Impact Factor ISSN: 1449-2288
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2015; 11(4):411-422. doi:10.7150/ijbs.10745 This issue Cite
Research Paper
MEK Inhibitor PD-0325901 Overcomes Resistance to CK2 Inhibitor CX-4945 and Exhibits Anti-Tumor Activity in Head and Neck Cancer
Yansong Bian1#, Jiawei Han1,2#, Vishnu Kannabiran1,3#, Suresh Mohan1,3, Hui Cheng1, Jay Friedman1, Luo Zhang2, Carter VanWaes1 ![]() , Zhong Chen1
, Zhong Chen1 ![]()
1. Tumor Biology Section, Head and Neck Surgery Branch, National Institute on Deafness and Other Communication Disorders, NIH, Bethesda, MD, USA;
2. Department of Otolaryngology Head and Neck Surgery, National Key Discipline, Key Laboratory of Otolaryngology Head and Neck Surgery of the Ministry of Education, Beijing Tongren Hospital, Capital Medical University, Beijing, China;
3. NIH Clinical Research Training Program-NIH Medical Research Scholars Program, Bethesda, MD, USA.
# Contribute equally as the first author.
Received 2014-10-7; Accepted 2015-1-17; Published 2015-2-23
Abstract

The serine-threonine kinase CK2 exhibits genomic alterations and aberrant overexpression in human head and neck squamous cell carcinomas (HNSCC). Here, we investigated the effects of CK2 inhibitor CX-4945 in human HNSCC cell lines and xenograft models. The IC50's of CX-4945 for 9 UM-SCC cell lines measured by MTT assay ranged from 3.4-11.9 μM. CX-4945 induced cell cycle arrest and cell death measured by DNA flow cytometry, and inhibited prosurvival mediators phospho-AKT and p-S6 in UM-SCC1 and UM-SCC46 cells. CX-4945 decreased NF-κB and Bcl-XL reporter gene activities in both cell lines, but upregulated proapoptotic TP53 and p21 reporter activities, and induced phospho-ERK, AP-1, and IL-8 activity in UM-SCC1 cells. CX-4945 exhibited modest anti-tumor activity in UM-SCC1 xenografts. Tumor immunostaining revealed significant inhibition of PI3K-Akt-mTOR pathway and increased apoptosis marker TUNEL, but also induced p-ERK, c-JUN, JUNB, FOSL1 and proliferation (Ki67) markers, as a possible resistance mechanism. To overcome the drug resistance, we tested MEK inhibitor PD-0325901 (PD-901), which inhibited ERK-AP-1 activation alone and in combination with CX-4945. PD-901 alone displayed significant anti-tumor effects in vivo, and the combination of PD-901 and CX-4945 slightly enhanced anti-tumor activity when compared with PD-901 alone. Immunostaining of tumor specimens after treatment revealed inhibition of p-AKT S129 and p-AKT T308 by CX-4945, and inhibition of p-ERK T202/204 and AP-1 family member FOSL-1 by PD-901. Our study reveals a drug resistance mechanism mediated by the MEK-ERK-AP-1 pathway in HNSCC. MEK inhibitor PD-0325901 is active in HNSCC resistant to CX-4945, meriting further clinical investigation.
Keywords: CX-4945, CK2 inhibitor, PD-0325901, MEK inhibitor, head and neck cancer.
Introduction
Protein Kinase CK2 (formerly Casein Kinase II) is a highly conserved and ubiquitous protein serine/threonine kinase located in both the cytoplasm and the nucleus. The CK2 holoenzyme consists of two catalytic subunits (α or α') and two regulatory subunits (β), existing as α2β2, αα'β2, α'2β2 tetrameric complexes, but CK2α/α' subunits are also found associated with nuclear chromatin 1. CK2 plays an important role in control of cell growth, proliferation and survival 1-4. Overexpression of CK2 has been observed in many types of solid tumors including prostate, breast, kidney, and lung, as well as in hematopoietic malignancies. In head and neck squamous cell carcinomas (HNSCC), which arise from the upper aerodigestive tract, increased CK2 activity is associated with the malignant transformation of normal mucosa and with poorer clinical outcomes 5, 6.
We previously identified upregulated CK2 as an upstream kinase activating NF-κB and repressing TP53 mediated signaling pathways in promoting the malignant phenotype of HNSCC 7, 8. The Nuclear Factor-κB signal pathway is constitutively activated and promotes cell survival, radiation and chemotherapy resistance of HNSCC 9, 10. CK2 is a key activator of IKK and NF-κB in HNSCC, by promoting IKKβ mediated phosphorylation of IκBα, in addition to the direct phosphorylation of IκBα and the RELA subunit 8, 11. In addition, TP53 is the most commonly mutated or inactivated tumor suppressor gene in HNSCC, as recently confirmed by The Cancer Genomic Atlas (TCGA) project12. CK2 was shown to repress TP53 mRNA and protein expression7. Together, our data suggested that targeting CK2 could modulate key signaling molecules, oncogenes, and tumor suppressor genes involved in critical regulatory pathways that promote tumor growth and malignant phenotypes.
CK2 has been proposed as a target for cancer therapy. Our laboratory showed that down regulation of CK2 in HNSCC models by anti-CK2 oligonucleotides via sub-50-nm nanocapsules exhibited antitumor effects as well as sensitization to cisplatin 7. Recently, a small molecule CK2 inhibitor has been developed, designated as CX-4945 [5‐(3‐chlorophenylamino)benzo[c][2,6]naphthyridine‐8‐carboxylic acid]13. It is an orally administered inhibitor selective for CK2 that effectively reduces CK2 enzymatic activity. It exhibited anti-proliferative activity against a broad range of cancer cell types, including breast, lung, and prostate cancer cells, in which CK2 overexpression was also observed14, 15. In addition, it has been shown that CX-4945 can attenuate PI3K-AKT signaling by inhibiting AKT serine129 phosphorylation, and modulate p21 stability in solid tumor models 13. Recently, TCGA data reveals that the PI3K-AKT pathway is one of the most commonly altered oncogenic signaling pathway in human cancers, including SCC of lung and head and neck, which are consistent with our previous studies 12, 16-20. In HNSCC, more than 60% of tumors have at least one PI3K-AKT-mTOR pathway alteration including gene mutations or amplifications of its components, but genomic alterations in CK2 have not been studied 21. Furthermore, our prior studies suggest that oncogenic PI3K-AKT activation promotes NF-κB activation and represses TP53 expression and function in HNSCC22. Together, these findings lead us to examine if and how CX-4945 modulates PI3K-AKT-NF-κB and TP53-p21 mediated signaling and malignant phenotypes in HNSCC.
Although we previously observed anti-tumor activity following inhibition of CK2 and modulation of these pathways by RNAi 7, this inhibition was incomplete, suggesting the hypothesis that other pathways may contribute to HNSCC survival and resistance. The MAPK (mitogen-activated protein kinase) signaling pathway is another critical pathway, that transduces signals from growth factors, cytokines and environmental stresses, and regulates diverse cellular activities, including proliferation, survival, differentiation, motility, and angiogenesis 23, 24. Activation of MAPK leads to distinct intracellular responses via a series of phosphorylation events and protein interaction, such as activation of AP-1 family members through phosphorylation, and translocation to the nucleus 16, 25-27. We and others have previously shown that EGFR and other growth factor receptors mediate activation of MAPK, ERK and AP-1 pathways that transduce important signals to promote tumor survival and drug resistance in HNSCC 16, 17, 20, 28. Currently, inhibitors of MEK have been developed that block MAPK/ERK/AP-1 signaling. PD-0325901 (PD-901) is one of the selective inhibitors of both MEK isoforms (MEK1/MEK2) that can prevent the activation of ERK as well as downstream AP-1 activity 29. Based on our previous investigations in HNSCC models, we hypothesize that MEK-ERK-AP-1 activation could contribute to resistance to CK2 inhibition, and combination of inhibitors targeting CK2 and MAPK could be the more effective strategy to blocking multiple pathways and enhance the anti-tumor activity.
