Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2015; 11(4):404-410. doi:10.7150/ijbs.10273 This issue Cite
Short Research Communication
Identification of Novel Focal Adhesion Kinase Substrates: Role for FAK in NFκB Signaling
Sheila Figel Dwyer, Lingqiu Gao, Irwin H. Gelman ![]()
Department of Cancer Genetics, Roswell Park Cancer Institute, USA.
Received 2014-8-5; Accepted 2014-11-10; Published 2015-2-17
Abstract

Focal adhesion kinase (FAK) is a major signaling molecule which functions downstream of integrins or in conjunction with mitogenic signaling pathways. FAK is overexpressed and/or activated in many types of human tumors, in which it promotes cell adhesion, survival, migration and invasion. In addition to FAK's ability to regulate signaling through its scaffolding activities, FAK encodes an intrinsic kinase activity. Although some FAK substrates have been identified, a more comprehensive analysis of substrates is lacking. In this study, we use a protein microarray to screen the human proteome for FAK substrates. We confirm that several of the proteins identified are bona fide in vitro FAK substrates, including several factors which are known to regulate the NFκB pathway. Finally, we identify a role for FAK's kinase activity in both canonical and non-canonical NFκB signaling. Our screen therefore represents the first high throughput screen for FAK substrates and provides the basis for future in-depth analysis of the role of FAK's kinase activity in the processes of tumorigenesis.
Keywords: FAK, substrates, phosphotyrosine, kinase, NFκB, IKKα.
Introduction
Focal adhesion kinase (FAK) is a major signaling molecule involved in the control of cell proliferation, survival, and motility. FAK functions in focal adhesions to facilitate intracellular signaling by transmembrane integrins during adhesion. In the simplest mechanism of activation, FAK is recruited to clustered integrins following cell attachment to extracellular matrix components [1]. Following recruitment, autophosphorylation on tyrosine 397 both activates FAK's kinase activity and creates a high-affinity SH2 binding site for cellular Src (c-Src) [2]. Binding to FAK in turn activates Src's kinase activity; consequently, Src phosphorylates additional tyrosines in FAK which stimulates maximal FAK kinase activity [3, 4].
Integrin clustering and Src recruitment serve to activate FAK's kinase activity, however little is known about the downstream effects of this. A few proteins, including paxillin [5], Shc [6], and p130Cas [7], have been shown to be phosphorylated by FAK in vitro. However, increased levels of phospho-paxillin and phospho-p130Cas are detected in FAK-null mouse embryonic fibroblasts [8], suggesting an overlap with other tyrosine kinases such as Src and the FAK family member, Pyk2 [9].
In an effort to identify FAK-specific substrates, we employed a novel in vitro approach in which a microarray with recombinant human proteins was subjected to an in situ kinase reaction with purified FAK. Among the many candidates identified, four proteins were confirmed in individual assays as novel in vitro FAK substrates. FAK was found to phosphorylate multiple molecules within the NFκB signaling pathway, including the major pathway component CHUK/IKKα, which was found to be phosphorylated by FAK both in vitro and in vivo. Further, we found that FAK kinase activity promotes both canonical and non-canonical NFκB signaling, partially through IKKα phosphorylation.
Materials and methods
Reagents
The following reagents/kits were used: ProtoArray Human Protein Microarray v5.0 Kinase Substrate Identification Complete Kit (Invitrogen), full-length active Src (SignalChem), GST-FAK (Invitrogen), baculovirus-encoded Pyk2 (gift of Vita Golubovskaya, Roswell Park Cancer Institute [RPCI]), purified vitronectin (Advanced BioMatrix), purified GST-PTPN5[a.a.17-565] (ProteinTech), FAK inhibitor PF-573,228 (Sigma), TNFα (Abcam), Recombinant Light (Enzo Life Sciences). The following antibodies (Ab) were used: HA (Abm), MAb-4G10 (anti-phosphotyrosine [PTyr]) and FAK (Millipore), poY397-FAK (BD Transduction), GAPDH (Santa Cruz), p100/p52 (Cell Signaling).
