Impact Factor ISSN: 1449-2288
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2014; 10(10):1193-1202. doi:10.7150/ijbs.11066 This issue Cite
Research Paper
SIRT1 Deacetylates TopBP1 and Modulates Intra-S-Phase Checkpoint and DNA Replication Origin Firing
Rui-Hong Wang1,4*, Tyler J. Lahusen1*, Qiang Chen1, Xiaoling Xu1,4, Lisa M. Miller Jenkins2, Elisabetta Leo3, Haiqing Fu3, Mirit Aladjem3, Yves Pommier3, Ettore Appella2, Chu-Xia Deng1,4 ![]()
1. Genetics of Development and Disease Branch, 10/9N105, National Institute of Diabetes, Digestive and Kidney Diseases, National Institutes of Health, Bethesda, Maryland MD 20892, USA;
2. Laboratory of Cell Biology, National Institutes of Health, Bethesda, Maryland MD 20892, USA;
3. Developmental Therapeutics Branch, National Cancer Institute, National Institutes of Health, Bethesda, Maryland MD 20892, USA;
4. Faculty of Health Sciences, University of Macau, Macau, SAR of People's Republic of China.
* Equal contribution
Received 2014-11-14; Accepted 2014-11-24; Published 2014-11-26
Abstract
SIRT1, the mammalian homolog of yeast Sir2, is a founding member of a family of 7 protein and histone deacetylases that are involved in numerous biological functions. Previous studies revealed that SIRT1 deficiency results in genome instability, which eventually leads to cancer formation, yet the underlying mechanism is unclear. To investigate this, we conducted a proteomics study and found that SIRT1 interacted with many proteins involved in replication fork protection and origin firing. We demonstrated that loss of SIRT1 resulted in increased replication origin firing, asymmetric fork progression, defective intra-S-phase checkpoint, and chromosome damage. Mechanistically, SIRT1 deacetylates and affects the activity of TopBP1, which plays an essential role in DNA replication fork protection and replication origin firing. Our study demonstrated that ectopic over-expression of the deacetylated form of TopBP1 in SIRT1 mutant cells repressed replication origin firing, while the acetylated form of TopBP1 lost this function. Thus, SIRT1 acts upstream of TopBP1 and plays an essential role in maintaining genome stability by modulating DNA replication fork initiation and the intra-S-phase cell cycle checkpoint.
Keywords: SIRT1, TopBP1, Intra-S-phase checkpoint, DNA replication fork, Genetic stability.
Introduction
The progression of cells through the cell cycle is precisely regulated by multiple checkpoints at different transition phases of the cycle [1-5]. Upon DNA damage, the intra-S-phase checkpoint is activated to ensure the accuracy of DNA replication by suppressing late replication origin activation to allow the DNA damage repair machinery to repair DNA [3-5]. Activation of the intra-S-phase checkpoint is largely mediated by upstream sensor kinases ATM (ataxia-mutated) and ATR (ataxia-telangiectasia and rad3-related), which are recruited to DNA double strand break or defective replication forks, respectively, where they phosphorylate downstream effector proteins. For example, the activation of ATM phosphorylates γH2AX and NBS1 (Nijmegen breakage syndrome), which forms a protein complex with MRE11 and the RAD50 (MRN) complex [6-10]. Consistently, both ATM- and NBS1-deficient cells are defective for the intra-S-phase checkpoint [11, 12]. It has been reported that SIRT1 interacts with MRN and deacetylates NBS1, which affects ionizing radiation-induced NBS1 Ser343 phosphorylation [13], yet the potential impact of SIRT1 on the intra-S-phase checkpoint and DNA replication is largely unknown.
SIRT1 is a member of the sirtuin family of type III histone deacetylases and plays a role in many biological processes, including cell death and survival, cell proliferation and differentiation, neurological functioning, metabolism, calorie restriction, aging, and cancer [14-23]. Using animal models carrying constitutive or tissue-specific loss-of-function mutations of SIRT1, we have previously demonstrated that liver-specific disruption of SIRT1 impairs mTorc2/Akt signaling and results in hyperglycemia, oxidative damage, and insulin resistance [20, 24]. Disruption of SIRT1 in the pancreas caused severe pancreas agenesis, characterized by markedly reduced β-cell formation and hyperglycemia [21].
The potential role of SIRT1 in tumor formation is complex and it may act either as a tumor suppressor or tumor promoter, depending on different tissue contexts and the distribution of SIRT1 downstream targets in specific signaling pathways [25, 26]. Our previous study suggested that expression of the SIRT1 gene is positively regulated by the breast cancer associated gene 1 (BRCA1) and over-expression of SIRT1 in BRCA1-deficient cancer cells inhibited their growth both in vitro and in vivo [27]. Furthermore, about 75% of SIRT1 and p53 double heterozygous (Sirt1+/-;p53+/-) mice developed tumors in multiple organs/tissues before 1 year of age [28]. Spectral karyotyping analysis of tumor cells revealed massive changes in chromosomal number and structure. Similar chromosome damage was also observed in Sirt1-/- embryos during embryonic developmental stages. Thus, it is conceivable that SIRT1 deficiency causes genetic instability, which eventually results in tumorigenesis when some permissive alterations occur, such as inactivation of tumor suppressor genes or activation of oncogenes. However, the exact mechanism by which SIRT1 deficiency causes DNA damage is unclear.
In this study, we investigated the role of SIRT1 in maintaining genome integrity, under base-line conditions and under replicative stress induced by two widely used anticancer drugs, the ribonucleotide reductase inhibitor hydroxyurea (HU) and the topoisomerase inhibitor camptothecin (CPT) [29, 30]. Our data indicate that SIRT1 interacts with multiple proteins that are involved in DNA replication origin firing and replication fork progression. By focusing on one of these proteins, TopBP1, we provide evidence that SIRT1 modulates the intra-S-phase checkpoint and DNA replication origin firing through deacetylating TopBP1.
Materials and Methods
Cell lines
293T cells were obtained from ATCC (Manassas, VA) and cultured with Dulbecco's Modified Eagle Medium (DMEM) (Invitrogen) (Grand Island, NY) supplemented with 10% fetal bovine serum (FBS) (Sigma, St. Louis, MO) and 1% L-glutamine (Invitrogen). MEF cells were obtained from the embryos of wild type and Sirt1-/- mice as described [28]. Cell transfections were done with X-tremeGENE9 (Roche Applied Science, Indianapolis, IN).