In the current study, we examined the therapeutic potential of CK2 inhibitor CX‐4945. While CX-4945 inhibited AKT and NF-κB prosurvival signaling, as well as promoting TP53 expression and cell death, we found it also enhanced activation of the ERK-AP-1 signal pathway, target gene expression, and compensatory proliferation in vivo. Thus, we combined CK2 inhibitor CX-4945 and MEK inhibitor PD-0325901 (PD-901) in the human HNSCC models in vitro and in vivo. MEK inhibitor PD-901 overcame drug resistance to CK2 inhibitor CX-4945, and exhibited anti-tumor efficacy in vivo to inhibit tumor growth and related signaling pathways.
Materials and Methods
Cell lines. A panel of nine HNSCC cell lines from the University of Michigan squamous cell carcinoma (UM-SCC) series was obtained from Dr. T.E. Carey (University of Michigan; Ann Arbor, MI, Additional file 1: Supplemental Table 1). The origin of these UM-SCC cell lines were authenticated by genotyping with 9 markers as listed and described previously30, 31, and preserved in frozen stocks that were used within 3 months of culture. The UM-SCC cells were cultured in MEM and 10% FCS.
Reagents. CX-4945 was provided by Cylene Pharmaceuticals, and PD-0325901 was obtained from SelleckChem. The CX-4945 was resuspended to a stock solution of 40 mM in DMSO for in vitro experiments and 25 mM sodium bisphosphate buffer to deliver 25 or 75 mg/kg for in vivo experiments. PD-0325901 was dissolved to a stock solution of 10 mM in DMSO for in vitro experiments and in 0.5% HPMC (hydroxylpropyl methylcellulose) and 0.2% Tween 80 in water at 1.5 mg/kg for in vivo experiments. Both drugs were delivered through oral gavage.
MTT cell proliferation assay. Cell lines were plated in 96-well plates and treated with CX-4945 at varying concentrations. Cell proliferation was measured using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) Cell Proliferation Kit (Roche Diagnostics). The optical density was measured under a wavelength of 570 nm by a μQuant microplate reader (Bio-Tek Instruments). Each sample was assayed in 6 replicates and data are presented as the mean plus standard deviation (SD).
Analysis of cell cycle and apoptosis by flow cytometry. UM-SCC 1 and UM-SCC 46 cells were plated in 6-well plates. At 24 hours, cells were treated with increasing concentrations of CX-4945 for another 24 hours, and then harvested, counted, and labeled using the Cycletest Plus DNA Reagent Kit (BD Biosciences) following standard protocol. Cells were measured by a FACS Canto machine and the data analyzed using Flow-Jo analysis software (Tree Star).
Reporter gene assay. UM-SCC 1 and UM-SCC 46 cells were cultured at approximately 70% confluency in a 24 well plate, and co-transfected with 0.15 μg of the reporter gene of interest and 0.02 μg RSV-LacZ reporter plasmid using Lipofectamine 2000 (Invitrogen) for 5 hours. The media was then replaced with MEM containing 10% FCS and varying concentrations of CX-4945, and cell lysates were harvested at 24, 48, and 72 hours. Reporter gene activity was assayed by the chemiluminescent detection protocol from the Dual-Light System Kit (Tropix/Applied Biosystems), using the Wallac VICTOR2 1420Multilabel Counter (PerkinElmer). Each sample was assayed in triplicate and data presented as the mean + standard deviation (SD).
In vivo HNSCC xenograft mouse model. All animal experiments were carried out under protocols approved by the Animal Care and Use Committee of the NIDCD. Four- to six-week-old female BALB/c severe combined immunodeficient (SCID) were obtained from Frederick Cancer Research and Development Center (National Cancer Institute) and housed in a specific pathogen-free animal facility. Mice were injected subcutaneously (s.c.) in right flanks with 5×106 UM-SCC 1 cells. Once the mice developed palpable tumors, they were randomized and treated. For the animal experiment treated with the single agent, CX-4945 was dosed at 25 and 75 mg/kg, by oral gavage twice daily (BID), for 5 weeks. Vehicle for CX-4945 delivery was 25 mM sodium bisphosphate buffer. Tumors were measured three days per week, harvested at day 13 (early time point) and day 33 (late time point), and measurements analyzed using GraphPad Prism. For the combination experiment, CX4945 was dosed at 75mg/kg by oral gavage twice daily (BID), and PD-901 was formulated as mentioned above for 1.5 mg/kg delivery by oral gavage once daily (QD), for 21 days. The tumor tissues were harvested at day 22 after treatment.
Western Blot. Western blots were performed as previously described 11,16, 30, using whole cell lysates and antibodies as described in Additional file 1: Supplemental Methods.
Immunohistochemical analyses of tumors. Immunohistochemical staining (IHC) and quantifications of slides with frozen tumors harvested from treated and vehicle control mice at early time points (13 days) and late time point (33 day) after treatment, for the single agent experiment. The tumors were harvested at day 14 after treatment for the combination experiment. The IHC was performed and quantified as described in Additional file 1: Supplemental Methods.
Results
Alterations in CK2 subunit expression and gene copy number in HNSCC tissues from The Cancer Genome Atlas (TCGA)
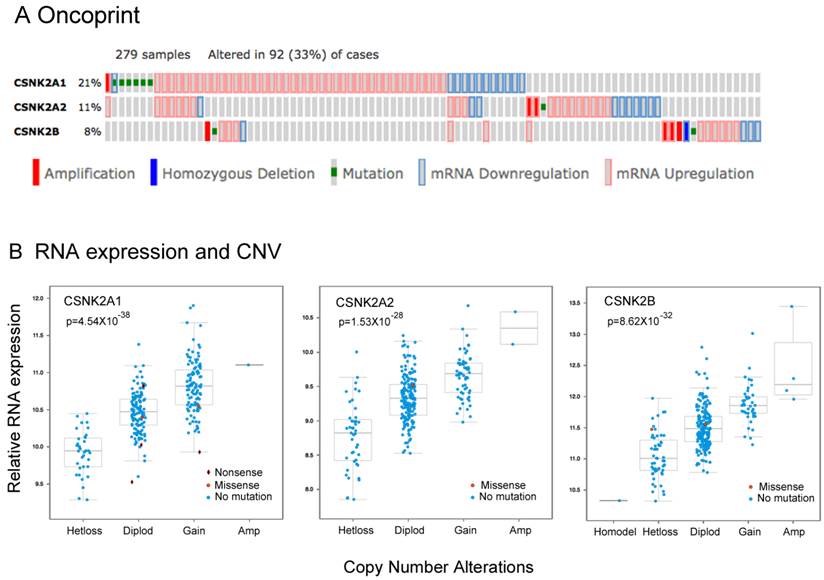
We previously showed that CK2 subunits, particularly CK2α, are overexpressed in HNSCC tumor tissue specimens and most of the HNSCC cell lines studied, and that CK2 promotes the malignant phenotype 7. Although more than 60%of HNSCC tumors have at least one PI3K-AKT-mTOR pathway alteration including gene mutations or amplifications, genomic alterations in CK2 have not been well studied previously. To explore the significance of the genomic alterations of CK2 subunits, we examined data from the HNSCC TCGA project, which recently completed a comprehensive genomic characterization of 279 human HNSCC samples including DNA and RNA sequencing, and SNP and protein microarrays 12. We extracted the genetic and expression data for CK2 subunits through the publically available database (www.cbioportal.org). In Figure 1A, we present the oncoprint for individual cases (each bar) displaying homozygous genetic and expression alterations of the CK2 subunits. Among 279 samples surveyed, 93 cases (33%) exhibited alterations in CK2 subunits, mainly increased mRNA expression. Consistent with prior studies, the highest percentage of cases with alterations (21%) were observed in CK2α (CSNK2A1), while CK2α' (CSNK2A2) alterations were observed in 11% cases, and 8% cases have CK2β (CSNK2B) alterations. The ratio of cases with overexpression and amplification of CK2 subunits could be under estimated, as expression data were compared with the mean value for tumors, and cbioportal only presents homozygous amplifications and deletions. Thus, we calculated the cases with heterozygous deletions and amplifications, and observed that 108 cases (38.7%) exhibited genetic gain of CK2α, 61 cases (21.8%) exhibited gain of CK2α', and 41 cases (14.7%) exhibited gain of CK2β (Additional file 1: Supplemental Table 2). Furthermore, the association of RNA expression and DNA copy number variation (CNV) of CK2 subunits were compared, and highly significant correlations were observed (Fig. 1B, Additional file 1: Supplemental Table 2). Our data provide evidence that genomic alterations can contribute to increased expression of CK2 subunit mRNA in HNSCC.