Cell culture
Spodoptera frugiperda Sf-9 cells (Xinjiang Wang, RPCI), HEK293T and MCF7 (ATCC, Manassas, VA), FAK+/+ and FAK-/- mouse embryonic fibroblasts (MEF) from a p53-/- background (T. Yamamoto and S. Aizawa, University of Tokyo), HeLa (A. Gudkov, RPCI) and MCF-7 (A. Bakin, RPCI). Sf9 were maintained in Grace's supplemented media with 10% fetal bovine serum, 1% Pluronic F-68 and penicillin/streptomycin; cultures were grown in glass flasks shaking at 165 rpm at 27°C. All other cells lines were maintained in DMEM plus 10% FBS and penicillin/streptomycin.
Plasmids
Mouse FAK cDNA was cloned into pFASTBac-HTb (Invitrogen) between BamHI and KpnI sites to form pFastFAK. GST-PDCD6 was generated by cloning the full-length open reading frame as an EcoRI fragment into pGEX-5X-2 (GE Healthcare). GST-paxillin (chicken; a.a.1-151) in pGEX-5X was described previously [5]; pCR-HA-IKKα (Addgene #15469), pLUdR-puro-YFP-FAK-Y180A/M183A (FAK180) (gift of M. Schaller, West Virginia University), NFκB-Luciferase (NK-Luc; gift of E. Kurenova, RPCI); pCMV-Renilla (gift of A. Bakin, RPCI), pCMV4-p100 (Addgene #23287). Y->F mutants in untagged IKKα (gift of E. Kurenova) were generated by site-directed mutagenesis using the primers msIKKa-Y187F: TGTGGGAACATTGCAGTTTTTGGCCCCAGAGCTCTTT, msIKKa-Y198F: CTTTGAAAATAAGCCGTTCACAGCCACTGTGGATTATTGG, and msIKKa-Y500F: GAGAGATATAGTGAGCAGATGACTTTTGGGATATCTTCAG.
Baculovirus purification of FAK
FAK bacmid DNA was generated by transformation of pFastFAK into DH10BacTM (Invitrogen). Recombinant FAK bacmid DNA was then transfected into Sf9 using Cellfectin (Invitrogen), and these cells were used to generate high-titer baculovirus. Sf9 were infected with high-titer virus for 48 h, pelleted by centrifugation at 1K for 10 min at 4°Cm resuspended in PBS, then lysed in 2X lysis buffer (20 mM Tris-Cl, pH 7.4, 500 mM NaCl, 10 mM imidazole, 2% Triton-X100, 2 mM phenylmethanesulfonyl fluoride, Roche cOmplete® protease inhibitor cocktail) on ice for 30 min. Cleared lysate was added 1:1 to equilibration buffer (20 mM Tris-Cl, pH 8.0, 500 mM NaCl, 10 mM imidazole) prior to binding to Ni2+ columns (Thermo Scientific) for 30 min at 4°C. Columns were washed thrice with 20 mM Tris-Cl, pH 8.0, 500 mM NaCl, 50 mM imidazole, and FAK was eluted in buffer containing 500 mM imidazole. Imidazole was removed by dialysis and His-FAK was concentrated using Amicon® Ultra (Millipore) spin columns- 125 kDa cutoff. FAK was stored at -20°C in 50 mM Tris-Cl, pH 7.4, 150 mM NaCl, 0.5 mM EDTA, and 1 mM DTT.
Protein microarray
The ProtoArray® Human Protein Microarray v5.0 was probed according to the manufacturer's instructions: slides blocked in 1% BSA in PBS for 1h at room temperature were incubated in Invitrogen Kinase Buffer containing 10µCi γ-33P-ATP (Perkin-Elmer) with or without 100 nM His-FAK for 1h at 30°C. Slides were washed twice each with 0.5% SDS and ultrapure water, dried and exposed to film. Images were scanned, saved as 16-bit TIF files and uploaded to the Protoarray Prospector Imager (Invitrogen), and using Protoarray Prospector Analyzer (Invitrogen) software, positive hits (>1.5-fold over control; coefficient of variation <0.5, Z-factor >0.5) were identified by comparing signals within duplicate spots, measurement of signal-to-noise ratio, and comparison of signals to negative controls.
