. 2010;107:163-224
19. Delmore JE, Issa GC, Lemieux ME. et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146(6):904-917
20. Sakamuro D, Prendergast GC. New Myc-interacting proteins: a second Myc network emerges. Oncogene. 1999;18(19):2942-2954
21. Ponzielli R, Katz S, Barsyte-Lovejoy D. et al. Cancer therapeutics: targeting the dark side of Myc. Eur J Cancer. 2005;41(16):2485-2501
22. Pelengaris S, Khan M. The many faces of c-MYC. Arch Biochem Biophys. 2003;416(2):129-136
23. Nie Z, Hu G, Wei G. et al. c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell. 2012;151(1):68-79
24. Cartwright P, McLean C, Sheppard A. et al. LIF/STAT3 controls ES cell self-renewal and pluripotency by a Myc-dependent mechanism. Development. 2005;132(5):885-896
25. Baudino TA, McKay C, Pendeville-Samain H. et al. c-Myc is essential for vasculogenesis and angiogenesis during development and tumor progression. Genes Dev. 2002;16(19):2530-2543
26. Cavalheiro GR, Matos-Rodrigues GE, Gomes AL. et al. c-myc regulates cell proliferation during Lens development. PloS one. 2014;9(2):e87182
27. Krysan K, Kusko R, Grogan T. et al. PGE2-driven expression of c-Myc and oncomiR-17-92 contributes to apoptosis resistance in NSCLC. Mol Cancer Res. 2014 molcanres-0377
28. Amati B. Integrating Myc and TGF-β signalling in cell-cycle control. Nat Cell Biol. 2001;3(5):E112-E113
29. Fernandez PC, Frank SR, Wang L. et al. Genomic targets of the human c-Myc protein. Genes Dev. 2003;17(9):1115-1129
30. Bouchard C, Dittrich O, Kiermaier A. et al. Regulation of cyclin D2 gene expression by the Myc/Max/Mad network: Myc-dependent TRRAP recruitment and histone acetylation at the cyclin D2 promoter. Genes Dev. 2001;15(16):2042-2047
31. Menssen A, Hermeking H. Characterization of the c-MYC-regulated transcriptome by SAGE: identification and analysis of c-MYC target genes. PNSA. 2002;99(9):6274-6279
32. Hermeking H, Rago C, Schuhmacher M. et al. Identification of CDK4 as a target of c-MYC. PNAS. 2000;97(5):2229-34
33. Gomez-Roman N, Grandori C, Eisenman R N. et al. Direct activation of RNA polymerase III transcription by c-Myc. Nature. 2003;421(6920):290-294
34. Steiner P, Philipp A, Lukas J. et al. Identification of a Myc-dependent step during the formation of active G1 cyclin-cdk complexes. EMBO J. 1995;14(19):4814-4836
35. Bouchard C, Thieke K, Maier A. et al. Direct induction of cyclin D2 by Myc contributes to cell cycle progression and sequestration of p27. EMBO J. 1999;18(19):5321-5333
36. Staller P, Peukert K, Kiermaier A. et al. Repression of p15INK4b expression by Myc through association with Miz-1. Nat Cell Biol. 2001;3(4):392-399
37. Wu S, Cetinkaya C, Munoz-Alonso M J. et al. Myc represses differentiation-induced p21CIP1 expression via Miz-1-dependent interaction with the p21 core promoter. Oncogene. 2003;22(3):351-360
38. Seoane J, Pouponnot C, Staller P. et al. TGF-β influences Myc, Miz-1 and Smad to control the CDK inhibitor p15INK4b. Nat Cell Biol. 2001;3(4):400-408
39. Seoane J, Le H V, Massagué J. Myc suppression of the p21Cip1 Cdk inhibitor influences the outcome of the p53 response to DNA damage. Nature. 2002;419(6908):729-734
40. Coller HA, Grandori C, Tamayo P. et al. Expression analysis with oligonucleotide microarrays reveals that MYC regulates genes involved in growth, cell cycle, signaling, and adhesion. PNAS. 2000;97(7):3260-3265
41. Chapman N, Webster G, Gillespie P. et al. A novel form of the RelA nuclear factor κB subunit is induced by and forms a complex with the proto-oncogene c-Myc. Biochem J. 2002;366:459-469