The genetic and expression alterations of CK2 subunits in HNSCC tissues from The Cancer Genome Atlas (TCGA). Data for genetic and expression alterations of CK2 subunits were extracted from TCGA HNSCC project through cbioportal. A, Oncoprint presents individual cases (each bar) with genetic and expression alteration of CK2 subunits. CSNK2A1: CK2α; CSNK2A2: CK2α'; CSNK2B: CK2β. Solid red: homozygous amplification; solid blue, homozygous deletion; green: mutation; grey bar with blue frame: mRNA down-regulation compared with tumor mean; grey bar with pink frame: mRNA up-regulation compared with tumor mean. % on the left represents the percentage of cases with alteration of CK2 subunits. B, RNA expression and DNA copy number variation (CNV) of CK2 subunits were correlated and presented. The statistical correlation of RNA expression with CNV was examined and presented as p value.
CK2 inhibitor CX-4945 inhibits cell growth, cell cycle, and survival of HNSCC in vitro
Next, we investigated the effects of pharmacological CK2 inhibitor CX-4945 on cell proliferation, measured by MTT assay in a panel of 9 UM-SCC cell lines, in which we previously demonstrated increased CK2 subunit expression 7. The IC50 in 9 UM-SCC cell lines ranged from 3.4 μM to 11.9 μM (Additional file 1: Supplemental Table 1). No correlation between sensitivity over this narrow range of IC50 with CK2 expression 7 was observed (data not shown). UM-SCC1 and UM-SCC46 cells were selected for further study of the inhibitory effects, with UM-SCC1 representing a subset with reduced wild type (wt) TP53, and UM-SCC 46 expressing a mutant (mt) TP53 32, 33. As shown in Figure 2A, both UM-SCC1 and 46 cell lines exhibited a dose dependent decrease in cell density with increasing CX-4945 concentrations between 0.1 μM and 30 μM over a five-day MTT assay. UM-SCC1 cells had a similar IC50 value of ~4.1 μM when compared with an IC50 of ~3.4 μM for UM-SCC46 cells.
CX-4945 inhibits proliferation and cell cycle and promotes death in HNSCC cells. (A) UM-SCC1 and UM-SCC46 cells were treated with varying concentrations of CX-4945 (0.1 μM to 30μM), and the effects on cell density was assessed in five-day MTT assay. Decreasing cell density was seen in both UM-SCC1 and UM-SCC46 cells with increasing CX-4945 concentrations, with IC50 values of 4.1 μM and 3.4 μM respectively. Data were calculated from six replicates of one representative experiment. (B) Cell cycle and DNA fragmentation were measured by DNA cytofluorometry for UM-SCC1 and UM-SCC46 cells 24 hours following CX-4945 treatment at varying concentrations. There was an increase in sub-G0 DNA fragmentation, and cells arresting in the S and G2/M phases when treated with CX-4945 at 10μM.
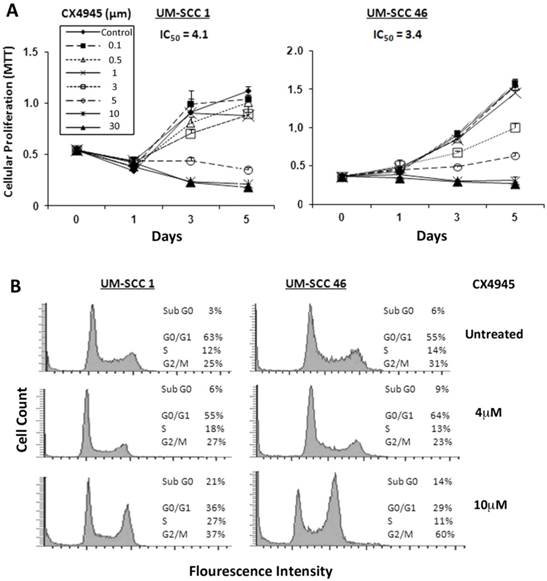
To evaluate the effects of CX-4945 on cell cycle and death, DNA cytofluorometric analysis of UM-SCC1 and UM-SCC46 cells was performed after treatment with CX-4945 at different doses of 4 and 10μM, approximating1-2.5X IC50 values. When treated with the higher concentration of 10μM CX-4945, UM-SCC1 cells showed notable changes in S (from 12% to 27%) and G2/M phase (from 25% to 37%) arrest, and increased sub-G0 DNA (from 3% to 21%), an indicator of cell death, by 24 hours. Under the lower concentration of 4μM CX-4945, UM-SCC1 cells did not exhibit significant alteration in percentages of cell cycle arrest and cell death. By contrast, treatment of UM-SCC46 cells with 10 µM CX-4945 showed a profound increase in G2/M (from 31% to 60%), and lesser increase in the sub-G0 fraction (from 6% to 14%) (Fig. 2B). These observations provide evidence that CK2 inhibitor predominantly induced block of G2/M phase of the cell cycle, and a modest increase in cell death, as indicated by sub-G0 fragmentation in HNSCC in vitro.
CX-4945 differentially modulates NF-κB, TP53 and AP-1 transcription activities, and Bcl-XL, p21, and IL-8 target gene promoter activities in HNSCC in vitro
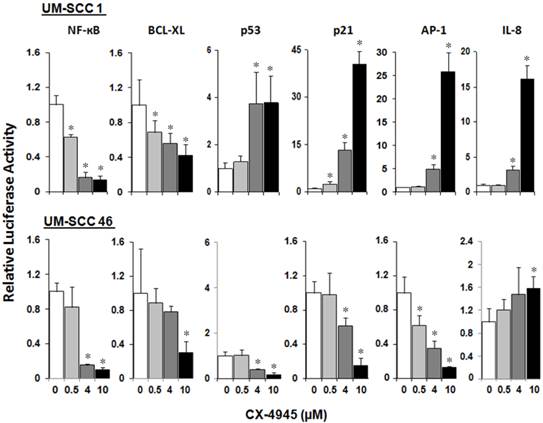
We have previously shown that inversely modulated NF-κB, AP-1 and TP53 activities, anti-apoptotic Bcl-XL, cell cycle inhibitor p21, and angiogenic cytokine IL-8 are important oncogenes and drug targets in UM-SCC cells 7. To study the modulatory effects of CX-4945 on the regulatory activities of these major transcription factors and effector genes altered in HNSCC, UM-SCC 1 and UM-SCC46 cells were transfected with reporter plasmids and treated with CX-4945 at varying concentrations for 72 hours. In both cell lines, CX-4945 treatment strongly inhibited NF-κB reporter activity, by more than 80% at the concentration of 4μM and 10μM. CX-4945 also inhibited Bcl-XL prosurvival gene promoter activity in a dose dependent manner. Reciprocal upregulation of the reporter activities of TP53 and cell cycle inhibitor gene p21 promoter were found in wtTP53 UM-SCC1 but not in mtTP53 UM-SCC46 cell line. In addition, a significant increase in AP-1 and target proinflammatory and angiogenic cytokine IL-8 activities was observed only in UM-SCC1 cells. Our data suggest that CK2 inhibitor CX-4945 can differentially modulate NF-κB, TP53 and AP-1 transcription activities and important target genes in HNSCC in vitro (Fig. 3).