Protein purification
GST, GST-paxillin and GST-PDCD6 were purified from BL21-pLysS as described previously [10].
In vitro kinase (IVK) assays
For FAK-IPs, lysates made from HEK293T cells expressing HA-FAK were incubated with αHA at 4°C overnight, then with 20 μl Protein A/G resin (Santa Cruz) for 1h. Beads were washed twice with RIPA buffer and twice with kinase buffer (20 mM Hepes pH 7.2, 5 mM MnCl2, and 5 mM MgCl2), then resuspended in 20 μl kinase buffer. 5 μl beads were reacted with substrate protein plus 10 μCi 32P-ATP at 30°C for 30 min, after which samples were subjected to SDS-PAGE. Dried gels were autoradiographed. Src, FAK, and Pyk2 IVK buffer contained 50 mM Tris-Cl pH 7.4, 10 mM MgCl2, 1 mM DTT, and 0.1 mM Na3VO4. For non-radioactive IVK assays, HA-IKKα-beads were incubated with GST-FAK and 10 mM ATP at 30°C for 30 min, and then analyzed for phosphate incorporation by PTyr immunoblot (IB).
Luciferase assays
Cells were transfected with NK-Luc and CMV-Renilla, and stimulated with TNFα where indicated. Luciferase activity was measured using the Promega Dual-Glo Luciferase Assay System according to manufacturer instructions. In FAK-/- cells stably expressing the pLA-NFκB-Luciferase reporter +/- FAK180, equal numbers of cells were assayed for luciferase activity using the Promega Luciferase Assay System according to manufacturer instructions.
Results and Discussion
Identification of FAK substrates using protein microarray
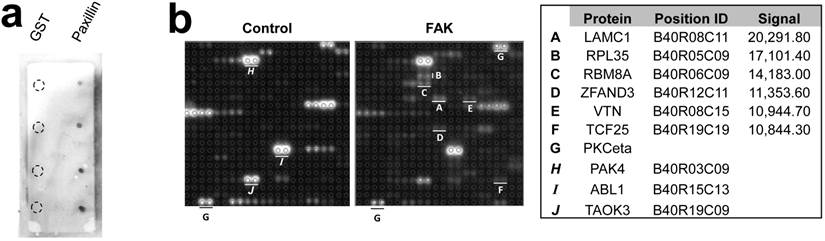
The Protoarray Human Protein Microarray, containing 9,483 purified human proteins printed in duplicate on a nitrocellulose-coated slide, was used to identify FAK substrates by incubating with either purified FAK plus 33P-ATP, or with 33P-ATP alone. The known FAK substrates, p130Cas and Shc, were absent from the array, and two paxillin sequences listed were either incomplete or incorrectly annotated. As a control, we showed that FAK could phosphorylate GST-paxillin, but not GST, in custom microarrays (Fig. 1a, right). Normalizing to the positive control, PKCeta, which binds 33P-ATP directly (Fig. 1b), 43 candidate FAK substrates were identified (Fig. 2a). Due to manufacturing inconsistencies (Invitrogen communication), the data from blocks 1-20 were excluded from analysis, thus limiting the analysis to 6,322 proteins.
Microarray screen for FAK substrates. a. Left: Autoradiograph image of control and FAK protein microarray slides. Right: Control microarray containing 1, 5, 10 and 25 ng/dot of GST or GST-Paxillin. b. Spot-pair signals from control and FAK slides block #40 (upper panels), with identified proteins, microarray positions and relative signal intensities (average pixels/pair) below. A-F, proteins with increased phosphorylation by FAK relative to control; G, positive control protein, PKCeta; H-J, proteins with equal or decreased phosphorylation levels in the FAK slide compared to control.