42. Gordan J D, Bertout J A, Hu C J. et al. HIF-2α promotes hypoxic cell proliferation by enhancing c-Myc transcriptional activity. Cancer cell. 2007;11(4):335-347
43. Van Riggelen J, Yetil A, Felsher DW. MYC as a regulator of ribosome biogenesis and protein synthesis. Nat Rev Cancer. 2010;10(4):301-309
44. Mateyak MK, Obaya AJ, Adachi S. et al. Phenotypes of c-Myc-deficient rat fibroblasts isolated by targeted homologous recombination. Cell Growth Differ. 1997;8(10):1039-1048
45. Orian A, van Steensel B, Delrow J. et al. Genomic binding by the Drosophila Myc, Max, Mad/Mnt transcription factor network. Genes Dev. 2003;17(9):1101-1014
46. Mao DY, Watson JD, Yan PS. et al. Analysis of Myc bound loci identified by CpG island arrays shows that Max is essential for Myc-dependent repression. Curr Biol. 2003;13(10):882-886
47. Grandori C, Gomez-Roman N, Felton-Edkins ZA. et al. c-Myc binds to human ribosomal DNA and stimulates transcription of rRNA genes by RNA polymerase I. Nature Cell Biol. 2005;7(3):311-318
48. Li Z, Hann SR. Nucleophosmin is essential for c-Myc nucleolar localization and c-Myc-mediated rDNA transcription. Oncogene. 2013;32(15):1988-1994
49. Schlosser I, Hölzel M, Mürnseer M. et al. A role for c-Myc in the regulation of ribosomal RNA processing. Nucleic Acids Res. 2003;31(21):6148-6156
50. Grewal SS, Li L, Orian A. et al. Myc-dependent regulation of ribosomal RNA synthesis during Drosophila development. Nat Cell Biol. 2005;7(3):295-302
51. Pourdehnad M, Truitt M L, Siddiqi I N. et al. Myc and mTOR converge on a common node in protein synthesis control that confers synthetic lethality in Myc-driven cancers. PNAS. 2013;110(29):11988-11993
52. Nie Z, Hu G, Wei G. et al. c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell. 2012;151(1):68-79
53. Betschinger J, Mechtler K, Knoblich JA. Asymmetric segregation of the tumor suppressor brat regulates self-renewal in Drosophila neural stem cells. Cell. 2006;124(6):1241-1253
54. Schwamborn JC, Berezikov E, Knoblich JA. The TRIM-NHL protein TRIM32 activates microRNAs and prevents self-renewal in mouse neural progenitors. Cell. 2009;136(5):913-925
55. Wumesh KC, Satpathy AT, Rapaport AS. et al. L-Myc expression by dendritic cells is required for optimal T-cell priming. Nature. 2014
56. Meyer N, Penn LZ. Reflecting on 25 years with MYC. Nat Rev Cancer. 2008(8):976-990
57. Larsson LG. Henriksson MA. The Yin and Yang functions of the Myc oncoprotein in cancer development and as targets for therapy. Exp Cell Res. 2010;316(8):1429-1437
58. Cotter TG. Apoptosis and cancer: the genesis of a research field. Nat Rev Cancer. 2009;9(7):501-507
59. Inoue S, Hao Z, Elia AJ. et al. Mule/Huwe1/Arf-BP1 suppresses Ras-driven tumorigenesis by preventing c-Myc/Miz1-mediated down-regulation of p21 and p15. Genes Dev. 2013;27(10):1101-1114
60. Herkert B, Eilers M. Transcriptional repression the dark side of Myc. Genes Cancer. 2010;1(6):580-586
61. Eischen CM, Woo D, Roussel MF. et al. Apoptosis triggered by Myc-induced suppression of Bcl-X(L) or Bcl-2 is bypassed during lymphomagenesis. Mol Cell Biol. 2001;21(15):5063-5070
62. Martinou JC, Green DR. Breaking the mitochondrial barrier. Nat Rev Mol Cell Biol. 2001;2(1):63-67
63. Hoffman B, Liebermann DA. Apoptotic signaling by c-MYC. Oncogene. 2008;27(50):6462-6472
64. O'Donnell KA, Wentzel EA, Zeller KI. et al. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005;435(7043):839-843
65. Ma L, Young J, Prabhala H. et al. miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat Cell Biol. 2010;12(3):247-256