DNA combing assay
Cells were incubated with or without 5 μM camptothecin (CPT) for 4 hours before they were pulse-labeled sequentially with 50 μM iododeoxyuridine (IdU) and 100 μM chlorodeoxyuridine (CldU) (Sigma) for 20 min each. Cells were then harvested for the DNA combing assay as described previously [31]. Images were scanned with an inverted fluorescence microscope using a 40X objective, and recorded using IPLab software (BD Biosciences, Rockville, MD). Fluorescent signals were measured using the ImageJ program (National Cancer Institute) and converted to base-pair values according to the criteria that 1 μm equals 2 kb and 1 pixel encompasses 340 base pairs. Statistical analysis was performed using Prism 5 software (GraphPad).
Preparation and transduction of lentiviral-delivered short-hairpin RNA (shRNA)
For transduction of lentiviral shRNA, pLKO.1 lentiviral vectors targeting SIRT1 were obtained from Sigma. The lentiviral SIRT1 shRNA clone, TRCN0000018979, targets the nucleotide sequence (5'- AAAGCCTTTCTGAATCTAT-3') of SIRT1 mRNA. Lentiviral particles expressing either scrambled or SIRT1 shRNA were produced in 293T cells after 48 hours with the pCMV-dR8.2 dvpr packaging vector and VSV-G envelope vector obtained from Addgene (Cambridge, MA). 293T cells were then transduced with lentiviral particles in the presence of polybrene (8 µg/ml) (Sigma).
Western blotting and Immunoprecipitation
Western blotting was carried out with a Licor (Lincoln, Nebraska) or using the conventional ECL system with antibodies against CHK1 (Cell signaling), CDC7 (Cell Signaling), SIRT1 (Millipore), TopBP1 (Bethyl Lab), PARP (Santa Cruz), Flag antibody (Sigma, M2) and acetyl-lysine (Millipore and Cell Signaling). Immunoprecipitation was carried out as described previously [32].
DNA synthesis analysis
DNA synthesis analysis was assessed as described [33]. Briefly, cells were cultured in the logarithmic phase and labeled with 10 nCi of 14C-thymidine (NEN) for 24 hours in DMEM. The medium containing 14C was then removed and replaced with normal DMEM culture medium for another 24 hours. After the cells were irradiated and incubated accordingly, they were pulse labeled with 3H-thymidine (NEN) at 2.5 mCi/ml for 15 min. Cells were harvested, washed twice with PBS, and fixed overnight at -20C with 70% ethanol. The samples then were transferred onto Whatman filters and washed with 70% ethanol and 90% methanol sequentially. The dried filters were assayed with a liquid scintillation counter (Beckman, LS6000IC). The resulting ratio of 3H counts per minute to 14C counts per minute, corrected for the counts per minute that were the result of channel crossover, was a measure of DNA synthesis.
BrdU incorporation assay
MEF cells were cultured overnight in 10 mm dishes. The cells were then either directly labeled with BrdU (BD), or treated with 10 mM hydroxyurea (HU) for 4 hours and released into medium containing BrdU, or reverse transfected by LipofectamineTM 2000 (Invitrogen) and then followed with the indicated treatment. The detailed FACS analysis of BrdU-containing samples was performed as described [27].
Protein pull-down and Mass spectrometry analysis
293T cells were transfected with different amounts of Flag-SIRT1 plasmid (0, 1, 5, 10 μg). Forty-eight hours post transfection, these cells were harvested and lysed. The lysates were immunoprecipitated with an anti-Flag antibody (Sigma, M2) overnight at 4° C. The immuno-complex containing agarose beads were thoroughly washed and the immunoprecipitated beads were used for mass spectrometry analysis as described [21].
Acetylation assay
Flag-TopBP1 was transfected into 293 cells. Forty-eight hours post transfection; the cells were treated with either 10 mM Nicotinamide or PBS for 14 hours. The cells were then collected and lysed. Flag-TopBP1 was immunoprecipitated by Flag antibody (Sigma, M2) and separated by SDS-PAGE (Invitrogen). The TopBP1 protein band was then in-gel digested with trypsin (Sigma) overnight at 37° C as described [34]. The peptides were extracted following cleavage and lyophilized. The dried peptides were solubilized in 2% acetonitrile, 0.5% acetic acid, 97.5% water. They were trapped on a trapping column and separated on a 75 µm x 15 cm, 2 µm Acclaim PepMap reverse phase column (Thermo Scientific) using an UltiMate 3000 RSLCnano HPLC (Thermo Scientific). Peptides were separated at a flow rate of 300 nL/min followed by online analysis by tandem mass spectrometry using a Thermo Orbitrap Fusion mass spectrometer. Peptides were eluted into the mass spectrometer using a linear gradient from 96% mobile phase A (0.1% formic acid in water) to 55% mobile phase B (20% water, 80% acetonitrile, 0.08% formic acid) over 30 minutes. Parent full-scan mass spectra were collected in the Orbitrap mass analyzer set to acquire data at 120,000 FWHM resolution; ions were then isolated in the quadrupole mass filter, fragmented within the HCD cell (HCD normalized energy 32%, stepped ± 3%), and the product ions were analyzed in the ion trap. The instrument was operated in top-speed mode in which the parental MS spectra were scheduled for collection at least every 3 s, with the embedded control system maximizing the number of MS/MS spectra collected during this period. Monoisotopic precursor selection and dynamic exclusion, with a 30 s duration and 10 ppm mass tolerance, were enabled. Proteome Discoverer 1.4 (Thermo) was used to search the data against human proteins from the UniProt database using SequestHT. The search was limited to tryptic peptides, with maximally two missed cleavages allowed. Cysteine carbamidomethylation was set as a fixed modification; methionine oxidation and lysine acetylation were allowed as variable modifications. The precursor mass tolerance was 10 ppm, and the fragment mass tolerance was 0.6 Da. The Percolator node was used to score and rank peptide matches using a 1% false discovery rate. The spectra for all modified peptides were manually validated.
Chromosome spread
Chromosome spread in MEFs was performed as described [35, 36]. Briefly, the MEF cells were incubated with 100 ng/ml colcemid for 2 hours. The hypotonic treatment was carried out for 20 min at RT in 0.56% KCl. The cells were then transferred to methanol:acetic acid (3:1) for fixation. The fixation was repeated twice. All the chromosome spreads were stained with Giemsa and chromosome number and structure were examined under a 100X lens with a Leica microscope, equipped with a DP72 Olympus camera and CellSense software.