Candidate identified FAK substrates. a. Identified FAK substrates categorized according to function groups. b. Percentage of FAK substrates based on functional grouping.
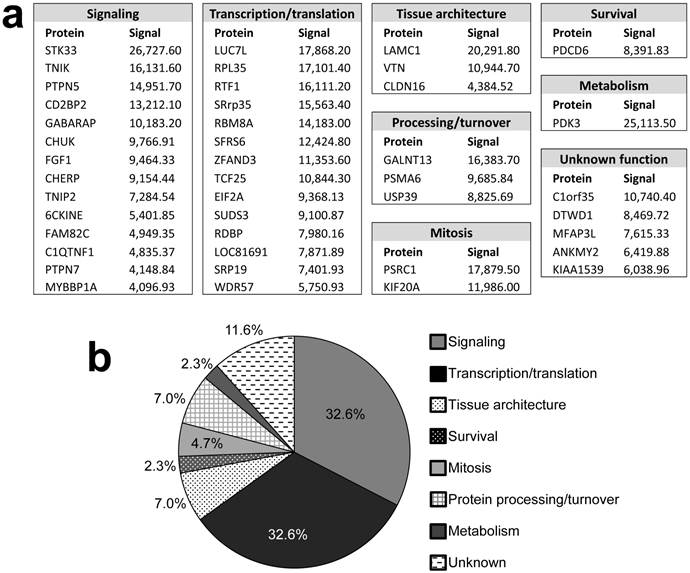
Pubmed and Gene (http://ncbi.nlm.nih.gov) searches were used to assign functions to the identified proteins (Fig. 2). Substrates were grouped into 7 categories: Signaling (32.6%), transcription/translation (32.6%), tissue architecture (7%), protein processing/turnover (7%), mitosis (4.7%), survival (2.3%), and metabolism (2.3%). The functions of 5 proteins (11.6%) could not be assigned. The identification of many signaling proteins was expected based on FAK's role in many pathways, yet it is surprising that few of these candidates are involved in the signaling pathways known to typically cooperate with or be regulated by FAK, such as PI3K [11] and ERK/MAPK [12]. Interestingly, FAK also showed a substrate preference for molecules involved in transcription and translation, processes in which FAK has not yet been implicated. If verified, phosphorylation of such factors may suggest entirely new FAK functions.
Vitronectin, PDCD6 and PTPN5 are novel FAK substrates
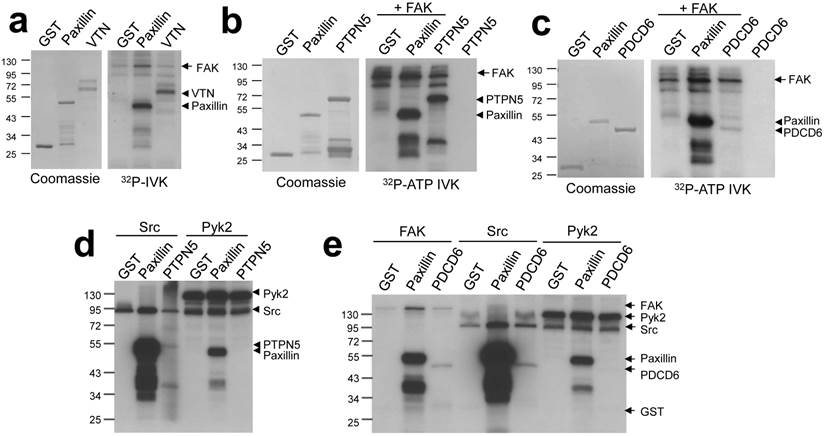
In order to validate screen hits, we performed individual kinase assays with several candidate proteins known to function in pathways relevant to FAK biology. The secreted glycoprotein vitronectin (VTN) is an extracellular matrix component which plays a role in maintaining tissue architecture by mediating interactions between cells and the extracellular matrix. Two other proteins identified, Programmed cell death 6 (PDCD6) and Protein tyrosine phosphatase non-receptor type 5 (PTPN5), modulate cell survival through the regulation of apoptosis. PTPN5 was shown to positively regulate apoptosis through its dephosphorylation of Bak [13]; PDCD6 has been shown to promote apoptosis in ovarian cancer cells [14]. Vitronectin, PDCD6 and PTPN5 were phosphorylated in FAK-IVK reactions (Fig. 3a - c). Src phosphorylated PDCD6 and PTPN5, although to lesser extents than FAK, and indeed, when normalized to Src or FAK autophosphorylation levels, PTPN5 seems to be a better substrate of FAK. In contrast, Pyk2 was unable to phosphorylate either protein in vitro (Fig. 3d - e) although it could phosphorylate paxillin. This suggests a degree of specificity for FAK within the FAK/Pyk2 kinase family. The relevance of vitronectin phosphorylation is less clear because these proteins are not found in the same cellular compartment (FAK: plasma membrane; vitronectin: extracellular matrix). However, at least two phosphotyrosine sites on vitronectin have been identified in cell/tissue lysates (http://www.phosphosite.org). It is unclear why FAK phosphorylates vitronectin, PDCD6 or PTPN5 with different efficiencies, yet we have noted that once normalized for equal protein concentrations, FAK phosphorylates many full-length proteins, such as paxillin, to a lesser extent than truncated versions of the substrates (e.g.- paxillin a.a. 1-151 used here). This suggests that in addition to motif preference, FAK phosphorylation might be affected by substrate conformation, specifically, by domains that affect substrate availability.