66. Chang TC, Yu D, Lee Y S. et al. Widespread microRNA repression by Myc contributes to tumorigenesis. Nat Genet. 2007;40(1):43-50
67. Gao P, Tchernyshyov I, Chang TC. et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. 2009;458(7239):762-765
68. Dews M, Homayouni A, Yu D. et al. Augmentation of tumor angiogenesis by a Myc-activated microRNA cluster. Nat Genet. 2006;38(9):1060-1065
69. Bui TV, Mendell JT. Myc Maestro of MicroRNAs. Genes & cancer. 2010;1(6):568-575
70. Wang X, Zhao X, Gao P. et al. c-Myc modulates microRNA processing via the transcriptional regulation of Drosha. Sci Rep. 2013;3:1-7
71. Davis BN, Hilyard AC, Lagna G. et al. SMAD proteins control DROSHA-mediated microRNA maturation. Nature. 2008;454(7200):56-61
72. Davis BN, Hilyard AC, Nguyen PH. et al. Smad proteins bind a conserved RNA sequence to promote microRNA maturation by Drosha. Mol Cell. 2010;39(3):373-384
73. Suzuki HI, Yamagata K, Sugimoto K. et al. Modulation of microRNA processing by p53. Nature. 2009;460(7254):529-533
74. Zhang X, Wan G, Berger FG. et al. The ATM kinase induces microRNA biogenesis in the DNA damage response. Mol Cell. 2011;41(4):371-383
75. Mao G, Lee S, Ortega J. et al. Modulation of microRNA processing by mismatch repair protein MutLα. Cell Res. 2012;22(6):973-985
76. Kawai S, Amano A. BRCA1 regulates microRNA biogenesis via the DROSHA microprocessor complex. J Cell Biol. 2012;197(2):201-208
77. Neil M, Keith IE. Targeting a c-MYC G-quadruplex DNA with a fragment library. Chem Commun. 2014;50(14):1704-1707
78. Halder K, Mathur V, Chugh D. et al. Quadruplex-duplex competition in the nuclease hypersensitive element of human c-myc promoter: C to T mutation in C-rich strand enhances duplex association. Biochem Biophys Res Commun. 2005;327:49-56
79. Li W, Miyoshi D, Nakano S. et al. Structural competition involving G-Quadruplex DNA and its complement. Biochemistry. 2003;42:11736-11744
80. Risitano A, Fox KR. Stability of intramolecular DNA quadruplexes: comparison with DNA duplexes. Biochemistry. 2003;42(21):6507-6513
81. Franceschin M. G-Quadruplex DNA structures and organic chemistry: more than one connection. Eur J Org Chem. 2009;2009(14):2225-2238
82. Steele NL, Plumb J A, Vidal L. et al. A phase I pharmacokinetic and pharmacodynamic study of the histone deacetylase inhibitor belinostat in patients with advanced solid tumors. Clin Cancer Res. 2008;14(3):804-810
83. Brooks TA, Hurley LH. Targeting MYC expression through G-quadruplexes. Genes & cancer. 2010;1(6):641-649
84. Kerwin SM, Chen G, Kern JT. et al. Perylene diimide G-Quadruplex DNA binding selectivity is mediated by ligand aggregation bioorg. Med Chem Lett. 2002;12(3):447-450
85. Li Q, Xiang JF, Zhang H. et al. Searching drug-like anti-cancer compound (s) based on G-quadruplex ligands. Curr Pharm Des. 2012;18(14):1973-1983
86. Pivetta C, Lucatello L, Krapcho AP. et al. Perylene side chains modulate G-quadruplex conformation in biologically relevant DNA sequences. Bioorg Med Chem. 2008;16(20):9331-9339
87. Sun D, Guo K, Rusche JJ. et al. Facilitation of a structural transition in the polypurine/polypyrimidine tract within the proximal promoter region of the human VEGF gene by the presence of potassium and G-quadruplex-interactive agents. Nucleic Acids Res. 2005;33(18):6070-6080
88. Kato GJ, Barrett J, Villa-Garcia M. et al. An amino-terminal c-myc domain required for neoplastic transformation activates transcription. Mol Cell Biol. 1990;10(11):5914-5920
89. Seenisamy J, Rezler EM, Powell TJ. et al. The dynamic character of the G-quadruplex element in the c-MYC promoter and modification by TMPyP4. J Am Chem Soc. 2004;126(28):8702-8709