Results
Loss of SIRT1 impairs intra-S-phase checkpoint
Previously we reported that embryos homozygous for a null mutation of SIRT1 (Sirt1-/-) displayed a higher rate of BrdU incorporation at embryonic day 9.5-14.5 (E10.5-14.5) as compared to control embryos at the same stage, yet the underlying mechanism remains elusive [28]. To investigate this, murine embryonic fibroblast cells (MEFs) from SIRT1 wild type (WT) and mutant (MT) embryos at passage 1 or 2 were utilized for BrdU labeling analysis. Under normal culture conditions, higher percentage of SIRT1-MT MEF cells incorporated BrdU compared with SIRT1-WT cells (Fig. 1A). When these MEF cells were treated with 10 mM hydroxyurea (HU) for 4 hours, and released into medium containing BrdU for 1 hour, SIRT1-MT cells still contained more BrdU positive cells than SIRT1-WT cells (Fig. 1B). Because similar numbers of SIRT1-WT and MT MEF cells were present at passages 1 and 2 (Fig. 1C), the increased BrdU incorporation is unlikely to be due to greater cell proliferation, but rather abnormalities associated with DNA replication.
Loss of SIRT1 impairs intra-S-phase checkpoint. (A, B) Percentage of BrdU incorporation of primary SIRT1-WT and SIRT1-MT MEF cells under regular culture condition (A) and upon HU treatment (B). (C) Number of MEF cells at passage 1-3. (D) Dose response of DNA synthesis 30 min post γ-irradiation, as detected by thymidine incorporation assay. (E) Time course response of DNA synthesis after 10 Gy γ-irradiation. Three pairs of primary MEF cells at passage 1 or 2 were used for this assay. (F) Response of immortalized SIRT1-WT or MT MEFs upon CPT treatment. Cells were exposed to 5 μM CPT for 2 and 4 hours, and DNA synthesis was assessed after 30 minutes of BrdU labeling by FACs analysis. * P<0.05 and ** P<0.01: comparison between SIRT1-MT and SIRT1-WT cells.
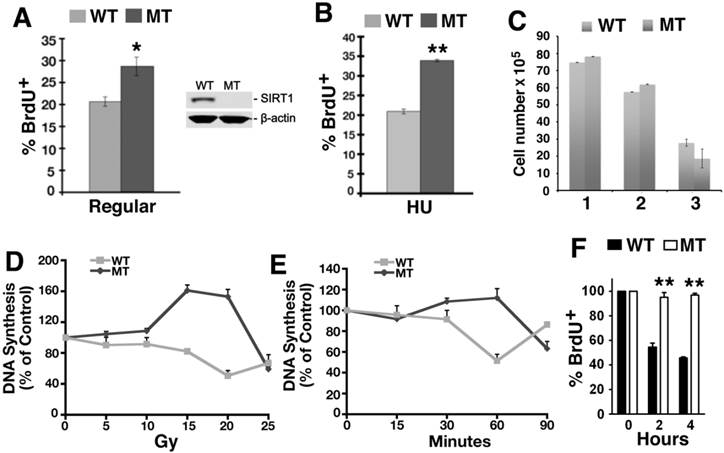
To investigate this, we determined if there is a role for SIRT1 in the intra-S-phase checkpoint by examining primary MEFs in a thymidine incorporation assay to measure DNA replication after γ-radiation. Thirty minutes after γ-radiation exposure ranging from 5 to 20 Gray (Gy), the SIRT1-MT MEFs continued to replicate DNA and even incorporated more thymidine than untreated cells, in contrast to the dose-dependent decline in thymidine incorporation exhibited by the SIRT1-WT MEFs (Fig. 1D). A time course to assess DNA synthesis demonstrated that SIRT1-MT MEF cells failed to reduce DNA replication within the first 60 min post 10 Gy irradiation, while about a 50% reduction was observed in SIRT1-WT cells (Fig. 1E). Also, immortalized SIRT1-MT MEFs were more resistant than WT MEFs to inhibition of DNA replication after exposure to the topoisomerase I inhibitor camptothecin (CPT) [29][37, 38] (Fig. 1F). Altogether, these data indicate that SIRT1 deficiency results in impaired intra-S-phase checkpoint.
SIRT1 regulates DNA replication fork velocity and origin firing
Next we investigated the potential effect of SIRT1 on DNA replication by using a DNA combing assay. SIRT1-WT and MT-MEFs were first treated with CPT for 1 hour and then pulsed with CldU for the last 20 minutes before changing the media to IdU for the second pulse (Supplementary Material: Figure S1). SIRT1-MT MEF cells displayed significantly reduced fork velocity as compared to wild type cells both in normal culture and replicative stress conditions, indicating a slower DNA synthesis rate in the MT cells (Fig. 2A). We also measured inter-origin distances (IODs) in both types of cells. SIRT1-MT cells showed significantly shorter IODs than WT cells, suggesting there is an increase in origin firing in SIRT1-MT cells (Fig. 2B). These data are consistent with the observed higher thymidine or BrdU incorporation in SIRT1-MT cells compared to WT cells.
SIRT1 modulates velocity and inter-origin distance of DNA replication. (A, B) DNA combing analysis of replication fork velocity (A) and inter-origin distance (B) in SIRT1-WT and SIRT1-MT MEFs with or without CPT treatment. (C-F) Scatter plots showing replication speed (Kb/min) of left and right arms of replication forks in SIRT1-WT and SIRT1-MT MEFs without (C, D) and with (E, F) 5 μM CPT for one hour.