CX-4945 significantly alters reporter gene activity in UM-SCC1 and UM-SCC46 cells. UM-SCC1 and UM-SCC46 cells were transfected with reporter and LacZ plasmids, and treated with CX-4945 at varying concentrations for 72 hours. Cells showed a concentration-dependent down-regulation of the reporter activities for prosurvival genes such as NF-ĸB, Bcl-XL in both cell lines, but up-regulation of the proapoptoticTP53 transcription factor, TP53 inducible cell cycle inhibitor p21 promoter activity, AP-1 transcriptional and IL-8 promoter activities in a concentration-dependent manner only in UM-SCC-1. * indicates a statistically significant difference between control group versus CX-4945 treated group (P < 0.05). Data were adjusted to β-Gal activity, and calculated from triplicates of a representative of repeated experiments.
CX-4945 altered protein expression and phosphorylation of molecules involved in AKT, ERK and TP53/p21 pathways in HNSCC
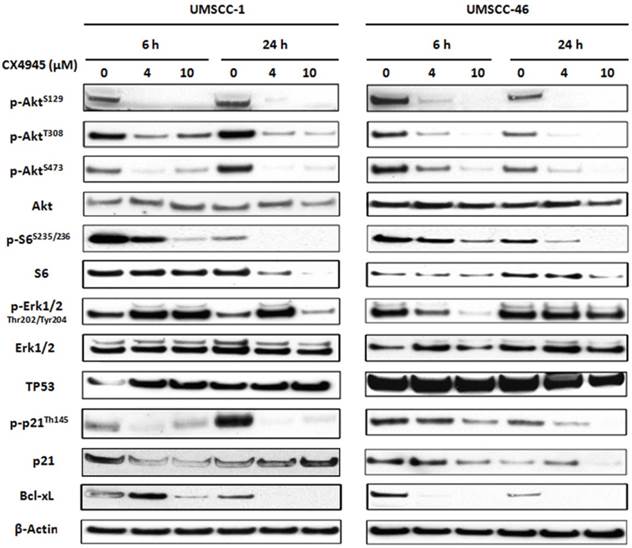
Previous studies in other solid tumors have indicated that CX-4945 modulated the downstream molecules of AKT pathway via phosphorylation of Akt serine129, and TP53 induced p21 via Thr145 13,14. We analyzed the protein expression of major components in PI3K/AKT, TP53 as well as ERK-AP-1 pathways (Fig. 4). Whole cell lysates from UM-SCC1 (left) and UM-SCC46 (right) were collected after treated with CX-4945 at 4 and 10 μM for 6 and 24 hours. In both cell lines, CX-4945 potently attenuated PI3K/AKT signal phosphorylation of AKT on the CK2-specific site (S129), while partially inhibiting phosphorylation of the canonical PDK-1 and mTORC phospho-acceptor regulatory sites (S308 and S473). CX-4945 also partially decreased downstream AKT-mTOR target S6 S235/236 phosphorylation and total S6 protein. However, in UM-SCC1, CX4945 treatment increased the phosphorylation of ERK1/2 at Thr202 and Tyr204 (Fig. 4), but partially inhibited ERK phosphorylation in UM-SCC 46 at early time point and higher concentration (Fig. 4). In addition, CX-4945 increased TP53 in UM-SCC1, but slightly decreased expression of TP53 in UM-SCC46 cells after 24-hour treatment. Although CX-4945 induced TP53 target p21 reporter gene activity in UM-SCC1 (Fig. 3), it inhibited Thr145 phosphorylation and total protein expression of cell cycle inhibitor p21 with different time course in UM-SCC1 and 46 cells. CX-4945 also inhibited Bcl-XL pro-survival protein expression in both cell lines. Thus, CK2 inhibitor CX-4945 has complex post-translational effects on AKT, ERK, TP53 and target effector molecules that could potentially attenuate their growth arrest and pro-apoptotic effects.
CX-4945 altered PI3K/AKT/Bcl-XL, ERK/AP-1 and TP53/p21proteins in HNSCC. UM-SCC-1 (left) and UM-SCC-46 (right) cell lines were treated with 4 and 10 μM CX-4945, whole cell lysates were harvested 6 and 24 hours after treatment, and subjected to SDS-PAGE and Western blot of indicated proteins. β-action was used as a loading control.
CX-4945 alone exhibited minimal anti-tumor activity in the UM-SCC1 xenograft model and opposing effects on target protein, apoptosis and proliferation markers in tumor specimens
Based on CX-4945 effects observed in in vitro experiments, we further evaluated the anti-tumor activity of CX-4945 in vivo in UM-SCC1 xenograft model of SCID mice. About 10 days after implantation, the tumors were palpable and treated with either 25 mg/kg or maximally tolerated dose of 75 mg/kg CX-4945 twice daily via oral gavage for 35 consecutive days (Fig. 5A, left). CX-4945 showed only moderate antitumor activity with the dosage of 75 mg/kg, and no significant improvement of survival was observed (Fig. 4A, right). All mice tolerated CX-4945 well without significant weight loss or other signs of toxicity (data not shown).
CX-4945 exhibited minimal anti-tumor activity and opposing effects on signaling, proliferation and apoptosis marker expression in UM-SCC1 xenografts. Human HNSCC xenografts were established utilizing 5X106 UM-SCC-1 cells implanted subcutaneously into the flank of SCID mice (n=12 or 13 mice per treatment group respectively). (A) Treatment was initiated when tumors were palpable as day 0, given 25 or 75 mg/kg of CX-4945, and control animals were given vehicle through oral gavage BID for 35 days. Statistical analysis performed on Day 26 shows a statistically significant difference between the control group and 75 mg/kg group, * (P < 0.05). (B) Kaplan Meier survival curves of control group versus treated animal groups until day 48. (C) Immunostaining was performed on tumors harvested from mice with CX-4945 treatment or vehicle control at early time points (13 days) or late time point (33 day). The images were acquired using an AperioScanscope at X20 magnification. Relevant areas were quantified as histological score using the Aperio Cell Quantification Software (Aperio, Vista, CA). TUNEL staining was presented as numbers of positive cells of the high power field (20X), and Ki67 was presented as the percentage of positive cells with respect to the total number of cells in each high power field. * Statistics significance (p<0.05, t-test).