FAK phosphorylates vitronectin, PTPN5, and PDCD6. FAK-IVK reactions with VTN (a), GST-PTPN5 (b), GST-PDCD6 (c and e), using GST or GST-paxillin as negative or positive controls, respectively. Left panels (a-c): stained proteins; right panels: autoradiographs of FAK-IVK. d. IVK with purified Src or Pyk2 using GST, GST-paxillin, or GST-PTPN5 as substrates. e. IVK reactions with purified FAK, Src or Pyk2 using GST, GST-paxillin or GST-PDCD6 as substrates.
FAK phosphorylation of CHUK/IKKα: role in NFκB signaling
We identified four potential substrates involved in NFκB signaling: MYB binding protein 1a (MYBBP1A/p160) [15], conserved helix-loop-helix ubiquitous kinase (CHUK/IKKα) [16], TNFAIP3-interacting protein 2 (TNIP2/Abin-2) [17], and TRAF2- and NCK-interacting kinase (TNIK) [18]. Previous studies have demonstrated a role for FAK kinase activity in NFκB signaling [19], including the ability of FAK to cooperate with NFκB to promote survival signaling [20]. Indeed, IPs containing HA-IKKα expressed in HEK293T cells could be tyrosine phosphorylated in FAK IVK assays (Fig. 4a), and as well, HA-IKKα could be tyrosine phosphorylated when co-expressed in HEK293T cells with a constitutively-active version of FAK, FAK180 [23] (Fig. 4b).
FAK phosphorylates CHUK/IKKα regulates NFκB signaling. a. FAK-IVK reaction using HA-IKKα IPs, identified by anti-PTyr IB. b. FAK180 phosphorylation of HA-IKKα in HEK293T cells. c. IKKα functions in both canonical (left side) and non-canonical (right side) NFκB pathways. d. Relative luciferase units (RLU) of NK-Luciferase reporter (normalized to CMV-Renilla) in FAK +/+ vs. FAK-/- MEF. e. RLU (NK-Luc vs CMV-Renilla) in FAK-/- cells transiently overexpressing FAK180. f. RLU (NK-Luc vs CMV-Renilla) in MCF7 cells stimulated with TNFα (20ng/mL for 16 h). IB for active and total FAK (upper panel). g. FAK induces cleavage of p100 to p52 in HEK293T cells. h. p100 to p52 cleavage in HeLa cells ectopically expressing p100, after stimulation with 100 or 200 ng/mL Light for 16 h in the presence of either DMSO or 10 μM PF-228. i. Decreased FAK180-induced pTyr of IKKα187F and IKKα198F relative to WT or IKKα500F in HEK293T cells ("-", vector). j. Decreased RLU (NK-Luc vs CMV-Renilla) in MCF7 transiently expressing IKKα187F and IKKα198F relative to WT or IKKα500F (V, vector). Error bars for all experiments: SEM, three independent experiments. *, p<0.05; **, p<0.005 as determined by unpaired t test.