90. González V, Hurley L H. The c-MYC NHE III1: function and regulation. Annu Rev Pharmacool Toxicol. 2010;50:111-129
91. Andrew EJ, Merchan S, Lawless C. et al. Pentose phosphate pathway function affects tolerance to the G-Quadruplex binder TMPyP4. PloS one. 2013;8(6):e66242
92. Verdun RE, Karlseder J. Replication and protection of telomeres. Nature. 2007;447(7147):924-931
93. Ou TM, Lu YJ, Zhang C. et al. Stabilization of G-quadruplex DNA and down-regulation of oncogene c-myc by quindoline derivatives. J Med Chem. 2007;50(7):1465-1474
94. Ou TM, Lin J, Lu YJ. et al. Inhibition of cell proliferation by quindoline derivative (SYUIQ-05) through its preferential interaction with c-myc promoter G-quadruplex. J Med Chem. 2011;54(16):5671-5679
95. Efferth T, Fu Y, Zu Y. et al. Molecular target-guided tumor therapy with natural products derived from traditional Chinese medicine. Curr Med Chem. 2007;14(19):2024-2032
96. Lee HM, Chan DSH, Yang F. et al. Identification of natural product Fonsecin B as a stabilizing ligand of c-myc G-quadruplex DNA by high-throughput virtual screening. Chem. Commun. 2010;46(26):4680-4682
97. Shin-ya K, Wierzba K, Matsuo K. et al. Telomestatin, a novel telomerase inhibitor from Streptomyces anulatus. J Am Chem Soc. 2001;123(6):1262-1263
98. Liu Y, Zheng B, Xu X. et al. Probing the binding affinity of small-molecule natural products to the G-quadruplex in C-myc oncogene by electrospray ionization mass spectrometry. Rapid Comm Mass Spectrom. 2010;24(20):3072-3075
99. Maiti M, Kumar GS. Protoberberine alkaloids: physicochemical and nucleic acid binding properties. Top Heterocycle Chem. 2007;10:155-209
100. Ji X, Sun H, Zhou H. et al. The interaction of telomeric DNA and c-Myc22 G-quadruplex with 11 natural alkaloids. Nucleic Acid Therapeutics. 2012;22(2):127-136
101. Zhang WJ, Ou TM, Lu YJ. et al. 9-Substituted berberine derivatives as G-quadruplex stabilizing ligands in telomeric DNA. Bioorg Med Chem. 2007;15(16):5493-501
102. Ma Y, Ou TM, Hou JQ. et al. 9-N-Substituted berberine derivatives: stabilization of G-quadruplex DNA and down-regulation of oncogene c-myc. Bioorg Med Chem. 2008;16(16):7582-7591
103. Vy Thi Le T, Han S, Chae J. et al. G-Quadruplex binding ligands: from naturally occurring to rationally designed molecules. Curr Pharm Des. 2012;18(14):1948-1972
104. Ou T, Lu Y, Tan J. et al. G-Quadruplexes: targets in anticancer drug design. ChemMedChem. 2008;3(5):690-713
105. Monchaud D, Teulade-Fichou M P. A hitchhiker's guide to G-quadruplex ligands. Org Biomol Chem. 2008;6(4):627-636
106. Ho YP, Au-Yeung SCF, To KKW. Platinum-based anticancer agents: innovative design strategies and biological perspectives. Med Res Rev. 2003;23(5):633-655
107. Kostova I. Platinum complexes as anticancer agents. Recent Pat Antican Drug Discov. 2006;1(1):1-22
108. Georgiades SN, Abd Karim NH, Suntharalingam K. et al. Interaction of metal complexes with G-quadruplex DNA. Angew Chem Int Ed. 2010;49(24):4020-4034
109. Zhang J, Zhang F, Li H. et al. Recent progress and future potential for metal complexes as anticancer drugs targeting G-quadruplex DNA. Curr Med Chem. 2012;19(18):2957-2975
110. Wang J, Lu K, Xuan S. et al. Pt (II)-Dip complex stabilizes parallel c-myc G-Quadruplexes. Chem Commun. 2013;49:4758-4760
111. Wu P, Ma DL, Leung CH, Yan SC. et al. Stabilization of G-Quadruplex DNA with Platinum(II) schiff base complexes: luminescent probe and down-regulation of c-myc oncogene expression. Chem Eur J. 2009;15(47):13008-13021