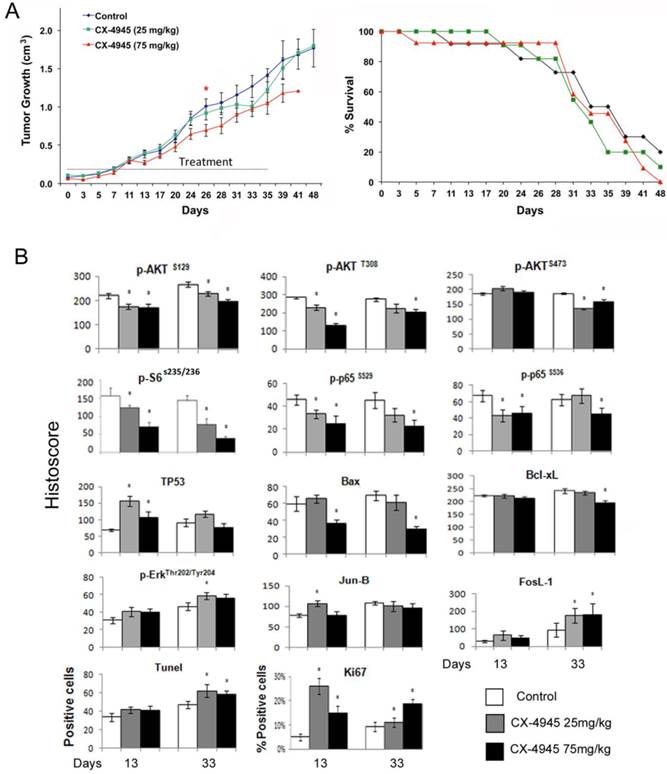
Next we examined the effects of CX-4945 treatment on the important signal, transcription factor, effector proteins and apoptosis and proliferation markers by immunostaining of tumors harvested at day 13 and 33 after treatment (Fig. 4B, and Additional file 1: supplemental Fig. 1). CX-4945 treatment incompletely inhibitedPI3K/AKT/mTOR pathway signaling as measured by decreased staining of p-AKT (S129) p-AKT (S308) and p-AKT (S473) (Fig. 5B, row 1). Signal phosphorylation of downstream molecules p-S6 (S235/236), NF-κB p-P65 (S529) and p-P65 (S536) staining was also decreased (Fig. 5B, row 2). Conversely, TP53 staining was significantly increased at early time points (13 days), but did not reach significance at late time points (33 days) after CX-4945 treatment (Fig. 5B, row 3). Pro-apoptotic protein BAX staining was decreased at the higher concentration, and anti-apoptotic protein Bcl-XL is also slightly decreased at the higher concentration and later time point. However, the molecules involved in MEK pathway, such as p-ERK, and AP-1 family member, Jun-B and FosL-1 were all increased. While TUNEL apoptosis marker staining was slightly increased in tumor specimens at the later time points following inhibition of prosurvival p-AKT, p-S6 and NF-κB p-65 markers, a significant increase in proliferation marker, Ki67 corresponding to increase in ERK-AP-1 proteins was observed at both time points. These data suggested the activation of MEK/ERK/AP-1 pathway could contribute to the drug resistance in vivo.
MEK inhibitor PD-0325901 overcomes CK2 drug resistance and exhibits anti-tumor activity in vivo
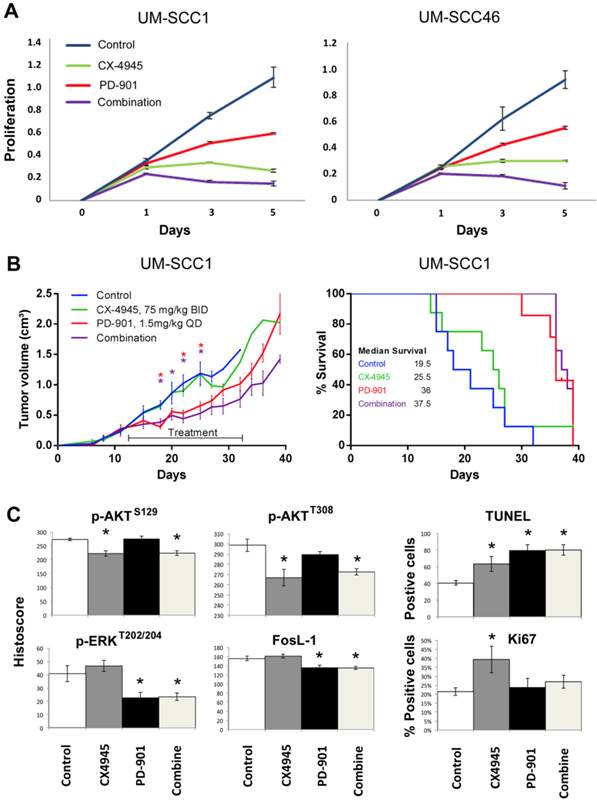
Next, we investigated the effects of MEK inhibitor PD-0325901 (PD-901) on cell proliferation, measured by MTT assay in UM-SCC1 and 46 cell lines (Fig. 6A). In preliminary studies we titrated the concentration of PD-901 single agent alone in the same UM-SCC cells as in Additional file 1: Supplemental Table 1, over nine concentrations ranging from 0.025μM to 10μM, following manufacturer's suggestions and literature 34-36. The panel of UM-SCC cells are relatively resistant to PD-901 in vitro, such that at the highest concentration of 10μM, 20-60% inhibition is observed in different lines, indicating the IC50 often exceeds 10μM. Such drug resistance in vitro has been observed in other solid tumor lines 34-36. We therefore tested increased PD-901 concentrations of 15, 30 and 60μM, alone and in combination with CX-4945 at 2, 4, or 8μM, which are in the range of 0.5, 1 or 2X IC50 for both cell lines (Supplemental Table 1). We present the data for 4μM CX-4945 and 30μM PD-901 approximating their IC50s alone and in combination (Fig. 6A). While PD-901 alone exhibited less potent inhibitory effect in in vitro proliferation assay, combination of PD-901 with CX-4945 exhibited additive effects in both cell lines in vitro. We further tested the anti-tumor activity of either drug alone or in combination in the UM-SCC1 xenograft model in vivo. Mice were treated with 75mg/kg of CX-4945 twice a day, 1.5mg/kg of PD-901 once a day, or in combination, by oral gavage for 21 consecutive days. The dose ranges of 1-2 mg/kg of PD-901 have been used in multiple preclinical studies, especially when testing drug combination 34-37, and is the tolerated dose without significant toxicity, that achieves concentrations comparable to those clinically achievable in humans. Tumor volume was reduced significantly by PD-901 alone, and the tumor growth was slightly further reduced with combination treatment (Fig. 6B, left panel). Mice bearing UM-SCC1 xenografts treated with PD-901 alone and in combination showed a median survival advantage of ~12 days (Fig. 6B, right panel, p=0.008, and p=0.002). Furthermore, we examined the tumor tissues harvested at day 22 after treatment (Fig. 6C), and examined p-AKT, p-ERK, FosL-1, TUNEL and Ki-67. Single agent CX-4945 or the combinatory treatment exhibited inhibition of p-AKT at S129 and T308. Single agent PD-901 and the combinatory treatment inhibited p-ERK and FosL-1. The combinatory treatment exhibited stronger induction of TUNEL staining, and increased Ki67 staining observed with CX-4945 was attenuated by PD-901. Our in vivo data demonstrated that the drug resistance to CX-4945 was overcome by the treatment with MEK inhibitor PD-901.
Combinatory effects of CX-4945 and PD-901 treatment on UM-SCC1 cells in vitro and in vivo. A.UM-SCC1 (left) and UM-SCC46 (right) were treated with 4µM CX-4945, or 30µM PD-901 alone, or in combination, and the effects on cell density were assessed in five-day MTT assay. Combination of MEK inhibitor PD-901 with CX-4945 exhibited additive effects on in vitro proliferation assay. (B, left) UM-SCC1 cells were implanted subcutaneously into the flank of SCID mice as described previously. Treatment was initiated when tumors were palpable, 12 days after implantation. Mice were either treated with 75mg/kg of CX-4945 twice (BID) a day, 1.5mg/kg of PD-901 once a day (QD), or the combination, by oral gavage for 21 consecutive days. Tumor volume was measured three times a week. Tumor volume was reduced by PD-901 alone and in combination. * indicates statistical significance, p<0.05. (B, right) Kaplan Meier survival curves of control group versus treated animal groups until day 38. Mice treated with PD-901 (p=0.0008) or combination (p=0.0002) showed a significantly increased median survival compared with controls. (C) Effects of CX-4945 on p-Akt, p-ERK, FosL-1, TUNEL and Ki67 expression in UM-SCC1 xenograft treated with PD-901 or in combination. Immunostaining was performed on tumors harvested from treated and vehicle control mice at 22 days after treatment. The images were acquired and the staining was quantified as previously described. * Statistics significance (p<0.05, t-test).