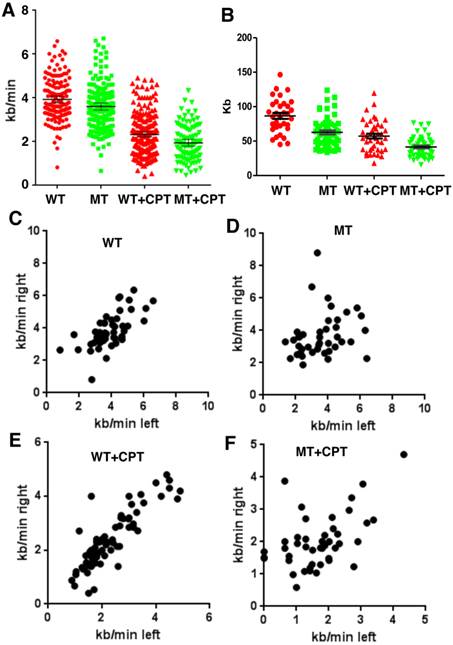
Further analysis of the rate of DNA synthesis indicated that SIRT1-MT cells displayed uncoordinated DNA replication. The speed of DNA synthesis in WT cells is equal for both right and left arms (Fig. 2C), but SIRT1-MT cells manifested asymmetric DNA synthesis rates, i.e. in SIRT1-MT cells, right arm synthesis is faster than left arm synthesis (Fig. 2D). CPT treatment further enhanced asymmetric DNA synthesis in SIRT1-MT cells as compared to WT cells (Fig. 2E, F). We reasoned that this uncoordinated DNA replication could reflect an accumulation of replication-dependent DNA damage due to the absence of SIRT1.
SIRT1 interacts with proteins associated with DNA replication and intra-S-phase checkpoint regulation
SIRT1 is a well-known histone and protein deacetylase [25, 39]. To identify potential SIRT1 target proteins, we transfected increasing amount of SIRT1 with a Flag epitope into 293T cells followed by mass spectrometric analysis of SIRT1 interacting proteins. We identified 16 proteins that are involved in DNA replication and replication fork protection (Table 1). Among the list are 4 previously reported SIRT1 deacetylase targets: MCM10 [40], NBS1 [41], PARP1 [42], and p53 [43, 44]; as well as 12 other interacting proteins that have not been reported previously. This includes components of the replisome, such as CDC7, MCM3, 5, and 7, as well as proteins in the replication protection complex, such as CHK1and TopBP1 [10]. To validate these interactions, 293T cells were transfected with a Flag-SIRT1 construct and then interacting proteins were verified by Flag immunoprecipitation (IP) and Western blot (WB). The results showed that Flag-SIRT1 could interact with both TopBP1 and CDC7 (Fig. 3A, B).
SIRT1 interacting proteins.
| 0 | 1 μg | 5 μg | 10 μg | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Protein | Gene symbol | T | U | T | U | T | U | T | U | Accession | Reference |
| Cell division cycle 7-related protein kinase | CDC7 | 14 | 8 | 15 | 6 | O00311 | CDC7 HUMAN | ||||
| Checkpoint kinase 1 | CHK1 | 6 | 3 | 15 | 4 | O14757 | CHK1 HUMAN | ||||
| Flap endonuclease 1 | FEN1 | 7 | 3 | 29 | 11 | P39748 | FEN1 HUMAN | ||||
| Histone acetyltransferase HBO1 | HBO1 | 8 | 3 | 11 | 7 | O95251 | HBO1 HUMAN | ||||
| DNA replication licensing factor | MCM3 | 8 | 5 | 5 | 3 | 28 | 15 | 43 | 22 | P25205 | MCM3 HUMAN |
| DNA replication licensing factor | MCM5 | 5 | 3 | 5 | 4 | 24 | 13 | 37 | 22 | P33992 | MCM5 HUMAN |
| DNA replication licensing factor | MCM7 | 3 | 3 | 1 | 1 | 19 | 12 | 26 | 16 | P33993 | MCM7 HUMAN |
| Protein MCM10 homolog | MCM10 | 6 | 3 | 5 | 5 | Q7L590 | MCM10 HUMAN | ||||
| Double-strand break repair protein 11A | MRE11A | 3 | 2 | 12 | 8 | P49959 | MRE11 HUMAN | ||||
| Nijmegen breakage syndrome protein 1 | NBS1 | 21 | 11 | 38 | 15 | O60934 | NBN HUMAN | ||||
| Poly [ADP-ribose] polymerase 1 | PARP1 | 14 | 9 | 10 | 7 | 51 | 22 | 65 | 26 | P09874 | PARP1 HUMAN |
| DNA polymerase delta catalytic subunit | POLD1 | 1 | 1 | 7 | 6 | 10 | 5 | P28340 | DPOD1 HUMAN | ||
| ATP-dependent DNA helicase Q4 | RECQL4 | 9 | 6 | 14 | 9 | O94761 | RECQ4 HUMAN | ||||
| Structural maintenance of chromosomes protein 3 | SMC3 | 1 | 1 | 3 | 2 | 5 | 3 | Q9UQE7 | SMC3 HUMAN | ||
| DNA topoisomerase 2-binding protein | TOPBP1 | 35 | 16 | 43 | 22 | Q92547 | TOPBP1HUMAN | ||||
| Cellular tumor antigen p53 | P53 | 1 | 1 | 12 | 3 | 10 | 3 | P04637 | P53 HUMAN |
T: total peptides, U: unique peptides.
FLAG-SIRT1 (0, 1, 5, 10 μg) transfected 293T cells.
SIRT1 interacts with proteins involved in the regulation of DNA replication. (A, B) Interaction between ectopically over-expressed Flag-SIRT1 with TopBP1 (A) and CDC7 (B) as detected by immuno-precipitation. (C-E) Reciprocal immuno-precipitation with endogenous proteins to confirm interaction between SIRT1 versus TopBP1,CHK1 and PARP1 (C); TopBP1 versus SIRT1 (D); and CHK1 versus SIRT1 (E). (F) DNA replication stress enhances interaction of SIRT1 with TopBP1 and CHK1. 293T cells were treated with different doses of HU for 4 hours. Then SIRT1 was immuno-precipitated and TopBP1 and CHK1 were detected by western blot.