112. Maita S, Chaudhury NK, Chowdhury S. Hoechst 33258 binds to G-quadruplex in the promoter region of human c-myc. Biochem Biophys Res Commun. 2003;310(2):505-512
113. Dash J, Shirude PS, Hsu STD. et al. Diarylethynyl amides that recognize the parallel conformation of genomic promoter DNA G-Quadruplexes. J Am Chem Soc. 2008;130(47):15950-15956
114. Chen Q, Kuntz ID, Shafer RH. Spectroscopic recognition of guanine dimeric hairpinquadruplexes by a carbocyanine dye. PNAS. 1996;93(7):2635-2639
115. Cheng JY, Lin SH, Chang TC. Vibrational investigation of DODC cation for recognition of guanine dimeric hairpin quadruplex studied by satellite holes. J Phys Chem B. 1998;102(28):5542-5546
116. White EW, Tanious F, Ismail MA. et al. Structure-specific recognition of quadruplex DNA by organic cations: Influence of shape, substituents and charge. Biophys Chem. 2007;126(1):140-153
117. Ma DL, Chan DSH, Fu WC. et al. Discovery of a natural product-like c-myc G-quadruplex DNA groove-binder by molecular docking. PloS one. 2012;7(8):e43278
118. Lemarteleur T, Gomez D, Paterski R. et al. Stabilization of the c-myc gene promoter quadruplex by specific ligands' inhibitors of telomerase. Biochemical and Biophysical Research Communications. 2004;323(3):802-808
119. Zhang S-L, Wu Y-L, Zhang W. G-Quadruplex structures and their interaction diversity with ligands. ChemMedChem. 2014;9:899-911
120. Gabelica V, Baker ES, Teulade-Fichou MP. et al. Stabilization and structure of telomeric and c-myc region intramolecular G-quadruplexes: the role of central cations and small planar ligands. J Am Chem Soc. 2007;129(4):895-904
121. Lüscher B, Vervoorts J. Regulation of gene transcription by the oncoprotein MYC. Gene. 2012;494(2):145-160
122. Amati B, Brooks M W, Levy N. et al. Oncogenic activity of the c-Myc protein requires dimerization with Max. Cell. 1993;72(2):233-245
123. Berg T. Inhibition of transcription factors with small organic molecules. Curr Opin Chem Biol. 2008;12(4):464-471
124. Wang H, Hammoudeh DI, Follis AV. et al. Improved low molecular weight Myc-Max inhibitors. Mol Cancer Ther. 2007;6(9):2399-2408
125. Kiessling A, Sperl B, Hollis A. et al. Selective inhibition of c-Myc/Max dimerization and DNA binding by small molecules. Chem Biol. 2006;13(7):745-751
126. Yap JL, Chauhan J, Jung KY. et al. Small-molecule inhibitors of dimeric transcription factors: Antagonism of protein-protein and protein-DNA interactions. Med Chem Comm. 2012;3(5):541-551
127. Berg T, Cohen SB, Desharnais J. et al. Small-molecule antagonists of Myc/Max dimerization inhibit Myc-induced transformation of chicken embryo fibroblasts. PNAS. 2002;99(6):3830-3835
128. Lu X, Vogt PK, Boger DL. et al. Disruption of the MYC transcriptional function by a small-molecule antagonist of MYC/MAX dimerization. Oncol Rep. 2008;19(3):825-830
129. Yin X, Giap C, Lazo J S. et al. Low molecular weight inhibitors of Myc-Max interaction and function. Oncogene. 2003;22(40):6151-6159
130. Xu Y, Shi J, Yamamoto N. et al. A credit-card library approach for disrupting protein-protein interactions. Bioorg Med Chem. 2006;14(8):2660-2673
131. Kiessling A, Wiesinger R, Sperl B. et al. Selective inhibition of c-Myc/Max dimerization by a pyrazolo [1,5-a] pyrimidine. ChemMedChem. 2007;2(5):627-630
132. Hammoudeh DI, Follis AV, Prochownik EV. et al. Multiple independent binding sites for small-molecule inhibitors on the oncoprotein c-Myc. J Am Chem Soc. 2009;131(21):7390-7401
133. Shi J, Stover JS, Whitby LR. et al. Small molecule inhibitors of Myc/Max dimerization and Myc-induced cell transformation. Bioorg Med Chem Lett. 2009;19(21):6038-6041