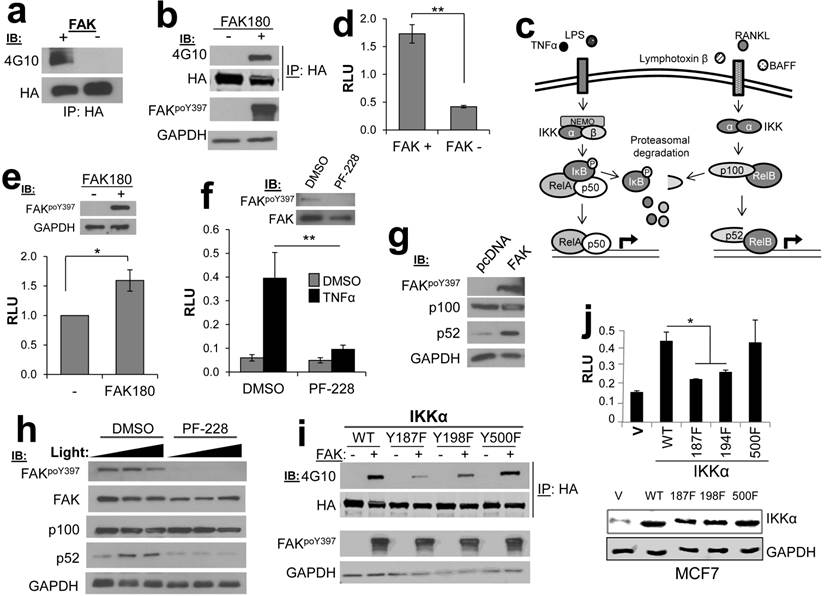
IKKα facilitates both canonical and non-canonical NFκB signaling (Fig. 4c), in the former case, by functioning within an IκB Kinase complex to directly phosphorylate the negative regulator, IκB [21], and in the latter case, by facilitating the cleavage p100 to produce a p52/RelB complex that controls the formation of tumor initiating cell subpopulations in breast cancer [22]. To dissect the role of FAK in NFκB signaling, we employed an NFκB-dependent luciferase reporter system. The level of NFκB activity was significantly decreased in FAK-null MEF (FAK-/-) as compared to MEF containing WT levels of FAK (FAK+/+) (Fig. 4d). Furthermore, re-expression of FAK180 could partially rescue NFκB activity in FAK-/- MEF (Fig. 4e). In the human breast cancer cell line, MCF7, treatment with the FAK kinase inhibitor PF-573,228 (PF-228) specifically inhibited induction of canonical NFκB signaling by TNFα (Fig. 4f). FAK also plays a regulatory role in the non-canonical NFκB pathway because its transient overexpression in HEK293T cells induced the cleavage of p100 to p52 (Fig. 4g). Consistent with this role, treatment of HeLa cells with PF-228 prevented p100 cleavage mediated by the non-canonical pathway inducer, Lymphotoxin β Receptor Ligand Light (heretofore called “Light”) [24] (Fig. 4h). Lastly, in order to address how FAK phosphorylation of IKKα facilitates NFκB signaling, the major FAK phosphorylation sites on IKKα were identified as Y187 and Y198, based on anti-PTyr IB analysis of HEK293T cells co-transfected with IKKα WT or individual site Y-->F variants plus FAK180 (Fig. 4i). In comparison to WT HA-IKKα or HA-IKKα500F (whose phosphorylation by FAK was not altered in Fig. 4i), HA-IKKα187F or HA-IKKα198F failed to induce NFκB signaling when transiently overexpressed in MCF-7 cells (Fig. 4j). Taken together, these data show that both canonical and non-canonical NFκB signaling are controlled by FAK, likely through the specific phosphorylation of IKKα.