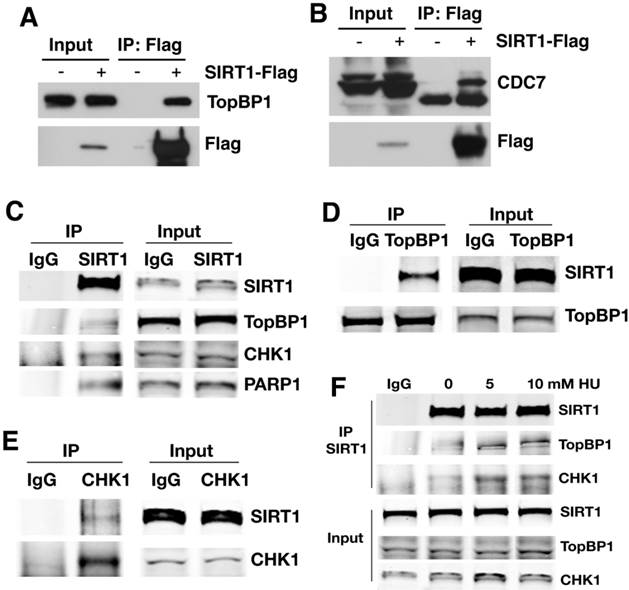
To further validate the interaction, antibodies for endogenous proteins were used for IP and WB. When endogenous SIRT1 was pulled down, TopBP1, CHK1, and PARP1 were all detected (Fig. 3C); at the same time, upon immunoprecipitation of endogenous TopBP1 (Fig. 3D) or CHK1 (Fig. 3E), SIRT1 was detected as well. Thus, SIRT1, TopBP1, and CHK1 were reciprocally interacting with one another. The interaction among SIRT1, TopBP1, and CHK1 is enhanced in the presence of hydroxyurea (HU) (Fig. 3F), suggesting that such interactions are enhanced by replication stress.
SIRT1 deacetylates TopBP1 and regulates its activity
Because SIRT1 is a NAD+ dependent protein deacetylase, we hypothesized that the identified SIRT1-interacting proteins could be deacetylated by SIRT1. To investigate if this is the case, we first tested whether SIRT1 could deacetylate TopBP1, which plays an essential role in the replication checkpoint response after exposure of cells to replication stress [39, 45]. The results demonstrated that TopBP1 is acetylated and its acetylation level decreased upon ectopic over-expression of wild type SIRT1 (Fig. 4A). Knocking down SIRT1 by shRNA with lentivirus or over-expressing a SIRT1 deacetylase-defective mutant (SIRT1-HY) enhanced the acetylation of TopBP1 (Fig. 4A).
SIRT1 deacetylates TopBP1 to affect DNA replication. (A). Acetylation of TopBP1 is regulated by SIRT1 activity. 293T cells were transfected with a Flag-TopBP1 plasmid in combination with GFP, SIRT1 (T1), SIRT1-HY (T1-HY) or shSIRT1 (shT1), respectively. TopBP1 was pulled down with anti-Flag antibody, and then the immuno complexes were western blotted with an antibody against acetyl-lysine. (B) Potential acetylation sites in TopBP1 as revealed by in-silico analysis in combination with mass spectrometry analysis. (C) MS/MS spectra for an acetylated peptide of TopBP1 containing acetylation of Lys475. (D) Effect of SIRT1 and SIRT1-HY on acetylation of TopBP1. (E) Effect of ectopic expression of TopBP1 constructs on DNA replication as determined by BrdU incorporation in primary SIRT1-MT and SIRT1-WT MEF cells after release from HU treatment. ## P<0.01: comparison between SIRT1-WT and MT cells with the same transfection condition (i.e. control vector vs control vector, and 3R vs 3R). * P<0.05 and ** P<0.01: comparison between control with other transfection conditions (i.e. control vs 3R, and control vs 3Q).
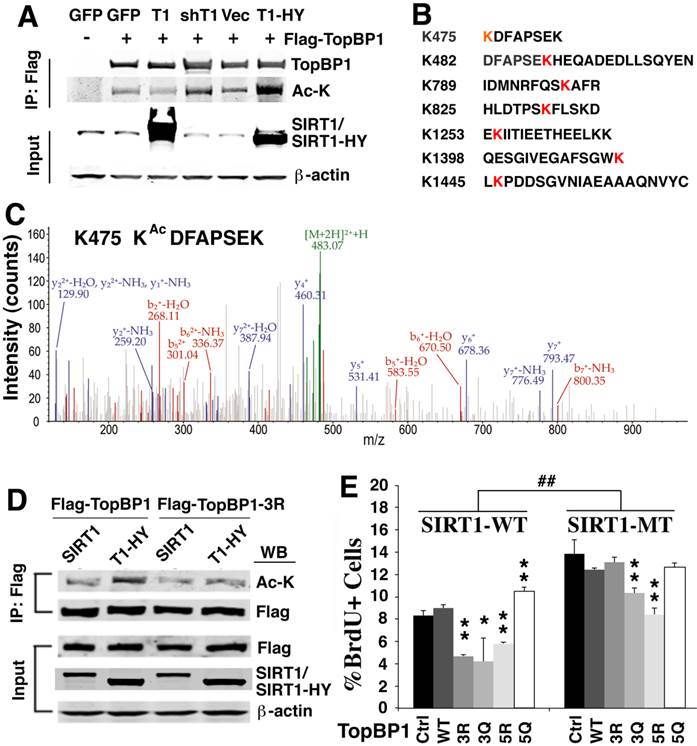
To identify the potential acetylation sites on TopBP1, we performed in-silico analysis in combination with mass spectrometry analysis. We detected 7 lysine sites within TopBP1 that could be acetylated (K475, K482, K789, K825, K1253, K1398 and K1445) (Fig. 4B, C and Supplementary Material: Figure S2). Because of a difficulty to predict which site is important for function of TopBP1, we performed site-directed mutagenesis to generate a TopBP1 expression construct containing three lysine residues mutated to arginine (TopBP1-3R: K475R, K789R and K825R) to mimic the deacetylated form. Comparison of the acetylation state of TopBP1 and TopBP1-3R revealed that these mutations exhibited a reduced acetylation level caused by SIRT1-HY over-expression (Fig. 4D). To further study the effect of these mutations on DNA replication, we transfected TopBP1-WT, TopBP1-3R (K475R, K789R and K825R), TopBP1-3Q (K475Q, K789Q and K825Q), TopBP1-5R (K475R, K789R, K825R, K1253R, and K1398R) and TopBP1-5Q (K475Q, K789Q, K825Q, K1253Q, and K1398Q), into SIRT1-WT and SIRT1-MT MEF cells and used them in a BrdU incorporation assay. The transfected cells were first treated with HU for 4 hours and then BrdU labeling was performed for 1 hour. As seen in Figure 4E, under all transfection conditions, SIRT1-MT cells exhibited a significantly higher percentage of BrdU+ cells than wild type cells, which is consistent with the view that SIRT1 regulates the activity of TopBP1 (Fig. 4E). In wild type MEF cells, over-expression of the deacetylated forms, TopBP1-3R and TopBP1-5R, significantly reduced the percentage of BrdU positive cells, whereas this effect was not observed when TopBP1-5Q was expressed. Instead, TopBP1-5Q significantly increased the fraction of BrdU incorporating cells in SIRT1-WT cells, mimicking the effect observed in SIRT1-MT MEF cells. Of note, there was minimal effect of these constructs in SIRT1-MT cells, except for TopBP1-5R, which significantly repressed BrdU incorporation. This result is consistent with the view that TopBP1-5R represents the highly activated form of TopBP1 that results from SIRT1 deacetylation.