134. Follis AV, Hammoudeh DI, Daab AT. et al. Small-molecule perturbation of competing interactions between c-Myc and Max. Bioorg Med Chem Lett. 2009;19(3):807-810
135. Guo J, Parise RA, Joseph E. et al. Efficacy, pharmacokinetics, tisssue distribution, and metabolism of the Myc-Max disruptor, 10058-F4 [Z, E]-5-[4-ethylbenzylidine]-2-thioxothiazolidin-4-one, in mice. Cancer Chemoth Pharm. 2009;63(4):615-625
136. Mo H, Henriksson M. Identification of small molecules that induce apoptosis in a Myc- dependent manner and inhibit Myc-driven transformation. PNAS. 2006;103(16):6344-6349
137. Mustata G, Follis AV, Hammoudeh DI. et al. Discovery of novel myc-max heterodimer disruptors with a three-dimensional pharmacophore model. J Med Chem. 2009;52(5):1247-1250
138. Wang H, Chauhan J, Hu A. et al. Disruption of Myc-Max Heterodimerization with Improved Cell-Penetrating Analogs of the Small Molecule 10074-G5. Oncotarget. 2013;4(6):936-947
139. Follis AV, Hammoudeh DI, Wang H. et al. Structural rationale for the coupled binding and unfolding of the c-Myc oncoprotein by small molecules. Chem Biol. 2008;15(11):1149-1155
140. Mo H, Vita M, Crespin M. et al. Myc overexpression enhances apoptosis induced by small molecules. Cell Cycle. 2006;5(19):2191-2194
Author biography
 Bing-Jia Chen is a postgraduate majoring in pharmacy. She obtained the Bachelor's Degree in Bio-pharmaceutical Engineering in 2012 from Zhejiang University of Technology, China. After that, she entered Prof. Zhang's group of College of Pharmaceutical Science, Zhejiang University of Technology, China, working with small molecules to regulate disease-related gene to explore gene-targeted drugs under the direction of Prof W. Zhang.
Bing-Jia Chen is a postgraduate majoring in pharmacy. She obtained the Bachelor's Degree in Bio-pharmaceutical Engineering in 2012 from Zhejiang University of Technology, China. After that, she entered Prof. Zhang's group of College of Pharmaceutical Science, Zhejiang University of Technology, China, working with small molecules to regulate disease-related gene to explore gene-targeted drugs under the direction of Prof W. Zhang.
 Dr. Yan-Ling Wu is a professor in Molecular Immunology and now heads the Cellular and Molecular Immunology Research Group. She received Master and Doctoral degrees in Applied Life Science in 2003 and in Medicine Science in 2006, respectively, from Tohoku University, Japan. After that, she entered to Professor Minato's group of School of Medicine, Kyoto University, Japan, as a senior researcher working in the field of molecular immunology. Her current researches focus on understanding the molecular mechanisms of gene regulation related to diseases by immune inhibitory receptors. Dr. Wu have given oral presentations in international conferences and published related papers.
Dr. Yan-Ling Wu is a professor in Molecular Immunology and now heads the Cellular and Molecular Immunology Research Group. She received Master and Doctoral degrees in Applied Life Science in 2003 and in Medicine Science in 2006, respectively, from Tohoku University, Japan. After that, she entered to Professor Minato's group of School of Medicine, Kyoto University, Japan, as a senior researcher working in the field of molecular immunology. Her current researches focus on understanding the molecular mechanisms of gene regulation related to diseases by immune inhibitory receptors. Dr. Wu have given oral presentations in international conferences and published related papers.