Discussion
In this pre-clinical study, we characterized the molecular, cellular and anti-tumor activity of a selective orally administered small molecule inhibitor of CK2, CX‐4945, in human HNSCC cell lines and tumor xenografts. Our data elucidates regulatory functions of CX-4945 in modulation of AKT, NF-κB, and TP53 signaling, expression of other key effector genes and proteins, which are implicated in control of cell cycle, survival and the malignant phenotype of HNSCC (Additional file 1: Supplemental Fig. 2). However, CX-4945 treatment induced compensatory MEK/ERK/AP-1 activation, which could be one of the mechanisms contributing to the drug resistance in vivo. Using MEK inhibitor PD-901, we demonstrated potent anti-tumor activity in the UM-SCC1 xenograft model that overcame CX-4945 drug resistance in vivo.
We and others previously showed that Ser/Thr protein kinase CK2 subunit RNA and proteins are overexpressed in HNSCC cell lines 7, and our current analysis of recent TCGA data indicates that genomic amplification contributes to increased mRNA expression of CK2 subunits in tumor specimens (Fig. 1). CK2 regulates multiple signaling pathways, that play important roles in the malignant phenotype as well as the sensitivity to cancer therapeutics1, 7. Using the small molecule inhibitor, CX-4945, we have shown that it inhibited cell growth and survival of HNSCC in vitro, caused S and G2/M cell cycle arrest, and induced sub-G0 DNA fragmentation and cell death. The tumor suppressive activities observed in vitro are consistent with our previous results with CK2 subunit knockdown using siRNA in HNSCC cell lines 7. In addition, here we show that the activity of CX-4945 in HNSCC cells involves attenuation of PI3K/AKT signaling pathway via phosphorylation of AKT on the CK2-specific S129 site, and the canonical S473 and T308 regulatory sites, consistent with other tumors demonstrating aberrant activation of AKT13. Based on our previous study of siRNA knockdown of CK2 subunits demonstrating inhibition of NF-κB and enhanced TP53 expression and activities 7, we further investigated CX-4945 effects on NF-κB and TP53 activity and effector target genes and proteins. We have shown that CX-4945 decreased NF-κB transcription and Bcl-XL promoter activity, as demonstrated by gene reporter assay in vitro (Fig. 3). Conversely, CX-4945 induced TP53 and p21 gene expression and activity in wtTP53 UM-SCC1 cells (Fig. 4 and 5), in which CX-4945 induced greater cell death in vitro (Fig. 2B). This could be due to the combination of decreased NF-κB transcription and Bcl-XL promoter activity and over-expressed TP53 and p21 demonstrated by gene reporter assay in vitro (Fig. 4 and 5). These results are consistent with our previous study with CK2 siRNA that inhibition of NF-κB and Bcl-XL and over-expression of TP53 together promoted cell death in HNSCC cell lines 18. Our data are also consistent with the study performed in glioblastoma where CK2 inhibitor DRB (5,6-Dichlorobenzimidazole 1-β-D-ribofuranoside) or Apigenin enhanced TP53 function in wtTP53 cells, and sensitization to TNF-α induced apoptosis 38.
Short duration exposure with CX-4945 inhibited cell proliferation of the UM-SCC panel over a narrow IC50 range in vitro. These observations are similar to our previous data showing that the levels of mRNA and protein expression of CK2 subunits did not directly correlate with drug sensitivity of cells to CK2 inhibitor DMAT, which is consistent with the reports from other studies 13. Surprisingly, as a single agent in vivo, a high dose regimen of 75mg/kg twice daily for 35 days only slightly attenuated tumor growth, and without survival benefit (Fig. 5A). Similar resistance to CX-4549 in vivo was also observed in other tumor models, such as A2780 ovarian cancer xenograft model39. The discrepancy between strong anti-proliferative activity in vitro by CX-4945 and the minimal anti-tumor activity in vivo could be due in part to its relative potency, solubility, and bioavailability. Concentrations of 4-10 μM were required to achieve cytostatic effects in vitro, which may not be readily obtained in vivo. CX-4945 is the first generation of orally available small molecule CK2 inhibitor available for human clinical trials, and has limited solubility affecting anti-tumor efficacy. In animals, dosing is effectively limited to 75mg/kg, and in humans dosing has been investigated up to 2 grams twice daily. Newer generation CK2 inhibitors with higher solubility could potentially overcome such problems.
The differences in cell sensitivity and resistance to CK2 inhibitors could also be due to tumor dependent differences in the downstream targets of CK2 or other pathways, as we explored in this study. When we assessed the targets of CX-4945 treatment in the tumor specimens harvested 13 days and 33 days after treatment (Fig. 5B), the drug partially decreased phosphorylated AKT, p-S6 S235/236, NF-κB p-p65 S536 and S529 and target effector protein Bcl-XL. This partial inhibition of prosurvival NF-κB and Bcl-XL and induction of TP53 could explain the slightly increased TUNEL apoptosis marker staining. However, while CX-4945 induced TP53 expression, it phosphorylated and attenuated p21 protein expression in vitro and decreased BAX in vivo, providing evidence for post-translational effects that potentially attenuate the effects of these TP53-inducible genes. Further, we observed increased phosphorylation of ERK1/2 at Thr202 and Tyr204, as well as increased reporter activation of AP-1 in vitro, and protein expression of AP-1 family members, JUNB and FOSL1 in vivo. Thus, the compensatory increase of MEK/ERK/AP-1 pathway could be one of the pathways contributing to the increased Ki67 proliferation marker, and drug resistance to CX-4945 treatment in vivo.
We previously have shown that MEK, ERK1/2 and AP-1 pathway is co-activated in HNSCC through signaling by EGFR, other growth factor receptors, and cross talk by IKK/NF-κB pathways 16, 17, 19, 20. Activation of MEK, ERK1/2 and AP-1 pathway contributes to the malignant phenotype and drug resistance in HNSCC16, 19, 20, 28. We used PD-901, a highly selective and potent small molecule inhibitor against MEK1/2, to examine the role of ERK-AP-1 activation in vitro and in vivo. Although PD-901 exhibited less anti-proliferative effects in vitro when compared with CX-4945 (Fig. 6A), it exhibited strong anti-tumor activity in this relatively resistant tumor model. Consistent with the anti-tumor activity, PD-901 and combination with CX-4945 also inhibited p-ERK and FOSL-1, induced apoptosis and inhibited proliferation marker Ki67 (Fig. 6C). Previous studies showed tumors carrying RAS or BRAF mutations exhibited higher sensitivity to PD-901 in vitro and in vivo40. In this study, we observed strong anti-tumor efficacy in vivo in HNSCC, which rarely display RAS or BRAF mutations. The sensitivity could due to the aberrant activation MAPK pathways transduced signaling from multiple growth factor receptors, including EGFR and family members, c-MET, and FGFRs 12, 16-20. We also tested the individual and combination effects of additional inhibitors with CX-4945, such as SP600125 for JNK, SB203580 for p38 in both UM-SCC1 and UM-SCC46 cells. Both cell lines were relatively resistant to either inhibitors alone when measured by MTT assay in vitro, where the IC50s ranged between ~13-18μM when treated alone. The combination of SP600125 or SB203580 with CX-4945 did not exhibit significant combinatory effects in vitro (data not shown).