SIRT1 mutant cells display abnormal DNA replication origin firing and chromosomal damage
Based on these observation, we monitored the progression of replication forks under replication stress after HU treatment. As shown in Figure 5A, in SIRT1-WT MEFs, the majority of the IdU+ (red) spots co-localized with the CIdU+ (green) spots, indicating HU successfully stalled the replication forks and DNA synthesis restarted from the stalled forks once the replicative stress was removed (Fig. 5A). In contrast, in HU treated SIRT1-MT cells, fewer IdU+ (red) dots and CIdU+ (green) dots were co-localized with each other, suggesting that in SIRT1-MT cells, HU was not able to efficiently arrest replication forks (Fig. 5B). Furthermore, there were many more CIdU+ (green) dots in SIRT1-MT cells compared with SIRT1-WT cells, which is consistent with our earlier observation that SIRT1-MT cells contain more active replication foci as compared with SIRT1-WT cells.
SIRT1 deficiency affects DNA replication and is critical for genome stability. (A, B) DNA replication analysis by IdU-CIdU labeling in SIRT1-WT (A) and SIRT1-MT (B) MEFs, as indicated on the top of the panel. Briefly, cells were labeled with IdU for 20 min followed by 10 mM HU treatment for 4 hours. After removing HU, cells were labeled with CIdU for 60 min before processing for imaging. Three pairs of MEF cells at passage 2 were utilized. (C) Chromosome spreads showing abnormal chromosome structures (arrows) in mutant but not in WT cells. (D) Percentage of abnormal chromosomes.
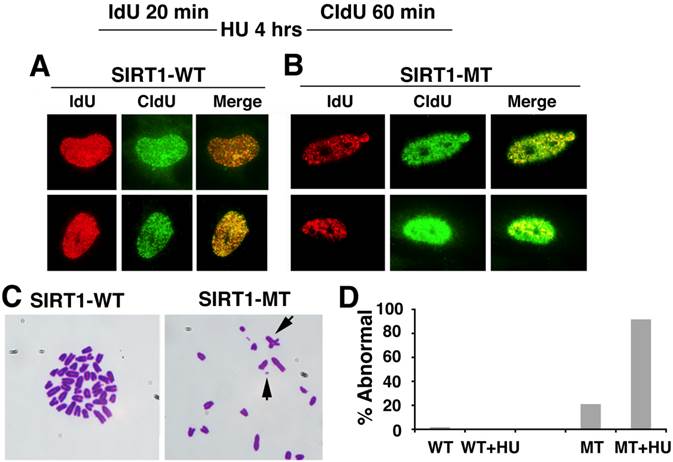
Next, we determined whether abnormal replication fork firing could lead to genomic instability by examining the chromosome spread. SIRT1-MT MEF cells displayed a high number of fragmented chromosomes, minute chromosomes, and chromosomes with quadro-crossover, while such abnormalities were minimal in WT cells (Fig. 5C, D). Altogether, these data uncover an essential role of SIRT1 in maintaining replication fork fidelity and genome stability.
Discussion
An important finding in the present study is that loss of SIRT1 impairs the intra-S-phase checkpoint, as revealed by a failure to arrest DNA replication under HU or CPT treatment conditions. Our analysis indicates that the effects of SIRT1 on the intra-S-phase checkpoint are, at least in part, mediated by TopBP1. Previous studies have demonstrated that TopBP1 plays a critical role in the control of the DNA replication checkpoint by activating ATR in two different ways. First, TopBP1 can directly interact with and activate ATR through its ATR-activation-domain (AAD) [46]. Second, TopBP1 can increase the binding of ATR to single-stranded DNA (SSD) through increased loading of RPA on SSD [39]. In both cases, activation of TopBP1 enhances ATR-dependent phosphorylation events in response to replication stress, leading to activation of the intra-S-phase checkpoint. The impact of SIRT1 on the intra-S-phase checkpoint might be much more profound, as our data also indicate that SIRT1 interacts with CHK1, which is phosphorylated and activated by ATR, MRE11A and NBS1, which are ATM targets [3-5, 47], and p53, which is an upstream regulator of both the ATM and ATR signaling pathways [1, 48, 49]. All these proteins are implicated in the intra-S-phase cell cycle checkpoint [3-5, 11-13]. The interaction of SIRT1 with these proteins and its consequences will be further investigated in our future studies.
We also made an intriguing observation that loss of SIRT1 enhances DNA replication origin firing. This could be related to our finding that SIRT1 interacts with three mini-chromosome maintenance proteins (MCM), i.e. MCM3, 5, and 7 that form a hexameric protein complex together with other three MCM proteins: MCM2, 4, and 6 [50]. Together with origin recognition complex proteins (ORC1-6), cell division cycle 6 (CDC6), and chromatin licensing and DNA replication factor 1 (CDT1), the MCM2-7 replicative helicase forms the pre-replication complex (pre-RC), which is required for DNA replication origin firing [50-52]. Our observation of the interaction of SIRT1 with the MCM2-7 helicase combined with our determination that the loss of SIRT1 caused increased replication origin firing suggests an inhibitory role of SIRT1 in this process. A recent study demonstrated that SIRT1 binds and deacetylates MCM10 and affects DNA replication fork initiation through MCM10 [40]. Thus, SIRT1 could play a role in DNA replication origin firing by regulating both MCM2-7 and MCM10. In addition, SIRT1 also interacts with CDC7 [10], FEN1 [53], HBO1 [54], PARP1 [55], POLD1 [56], RECQL4 [57], and SMC3 [58], which are involved in various aspects of DNA replication, such as activation, progression, and protection of replication forks. SIRT1 might modulate DNA replication through some of these interacting proteins, although details regarding these interactions remain elusive.