Conclusion
The in situ FAK kinase assay using protein microarrays is a novel and viable means to identify FAK substrates. Using this technology we have confirmed phosphorylation of four novel substrates of FAK, demonstrated specificity among FAK family members, and implicated FAK in regulation of two major signaling pathways. This screen represents a major starting point for an in-depth understanding of FAK's kinase activity and its role in tumorigenesis.
Acknowledgements
We thank Vita Golubovskaya, Xinjiang Wang, Andrei Gudkov, Andrei Bakin, Michael Schaller, Elena Kurenova for sharing reagents and cells. This work was supported by grants (I.H.G.) CA94108, CA116430 (National Institutes of Health/National Cancer Institute), PC074228, PC101210 (Department of Defense), and in part, through National Cancer Institute Comprehensive Cancer funds (P30-CA016056).
Conflict of Interest
The authors have no conflicts of interest to report.
References
1. Hao H, Naomoto Y, Bao X, Watanabe N, Sakurama K, Noma K. et al. Focal adhesion kinase as potential target for cancer therapy (Review). Oncol Rep. 2009;22(5):973-9
2. Schaller MD, Hildebrand JD, Shannon JD, Fox JW, Vines RR, Parsons JT. Autophosphorylation of the focal adhesion kinase, pp125FAK, directs SH2-dependent binding of pp60src. Mol Cell Biol. 1994;14(3):1680-8
3. Calalb MB, Polte TR, Hanks SK. Tyrosine phosphorylation of focal adhesion kinase at sites in the catalytic domain regulates kinase activity: a role for Src family kinases. Mol Cell Biol. 1995;15(2):954-63
4. Owen JD, Ruest PJ, Fry DW, Hanks SK. Induced focal adhesion kinase (FAK) expression in FAK-null cells enhances cell spreading and migration requiring both auto- and activation loop phosphorylation sites and inhibits adhesion-dependent tyrosine phosphorylation of Pyk2. Mol Cell Biol. 1999;19(7):4806-18
5. Bellis SL, Miller JT, Turner CE. Characterization of tyrosine phosphorylation of paxillin in vitro by focal adhesion kinase. J Biol Chem. 1995;270(29):17437-41
6. Schlaepfer DD, Hunter T. Integrin signalling and tyrosine phosphorylation: just the FAKs? Trends Cell Biol. 1998;8(4):151-7
7. Tachibana K, Urano T, Fujita H, Ohashi Y, Kamiguchi K, Iwata S. et al. Tyrosine phosphorylation of Crk-associated substrates by focal adhesion kinase. A putative mechanism for the integrin-mediated tyrosine phosphorylation of Crk-associated substrates. J Biol Chem. 1997;272(46):29083-90
8. Ilic D, Furuta Y, Kanazawa S, Takeda N, Sobue K, Nakatsuji N. et al. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature. 1995;377(6549):539-44
9. Sieg DJ, Ilic D, Jones KC, Damsky CH, Hunter T, Schlaepfer DD. Pyk2 and Src-family protein-tyrosine kinases compensate for the loss of FAK in fibronectin-stimulated signaling events but Pyk2 does not fully function to enhance FAK- cell migration. EMBO J. 1998;17(20):5933-47
10. Lin X, Tombler E, Nelson PJ, Ross M, Gelman IH. A novel src- and ras-suppressed protein kinase C substrate associated with cytoskeletal architecture. J Biol Chem. 1996;271(45):28430-8