Development of effective combination strategies is desired to reduce the incidence of clinical resistance that is common with single-agent targeted therapies. In the current study, relatively low response was observed in the in vivo HNSCC xenograft model when single agent CX-4945 was used, indicating the complexity of the modulation of networks involving prosurvival or proapoptotic pathways. Based on the in vitro observations and using immunohistochemistry of tumor specimens harvested from in vivo study, we identified potential mechanisms for drug resistance related to the activation of MER/ERK/AP-1 signaling. Our current study illustrates the potential usefulness of analyzing signal molecules by reporter and Western blots in vitro, as well as immunostaining in vivo to identify the drug resistance mechanism, which could be useful in preclinical cell line and patient derived xenograft models. These targets merit further investigation as biomarkers for selection of the effective targeted therapy for MEK/ERK/AP-1, PI3K and CK2-AKT pathways in this HNSCC xenograft model. In conclusion, we found that inhibition of CK2 via CX-4945 revealed modulation of PI3K/AKT, NF-κB, TP53 and AP-1, and pro-apoptotic activities in HNSCC cell lines in vitro and in vivo. Compensatory activation of MEK-ERK-AP-1 pathway leads to enhanced proliferation and drug resistance, and combinatory therapy with PD-901 to target MEK/ERK/AP-1 pathway improved anti-tumor effects observed with CK2 inhibition alone.
Supplementary Material
Additional File 1Supplemental Methods, Supplemental Tables 1-2 and Figures 1-2.
Acknowledgements
The authors wish to thank Drs. Barbara Conley, MD, Cancer Diagnosis Program, and James Mitchell, PhD, Radiation Biology Branch, Center for Cancer Research, National Cancer Institute, National Institutes of Health, for their reading and helpful comments on the manuscript; and Ms. Jamie Coupar for her technical support.
Grant information
Supported by NIDCD intramural projects ZIA-DC-000073 and ZIA-DC-000074 (YB, JH, VK, SM, JF, ZC, CVW).
Competing Interests
Carter Van Waes is recipient of Cylene and Pfizer pharmaceuticals for research under Material Transfer Agreements with NIDCD, but otherwise holds no financial interests.
Other authors have declared that no competing interest exists.
References
1. Trembley JH, Chen Z, Unger G, Slaton J, Kren BT, Van Waes C, Ahmed K. Emergence of protein kinase CK2 as a key target in cancer therapy. Biofactors. 2010;36:187-95
2. Hessenauer A, Schneider CC, Gotz C, Montenarh M. CK2 inhibition induces apoptosis via the ER stress response. Cellular signalling. 2011;23:145-51
3. Homma MK, Homma Y. Cell cycle and activation of CK2. Molecular and cellular biochemistry. 2008;316:49-55
4. Ruzzene M, Pinna LA. Addiction to protein kinase CK2: a common denominator of diverse cancer cells? Biochim Biophys Acta. 2010;1804:499-504
5. Faust RA, Gapany M, Tristani P, Davis A, Adams GL, Ahmed K. Elevated protein kinase CK2 activity in chromatin of head and neck tumors: association with malignant transformation. Cancer Lett. 1996;101:31-5
6. Gapany M, Faust RA, Tawfic S, Davis A, Adams GL, Ahmed K. Association of elevated protein kinase CK2 activity with aggressive behavior of squamous cell carcinoma of the head and neck. Mol Med. 1995;1:659-66
7. Brown MS, Diallo OT, Hu M, Ehsanian R, Yang X, Arun P, Lu H, Korman V, Unger G, Ahmed K, Van Waes C, Chen Z. CK2 modulation of NF-kappaB, TP53, and the malignant phenotype in head and neck cancer by anti-CK2 oligonucleotides in vitro or in vivo via sub-50-nm nanocapsules. Clinical Cancer Res. 2010;16:2295-307
8. Yu M, Yeh J, Van Waes C. Protein kinase casein kinase 2 mediates inhibitor-kappaB kinase and aberrant nuclear factor-kappaB activation by serum factor(s) in head and neck squamous carcinoma cells. Cancer Res. 2006;66:6722-31
9. Van Waes C. Nuclear factor-kappaB in development, prevention, and therapy of cancer. Clinical Cancer Res. 2007;13:1076-82
10. Brown M, Cohen J, Arun P, Chen Z, Van Waes C. NF-kappaB in carcinoma therapy and prevention. Expert Opin Ther Targets. 2008;12:1109-22
11. Wang D, Westerheide SD, Hanson JL, Baldwin AS Jr. Tumor necrosis factor alpha-induced phosphorylation of RelA/p65 on Ser529 is controlled by casein kinase II. J Biol Chem. 2000;275:32592-7
12. The Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinima. Nature. 2015;517:576-582
13. Siddiqui-Jain A, Drygin D, Streiner N, Chua P, Pierre F, O'Brien SE, Bliesath J, Omori M, Huser N, Ho C, Proffitt C, Schwaebe MK. et al. CX-4945, an orally bioavailable selective inhibitor of protein kinase CK2, inhibits prosurvival and angiogenic signaling and exhibits antitumor efficacy. Cancer Res. 2010;70:10288-98
14. Pierre F, Chua PC, O'Brien SE, Siddiqui-Jain A, Bourbon P, Haddach M, Michaux J, Nagasawa J, Schwaebe MK, Stefan E, Vialettes A, Whitten JP. et al. Pre-clinical characterization of CX-4945, a potent and selective small molecule inhibitor of CK2 for the treatment of cancer. Mol Cell Biochem. 2011;356:37-43
15. Pierre F, Chua PC, O'Brien SE, Siddiqui-Jain A, Bourbon P, Haddach M, Michaux J, Nagasawa J, Schwaebe MK, Stefan E, Vialettes A, Whitten JP. et al. Discovery and SAR of 5-(3-chlorophenylamino)benzo[c][2,6]naphthyridine-8-carboxylic acid (CX-4945), the first clinical stage inhibitor of protein kinase CK2 for the treatment of cancer. J Med Chem. 2011;54:635-54
16. Bancroft CC, Chen Z, Yeh J, Sunwoo JB, Yeh NT, Jackson S, Jackson C, Van Waes C. Effects of pharmacologic antagonists of epidermal growth factor receptor, PI3K and MEK signal kinases on NF-kappaB and AP-1 activation and IL-8 and VEGF expression in human head and neck squamous cell carcinoma lines. Int J Cancer. 2002;99:538-48
17. Dong G, Chen Z, Li ZY, Yeh NT, Bancroft CC, Van Waes C. Hepatocyte growth factor/scatter factor-induced activation of MEK and PI3K signal pathways contributes to expression of proangiogenic cytokines interleukin-8 and vascular endothelial growth factor in head and neck squamous cell carcinoma. Cancer Res. 2001;61:5911-8
18. Lee TL, Yeh J, Friedman J, Yan B, Yang X, Yeh NT, Van Waes C, Chen Z. A signal network involving coactivated NF-kappaB and STAT3 and altered p53 modulates BAX/BCL-XL expression and promotes cell survival of head and neck squamous cell carcinomas. Int J Cancer. 2008;122:1987-98