In summary, we have shown that SIRT1-MT cells incorporate more BrdU than control cells under both normal and replicative stress conditions. Further analysis revealed that this is primarily due to the fact that the SIRT1-MT cells contain more active DNA replication origins and fail to arrest DNA synthesis even under replicative stress conditions. By analyzing SIRT1 interacting proteins as revealed by mass spectrometry analysis, we demonstrated that SIRT1 plays an essential role in replication fork firing and intra-S-phase checkpoint through deacetylating and regulating TopBP1. This study provides a molecular basis for the underlying mechanism that SIRT1 deficiency results in genetic instability.
Supplementary Material
Figures S1 - S2.
Acknowledgements
We thank the members of the Deng lab for critical review of the manuscript. This research was supported by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases and the National Cancer Institute, National Institutes of Health, USA, and by Faculty of Health Sciences, University of Macau.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Deng CX. BRCA1: cell cycle checkpoint, genetic instability, DNA damage response, and cancer evolution. Nucleic Acids Res. 2006;34:1416-26
2. Boye E, Skjolberg HC, Grallert B. Checkpoint regulation of DNA replication. Methods in molecular biology. 2009;521:55-70
3. Labib K, De Piccoli G. Surviving chromosome replication: the many roles of the S-phase checkpoint pathway. Philosophical transactions of the Royal Society of London. Series B, Biological sciences. 2011;366:3554-61
4. Errico A, Costanzo V. Mechanisms of replication fork protection: a safeguard for genome stability. Critical reviews in biochemistry and molecular biology. 2012;47:222-35
5. Roseaulin LC, Noguchi C, Noguchi E. Proteasome-dependent degradation of replisome components regulates faithful DNA replication. Cell cycle. 2013;12:2564-9
6. Shiloh Y. ATM and ATR: networking cellular responses to DNA damage. Curr Opin Genet Dev. 2001;11:71-7
7. Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. The Journal of biological chemistry. 1998;273:5858-68
8. Luo Y, Deng X, Cheng F, Li Y, Qiu J. SMC1-mediated intra-S-phase arrest facilitates bocavirus DNA replication. Journal of virology. 2013;87:4017-32
9. Aves SJ, Liu Y, Richards TA. Evolutionary diversification of eukaryotic DNA replication machinery. Subcell Biochem. 2012;62:19-35
10. Tsuji T, Lau E, Chiang GG, Jiang W. The role of Dbf4/Drf1-dependent kinase Cdc7 in DNA-damage checkpoint control. Molecular cell. 2008;32:862-9
11. Antoccia A, Kobayashi J, Tauchi H, Matsuura S, Komatsu K. Nijmegen breakage syndrome and functions of the responsible protein, NBS1. Genome Dyn. 2006;1:191-205
12. Williams BR, Mirzoeva OK, Morgan WF, Lin J, Dunnick W, Petrini JH. A murine model of Nijmegen breakage syndrome. Current biology: CB. 2002;12:648-53
13. Yuan Z, Seto E. A functional link between SIRT1 deacetylase and NBS1 in DNA damage response. Cell Cycle. 2007;6:2869-71
14. Lavu S, Boss O, Elliott PJ, Lambert PD. Sirtuins--novel therapeutic targets to treat age-associated diseases. Nat Rev Drug Discov. 2008;7:841-53
15. Guarente L. Sirtuins in aging and disease. Cold Spring Harb Symp Quant Biol. 2007;72:483-8
16. Haigis MC, Sinclair DA. Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol. 2010;5:253-95
17. Herranz D, Serrano M. SIRT1: recent lessons from mouse models. Nat Rev Cancer. 2010;10:819-23
18. Morris BJ. Seven sirtuins for seven deadly diseases of aging. Free radical biology & medicine. 2013;56:133-71
19. Li X, Kazgan N. Mammalian sirtuins and energy metabolism. International journal of biological sciences. 2011;7:575-87
20. Wang RH, Kim HS, Xiao C, Xu X, Gavrilova O, Deng CX. Hepatic Sirt1 deficiency in mice impairs mTorc2/Akt signaling and results in hyperglycemia, oxidative damage, and insulin resistance. The Journal of clinical investigation. 2011;121:4477-90
21. Wang RH, Xu X, Kim HS, Xiao Z, Deng CX. SIRT1 deacetylates FOXA2 and is critical for Pdx1 transcription and beta-cell formation. International journal of biological sciences. 2013;9:934-46
22. Gonfloni S, Iannizzotto V, Maiani E, Bellusci G, Ciccone S, Diederich M. P53 and Sirt1: Routes of metabolism and genome stability. Biochemical pharmacology. 2014
23. Finkel T, Deng CX, Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature. 2009;460:587-91
24. Wang RH, Li C, Deng CX. Liver Steatosis and Increased ChREBP Expression in Mice Carrying a Liver Specific SIRT1 Null Mutation under a Normal Feeding Condition. Int J Biol Sci. 2010;6:682-90
25. Deng CX. SIRT1, is it a tumor promoter or tumor suppressor? Int J Biol Sci. 2009;5:147-52
26. Lin Z, Fang D. The Roles of SIRT1 in Cancer. Genes Cancer. 2013;4:97-104
27. Wang RH, Zheng Y, Kim HS, Xu X, Cao L, Luhasen T, Lee MH, Xiao C, Vassilopoulos A, Chen W, Gardner K, Man YG, Hung MC, Finkel T, Deng CX. Interplay among BRCA1, SIRT1, and Survivin during BRCA1-associated tumorigenesis. Mol Cell. 2008;32:11-20
28. Wang RH, Sengupta K, Li C, Kim HS, Cao L, Xiao C, Kim S, Xu X, Zheng Y, Chilton B, Jia R, Zheng ZM, Appella E, Wang XW, Ried T, Deng CX. Impaired DNA damage response, genome instability, and tumorigenesis in SIRT1 mutant mice. Cancer Cell. 2008;14:312-23
29. Seiler JA, Conti C, Syed A, Aladjem MI, Pommier Y. The Intra-S-Phase Checkpoint Affects both DNA Replication Initiation and Elongation: Single-Cell and -DNA Fiber Analyses. Mol Cell Biol. 2007;27:5806-18