11. Sonoda Y, Watanabe S, Matsumoto Y, Aizu-Yokota E, Kasahara T. FAK is the upstream signal protein of the phosphatidylinositol 3-kinase-Akt survival pathway in hydrogen peroxide-induced apoptosis of a human glioblastoma cell line. J Biol Chem. 1999;274(15):10566-70
12. Schlaepfer DD, Jones KC, Hunter T. Multiple Grb2-mediated integrin-stimulated signaling pathways to ERK2/mitogen-activated protein kinase: summation of both c-Src- and focal adhesion kinase-initiated tyrosine phosphorylation events. Molec Cell Biol. 1998;18(5):2571-85
13. Fox JL, Ismail F, Azad A, Ternette N, Leverrier S, Edelmann MJ. et al. Tyrosine dephosphorylation is required for Bak activation in apoptosis. EMBO J. 2010;29(22):3853-68
14. Huang Y, Jin H, Liu Y, Zhou J, Ding J, Cheng KW. et al. FSH inhibits ovarian cancer cell apoptosis by up-regulating survivin and down-regulating PDCD6 and DR5. Endocr Relat Cancer. 2011;18(1):13-26
15. Owen HR, Elser M, Cheung E, Gersbach M, Kraus WL, Hottiger MO. MYBBP1a is a novel repressor of NF-kappaB. J Mol Biol. 2007;366(3):725-36
16. DiDonato JA, Hayakawa M, Rothwarf DM, Zandi E, Karin M. A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature. 1997;388(6642):548-54
17. Leotoing L, Chereau F, Baron S, Hube F, Valencia HJ, Bordereaux D. et al. A20-binding inhibitor of nuclear factor-kappaB (NF-kappaB)-2 (ABIN-2) is an activator of inhibitor of NF-kappaB (IkappaB) kinase alpha (IKKalpha)-mediated NF-kappaB transcriptional activity. J Biol Chem. 2011;286(37):32277-88
18. Shkoda A, Town JA, Griese J, Romio M, Sarioglu H, Knofel T. et al. The germinal center kinase TNIK is required for canonical NF-kappaB and JNK signaling in B-cells by the EBV oncoprotein LMP1 and the CD40 receptor. PLoS Biol. 2012;10(8):e1001376
19. Funakoshi-Tago M, Sonoda Y, Tanaka S, Hashimoto K, Tago K, Tominaga S. et al. Tumor necrosis factor-induced nuclear factor kappaB activation is impaired in focal adhesion kinase-deficient fibroblasts. J Biol Chem. 2003;278(31):29359-65
20. Zhang HM, Keledjian KM, Rao JN, Zou T, Liu L, Marasa BS. et al. Induced focal adhesion kinase expression suppresses apoptosis by activating NF-kappaB signaling in intestinal epithelial cells. Am J Physiol Cell Physiol. 2006;290(5):C1310-20
21. Karin M, Yamamoto Y, Wang QM. The IKK NF-kappa B system: a treasure trove for drug development. Nat Rev Drug Discov. 2004;3(1):17-26
22. Kendellen MF, Bradford JW, Lawrence CL, Clark KS, Baldwin AS. Canonical and non-canonical NF-kappaB signaling promotes breast cancer tumor-initiating cells. Oncogene. 2014;33(10):1297-305
23. Lietha D, Cai X, Ceccarelli DF, Li Y, Schaller MD, Eck MJ. Structural basis for the autoinhibition of focal adhesion kinase. Cell. 2007;129(6):1177-87
24. Mordmuller B, Krappmann D, Esen M, Wegener E, Scheidereit C. Lymphotoxin and lipopolysaccharide induce NF-kappaB-p52 generation by a co-translational mechanism. EMBO Rep. 2003;4(1):82-7
Author contact
![]() Corresponding author: Irwin H. Gelman, Department of Cancer Genetics, Roswell Park Cancer Institute, Elm and Carlton Streets Buffalo, NY 14263, 716-845-7681, Irwin.gelmanorg.
Corresponding author: Irwin H. Gelman, Department of Cancer Genetics, Roswell Park Cancer Institute, Elm and Carlton Streets Buffalo, NY 14263, 716-845-7681, Irwin.gelmanorg.