19. Nottingham LK, Yan CH, Yang X, Si H, Coupar J, Bian Y, Cheng TF, Allen C, Arun P, Gius D, Dang L, Van Waes C. et al. Aberrant IKKalpha and IKKbeta cooperatively activate NF-kappaB and induce EGFR/AP1 signaling to promote survival and migration of head and neck cancer. Oncogene. 2013
20. Pernas FG, Allen CT, Winters ME, Yan B, Friedman J, Dabir B, Saigal K, Mundinger GS, Xu X, Morris JC, Calvo KR, Van Waes C. et al. Proteomic signatures of epidermal growth factor receptor and survival signal pathways correspond to gefitinib sensitivity in head and neck cancer. Clinical Cancer Res. 2009;15:2361-72
21. Broek RV, Mohan S, Eytan DF, Chen Z, Van Waes C. The PI3K/Akt/mTOR axis in head and neck cancer: functions, aberrations, crosstalk, and therapies. Oral diseases. 2013
22. Herzog A, Bian Y, Vander Broek R, Hall B, Coupar J, Cheng H, Sowers AL, Cook JD, Mitchell JB, Chen Z, Kulkarni AB, Van Waes C. PI3K/mTOR inhibitor PF-04691502 antitumor activity is enhanced with induction of wild-type TP53 in human xenograft and murine knockout models of head and neck cancer. Clinical Cancer Res. 2013;19:3808-19
23. Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, Van Dyk DE, Pitts WJ, Earl RA, Hobbs F, Copeland RA, Magolda RL. et al. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem. 1998;273:18623-32
24. Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410:37-40
25. Ondrey FG, Dong G, Sunwoo J, Chen Z, Wolf JS, Crowl-Bancroft CV, Mukaida N, Van Waes C. Constitutive activation of transcription factors NF-(kappa)B, AP-1, and NF-IL6 in human head and neck squamous cell carcinoma cell lines that express pro-inflammatory and pro-angiogenic cytokines. Mol Carcinog. 1999;26:119-29
26. Bancroft CC, Chen Z, Dong G, Sunwoo JB, Yeh N, Park C, Van Waes C. Coexpression of proangiogenic factors IL-8 and VEGF by human head and neck squamous cell carcinoma involves coactivation by MEK-MAPK and IKK-NF-kappaB signal pathways. Clinical Cancer Res. 2001;7:435-42
27. Shaulian E, Karin M. AP-1 as a regulator of cell life and death. Nat Cell Biol. 2002;4:E131-6
28. Chen Z, Ricker JL, Malhotra PS, Nottingham L, Bagain L, Lee TL, Yeh NT, Van Waes C. Differential bortezomib sensitivity in head and neck cancer lines corresponds to proteasome, nuclear factor-kappaB and activator protein-1 related mechanisms. Mol Cancer Ther. 2008;7:1949-60
29. LoRusso PM, Krishnamurthi SS, Rinehart JJ, Nabell LM, Malburg L, Chapman PB, DePrimo SE, Bentivegna S, Wilner KD, Tan W, Ricart AD. Phase I pharmacokinetic and pharmacodynamic study of the oral MAPK/ERK kinase inhibitor PD-0325901 in patients with advanced cancers. Clinical Cancer Res. 2010;16:1924-37
30. Brenner JC, Graham MP, Kumar B, Saunders LM, Kupfer R, Lyons RH, Bradford CR, Carey TE. Genotyping of 73 UM-SCC head and neck squamous cell carcinoma cell lines. Head & neck. 2010;32:417-26
31. Zhao M, Sano D, Pickering CR, Jasser SA, Henderson YC, Clayman GL, Sturgis EM, Ow TJ, Lotan R, Carey TE, Sacks PG, Grandis JR. et al. Assembly and initial characterization of a panel of 85 genomically validated cell lines from diverse head and neck tumor sites. Clinical Cancer Res. 2011;17:7248-64
32. Friedman J, Nottingham L, Duggal P, Pernas FG, Yan B, Yang XP, Chen Z, Van Waes C. Deficient TP53 Expression, Function, and Cisplatin Sensitivity Are Restored by Quinacrine in Head and Neck Cancer. Clinical Cancer Res. 2007;13:6568-78
33. Cohen J, Chen Z, Lu SL, Yang XP, Arun P, Ehsanian R, Brown MS, Lu H, Yan B, Diallo O, Wang XJ, Van Waes C. Attenuated transforming growth factor beta signaling promotes nuclear factor-kappaB activation in head and neck cancer. Cancer Res. 2009;69:3415-24
34. Cirone P, Andresen CJ, Eswaraka JR, Lappin PB, Bagi CM. Patient-derived xenografts reveal limits to PI3K/mTOR- and MEK-mediated inhibition of bladder cancer. Cancer chemotherapy and pharmacology. 2014;73:525-38
35. Pitts TM, Newton TP, Bradshaw-Pierce EL, Addison R, Arcaroli JJ, Klauck PJ, Bagby SM, Hyatt SL, Purkey A, Tentler JJ, Tan AC, Messersmith WA. et al. Dual Pharmacological Targeting of the MAP Kinase and PI3K/mTOR Pathway in Preclinical Models of Colorectal Cancer. PloS one. 2014;9:e113037
36. Sheppard KE, Cullinane C, Hannan KM, Wall M, Chan J, Barber F, Foo J, Cameron D, Neilsen A, Ng P, Ellul J, Kleinschmidt M. et al. Synergistic inhibition of ovarian cancer cell growth by combining selective PI3K/mTOR and RAS/ERK pathway inhibitors. European journal of cancer. 2013;49:3936-44
37. Simmons BH, Lee JH, Lalwani K, Giddabasappa A, Snider BA, Wong A, Lappin PB, Eswaraka J, Kan JL, Christensen JG, Shojaei F. Combination of a MEK inhibitor at sub-MTD with a PI3K/mTOR inhibitor significantly suppresses growth of lung adenocarcinoma tumors in Kras(G12D-LSL) mice. Cancer chemotherapy and pharmacology. 2012;70:213-20
38. Dixit D, Sharma V, Ghosh S, Mehta VS, Sen E. Inhibition of Casein kinase-2 induces p53-dependent cell cycle arrest and sensitizes glioblastoma cells to tumor necrosis factor (TNFalpha)-induced apoptosis through SIRT1 inhibition. Cell Death Dis. 2012;3:e271
39. Siddiqui-Jain A, Bliesath J, Macalino D, Omori M, Huser N, Streiner N, Ho CB, Anderes K, Proffitt C, O'Brien SE, Lim JK, Von Hoff DD. et al. CK2 inhibitor CX-4945 suppresses DNA repair response triggered by DNA-targeted anticancer drugs and augments efficacy: mechanistic rationale for drug combination therapy. Mol Cancer Ther. 2012;11:994-1005
40. Henderson YC, Chen Y, Frederick MJ, Lai SY, Clayman GL. MEK inhibitor PD0325901 significantly reduces the growth of papillary thyroid carcinoma cells in vitro and in vivo. Mol Cancer Ther. 2010;9:1968-76
Author contact
![]() Corresponding authors: Zhong Chen, MD, PhD, NIDCD/NIH, Building 10/5D55, 10 Center Drive, Bethesda, MD 20892. Email: chenznih.gov. Phone: 301-435-2073. Fax: 301-594-4643. Or Carter Van Waes, MD, PhD, NIDCD/NIH, Building 10/CRC, 4-2732, 10 Center Drive, Bethesda, MD 20892. Email: vanwaescnih.gov. Phone: 301-402-4216. Fax: 301-402-1140.
Corresponding authors: Zhong Chen, MD, PhD, NIDCD/NIH, Building 10/5D55, 10 Center Drive, Bethesda, MD 20892. Email: chenznih.gov. Phone: 301-435-2073. Fax: 301-594-4643. Or Carter Van Waes, MD, PhD, NIDCD/NIH, Building 10/CRC, 4-2732, 10 Center Drive, Bethesda, MD 20892. Email: vanwaescnih.gov. Phone: 301-402-4216. Fax: 301-402-1140.