 Yoshimasa Tanaka received his Ph. D in Hokkaido University Graduate School of Agriculture with a specialization in Enzymology and Biochemistry. After graduation, he continued his research in the field of Immunobiology. Since 2008, he is an associate professor and works in the Center for Innovation in Immunoregulative Immunology and Therapeutics that belongs to the Kyoto University Graduate School of Medicine.
Yoshimasa Tanaka received his Ph. D in Hokkaido University Graduate School of Agriculture with a specialization in Enzymology and Biochemistry. After graduation, he continued his research in the field of Immunobiology. Since 2008, he is an associate professor and works in the Center for Innovation in Immunoregulative Immunology and Therapeutics that belongs to the Kyoto University Graduate School of Medicine.
 Dr. Wen Zhang is a full professor with 25 years of research and teaching experience in Bioorganic Chemistry and Chemical Biology. Dr. Zhang earned his doctorate degree in Bioorganic Chemistry from East China University of Science and Technology, China. Then, he entered Professor Ohrui's Lab of Tohoku University, Japan, working in the field of molecular recognition as a JSPS postdoctoral fellow. After that, he moved to Kyoto University, Japan, to join Professor Sugiyama's Chemical Biology group as a COE and JST research fellow working on biology and chemistry of polyamide-nucleic acids interaction. Now, Dr. Zhang has a special interest in elucidating the gene regulation mechanisms with small organic molecules and the development of gene-targeted drug. His group formed in 2008 and established an extremely fruitful collaboration with Prof. Sugiyama's Group in order to better pursue aspects of gene-targeted drug research. To date, Dr. Zhang has published better papers as the first/corresponding author in excellent Journals including JACS, JASN, ChemBioChem, Chem & Biol, ChemMedChem, Int J Biol Sci, etc.
Dr. Wen Zhang is a full professor with 25 years of research and teaching experience in Bioorganic Chemistry and Chemical Biology. Dr. Zhang earned his doctorate degree in Bioorganic Chemistry from East China University of Science and Technology, China. Then, he entered Professor Ohrui's Lab of Tohoku University, Japan, working in the field of molecular recognition as a JSPS postdoctoral fellow. After that, he moved to Kyoto University, Japan, to join Professor Sugiyama's Chemical Biology group as a COE and JST research fellow working on biology and chemistry of polyamide-nucleic acids interaction. Now, Dr. Zhang has a special interest in elucidating the gene regulation mechanisms with small organic molecules and the development of gene-targeted drug. His group formed in 2008 and established an extremely fruitful collaboration with Prof. Sugiyama's Group in order to better pursue aspects of gene-targeted drug research. To date, Dr. Zhang has published better papers as the first/corresponding author in excellent Journals including JACS, JASN, ChemBioChem, Chem & Biol, ChemMedChem, Int J Biol Sci, etc.
![]() Corresponding authors: Wen Zhang, Department of Biopharmaceutical Science, College of Pharmaceutical Sciences, Zhejiang University of Technology, 18 Chaowang Road, Hangzhou, 310014, PR China; Tel: +86-571-88871507; Fax: +86-571-88871507; e-mail: wzhang63edu.cn. Yan-Ling Wu, Lab of Molecular Immunology, Virus Inspection Department, Zhejiang Provincial Center for Disease Control and Prevention, 630 Xincheng Road, Hangzhou, 310051, PR China; Tel: +86-571-87115282; Fax: +86-571-87115282; e-mail: ylwuzj.cn; Both contributed equally to this work.
Corresponding authors: Wen Zhang, Department of Biopharmaceutical Science, College of Pharmaceutical Sciences, Zhejiang University of Technology, 18 Chaowang Road, Hangzhou, 310014, PR China; Tel: +86-571-88871507; Fax: +86-571-88871507; e-mail: wzhang63edu.cn. Yan-Ling Wu, Lab of Molecular Immunology, Virus Inspection Department, Zhejiang Provincial Center for Disease Control and Prevention, 630 Xincheng Road, Hangzhou, 310051, PR China; Tel: +86-571-87115282; Fax: +86-571-87115282; e-mail: ylwuzj.cn; Both contributed equally to this work.