30. Pommier Y, Leo E, Zhang H, Marchand C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chemistry & biology. 2010;17:421-33
31. Conti C, Caburet S, Schurra C, Bensimon A. Molecular combing. Curr Protoc Cytom. 2001 Chapter 8:Unit 8 10
32. Wang RH, Yu H, Deng CX. A requirement for breast-cancer-associated gene 1 (BRCA1) in the spindle checkpoint. Proc Natl Acad Sci U S A. 2004;101:17108-13
33. Xu B, Kim S, Kastan MB. Involvement of Brca1 in S-phase and G(2)-phase checkpoints after ionizing irradiation. Mol Cell Biol. 2001;21:3445-50
34. Shevchenko A, Tomas H, Havlis J, Olsen JV, Mann M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nature protocols. 2006;1:2856-60
35. Deng CX, Xu X. Generation and analysis of Brca1 conditional knockout mice. Methods Mol Biol. 2004;280:185-200
36. Shen SX, Weaver Z, Xu X, Li C, Weinstein M, Chen L, Guan XY, Ried T, Deng CX. A targeted disruption of the murine Brca1 gene causes gamma-irradiation hypersensitivity and genetic instability. Oncogene. 1998;17:3115-24
37. Strumberg D, Pilon AA, Smith M, Hickey R, Malkas L, Pommier Y. Conversion of topoisomerase I cleavage complexes on the leading strand of ribosomal DNA into 5'-phosphorylated DNA double-strand breaks by replication runoff. Mol. Cell. Biol. 2000;20:3977-87
38. Conti C, Sacca B, Herrick J, Lalou C, Pommier Y, Bensimon A. Replication fork velocities at adjacent replication origins are coordinately modified during DNA replication in human cells. Molecular biology of the cell. 2007;18:3059-67
39. Gong Z, Kim JE, Leung CC, Glover JN, Chen J. BACH1/FANCJ acts with TopBP1 and participates early in DNA replication checkpoint control. Molecular cell. 2010;37:438-46
40. Fatoba ST, Tognetti S, Berto M, Leo E, Mulvey CM, Godovac-Zimmermann J, Pommier Y, Okorokov AL. Human SIRT1 regulates DNA binding and stability of the Mcm10 DNA replication factor via deacetylation. Nucleic acids research. 2013;41:4065-79
41. Yuan Z, Zhang X, Sengupta N, Lane WS, Seto E. SIRT1 regulates the function of the Nijmegen breakage syndrome protein. Mol Cell. 2007;27:149-62
42. Rajamohan SB, Pillai VB, Gupta M, Sundaresan NR, Birukov KG, Samant S, Hottiger MO, Gupta MP. SIRT1 promotes cell survival under stress by deacetylation-dependent deactivation of poly(ADP-ribose) polymerase 1. Mol Cell Biol. 2009;29:4116-29
43. Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, Guarente L, Gu W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001;107:137-48
44. Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149-59
45. Unsal-Kacmaz K, Chastain PD, Qu PP, Minoo P, Cordeiro-Stone M, Sancar A, Kaufmann WK. The human Tim/Tipin complex coordinates an Intra-S checkpoint response to UV that slows replication fork displacement. Molecular and cellular biology. 2007;27:3131-42
46. Zhou ZW, Liu C, Li TL, Bruhn C, Krueger A, Min W, Wang ZQ, Carr AM. An essential function for the ATR-activation-domain (AAD) of TopBP1 in mouse development and cellular senescence. PLoS genetics. 2013;9:e1003702
47. Zhang P, Wei Y, Wang L, Debeb BG, Yuan Y, Zhang J, Yuan J, Wang M, Chen D, Sun Y, Woodward WA, Liu Y, Dean DC, Liang H, Hu Y, Ang KK, Hung MC, Chen J, Ma L. ATM-mediated stabilization of ZEB1 promotes DNA damage response and radioresistance through CHK1. Nature cell biology. 2014;16:864-75
48. Chen Y, Poon RY. The multiple checkpoint functions of CHK1 and CHK2 in maintenance of genome stability. Frontiers in bioscience: a journal and virtual library. 2008;13:5016-29
49. Sperka T, Wang J, Rudolph KL. DNA damage checkpoints in stem cells, ageing and cancer. Nature reviews. Molecular cell biology. 2012;13:579-90
50. Simon NE, Schwacha A. The Mcm2-7 Replicative Helicase: A Promising Chemotherapeutic Target. Biomed Res Int. 2014;2014:549719
51. Niida H, Kitagawa M. Regulation of DNA replication licensing. Current drug targets. 2012;13:1588-92
52. Duncker BP, Chesnokov IN, McConkey BJ. The origin recognition complex protein family. Genome biology. 2009;10:214
53. Saharia A, Teasley DC, Duxin JP, Dao B, Chiappinelli KB, Stewart SA. FEN1 ensures telomere stability by facilitating replication fork re-initiation. The Journal of biological chemistry. 2010;285:27057-66
54. Miotto B, Struhl K. HBO1 histone acetylase activity is essential for DNA replication licensing and inhibited by Geminin. Molecular cell. 2010;37:57-66
55. Tempera I, Deng Z, Atanasiu C, Chen CJ, D'Erme M, Lieberman PM. Regulation of Epstein-Barr virus OriP replication by poly(ADP-ribose) polymerase 1. Journal of virology. 2010;84:4988-97
56. Maga G, van Loon B, Crespan E, Villani G, Hubscher U. The block of DNA polymerase delta strand displacement activity by an abasic site can be rescued by the concerted action of DNA polymerase beta and Flap endonuclease 1. The Journal of biological chemistry. 2009;284:14267-75
57. Keller H, Kiosze K, Sachsenweger J, Haumann S, Ohlenschlager O, Nuutinen T, Syvaoja JE, Gorlach M, Grosse F, Pospiech H. The intrinsically disordered amino-terminal region of human RecQL4: multiple DNA-binding domains confer annealing, strand exchange and G4 DNA binding. Nucleic acids research. 2014
58. Song J, Lafont A, Chen J, Wu FM, Shirahige K, Rankin S. Cohesin acetylation promotes sister chromatid cohesion only in association with the replication machinery. The Journal of biological chemistry. 2012;287:34325-36
Author contact
![]() Corresponding author: Tel: (301) 402-7225; Fax: (301) 480-1135 Email: chuxiadniddk.nih.gov and cxdengmo.
Corresponding author: Tel: (301) 402-7225; Fax: (301) 480-1135 Email: chuxiadniddk.nih.gov and cxdengmo.