International Journal of Biological Sciences
ISSN: 1449-2288
10
Impact Factor
ISSN: 1449-2288
- Current Issue
- Volume 21; 2025
- Volume 20; 2024
- Volume 19; 2023
- Volume 18; 2022
- Volume 17; 2021
- Archive
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Top
Introduction
Material and Methods
Statistical analysis
Results
Discussion
Supplementary Material
Acknowledgements
References
Introduction
Material and Methods
Statistical analysis
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2015; 11(4):434-447. doi:10.7150/ijbs.9311 This issue Cite
Research Paper
Tetratricopeptide Repeat Domain 9A Negatively Regulates Estrogen Receptor Alpha Activity
Smeeta Shrestha1, Yang Sun1, Thomas Lufkin2, Petra Kraus2, Yuzuan Or1, Yenni A. Garcia3, Naihsuan Guy3, Paola Ramos3, Marc B. Cox3, Fiona Tay1, Valerie CL Lin1 ![]()
1. School of Biological Sciences, Nanyang Technological University, Singapore;
2. Genome Institute of Singapore, Singapore;
3. Department of Biological Sciences, University of Texas at El Paso, USA.
Received 2014-4-7; Accepted 2014-12-29; Published 2015-2-27
Citation:
Shrestha S, Sun Y, Lufkin T, Kraus P, Or Y, Garcia YA, Guy N, Ramos P, Cox MB, Tay F, Lin VCL. Tetratricopeptide Repeat Domain 9A Negatively Regulates Estrogen Receptor Alpha Activity. Int J Biol Sci 2015; 11(4):434-447. doi:10.7150/ijbs.9311. https://www.ijbs.com/v11p0434.htm
Other stylesAbstract

Tetratricopeptide repeat domain 9A (TTC9A) is a target gene of estrogen and progesterone. It is over-expressed in breast cancer. However, little is known about the physiological function of TTC9A. The objectives of this study were to establish a Ttc9a knockout mouse model and to study the consequence of Ttc9a gene inactivation. The Ttc9a targeting vector was generated by replacing the Ttc9a exon 1 with a neomycin cassette. The mice homozygous for Ttc9a exon 1 deletion appear to grow normally and are fertile. However, further characterization of the female mice revealed that Ttc9a deficiency is associated with greater body weight, bigger thymus and better mammary development in post-pubertal mice. Furthermore, Ttc9a deficient mammary gland was more responsive to estrogen treatment with greater mammary ductal lengthening, ductal branching and estrogen target gene induction. Since Ttc9a is induced by estrogen in estrogen target tissues, these results suggest that Ttc9a is a negative regulator of estrogen function through a negative feedback mechanism. This is supported by in vitro evidence that TTC9A over-expression attenuated ERα activity in MCF-7 cells. Although TTC9A does not bind to ERα or its chaperone protein Hsp90 directly, TTC9A strongly interacts with FKBP38 and FKBP51, both of which interact with ERα and Hsp90 and modulate ERα activity. It is plausible therefore that TTC9A negatively regulates ERα activity through interacting with co-chaperone proteins such as FKBP38 and FKBP51.
Keywords: TTC9A, Knockout Mouse, ERα, Mammary gland, Co-chaperone.
Introduction
Tetratricopeptide repeat domain 9A (TTC9A) was first reported as a hypothetical protein based on the sequence analysis of a cDNA clone from a brain cDNA library [1]. It is now referred to as TTC9A because of its sequence homology to a family of TTC9 proteins that also include TTC9B and TTC9C, which share 46% and 35% homology with TTC9A in amino acids sequence, respectively [2]. TTC9A also shares significant homology with FK506 binding proteins cyclophilin 40, FKBP51, FKBP52 and FKBP38 that are steroid receptor co-chaperones involved in modulating steroid receptor assembly and maturation through interaction with Hsp90 [3-7].
TTC9A protein contains 3 tetratricopeptide repeat (TPR) domains at its carboxyl terminus [8]. TPR domain is a 34 amino acid (aa) consensus motif present in varying numbers in a large family of TPR-containing proteins [9-11]. These TPR motifs in tandem arrays form antiparallel α-helical hairpins that function as an important protein interaction interface and are involved in regulating diverse biological processes including steroid receptor signaling [11-14].
TTC9A is expressed in the mouse embryonic stem cells [15] and TTC9A protein is detected in all tissues after birth, with the highest expression in the brain and the lowest expression in the liver [15, 16]. Spleen and the reproductive tissues also express significantly high levels of TTC9A.
TTC9A expression is regulated by both estrogen and progesterone in breast cancer cells. It is drastically induced by progesterone in progesterone receptor (PR)-transfected breast cancer cells MDA-MB-231 [16], and the induction is associated with progesterone-induced growth inhibition and focal adhesion in these cells [17, 18]; its expression is down-regulated by the growth-stimulating hormone estrogen in breast cancer cells MCF-7 [16]. The data seems to suggest a negative association between TTC9A expression, cell growth and the involvement of TTC9A in ovarian steroid hormone signaling in breast cancer cells. However, studies in mice revealed that estrogen significantly up-regulated TTC9A expression in estrogen target tissues including the uterus and mammary gland [15]. The significance of the disparate observations in normal and malignant mammary cells is not clear.
To elucidate the physiological role of TTC9A in ovarian steroid hormone signaling, we generated Ttc9a knockout mice and characterized the consequence of Ttc9a deficiency in the female mice. Our study reveals that TTC9A gene deletion is associated with better development in terms of body weight, thymus size and mammary ductal side branching in post-pubertal female mice. We also provide evidence that estrogen can elicit greater response in the mammary glands of Ttc9a-/- mice than the Ttc9a+/+ mice. We propose that TTC9A is a negative regulator of estrogen activity through interacting with co-chaperone proteins such as FKBP38 and FKBP51.
Material and Methods
Ethical Statement
All in vivo procedures and animal care followed the Institutional Animal Care and Use Committee (IACUC) guidelines set by National Advisory Committee for Laboratory Animal Research (NACLAR) in Singapore. The IACUC protocols employed were reviewed and approved by the IACUC committees of the A*STAR Biopolis Biological Resource Center and Nanyang Technological University before any animal procedures were undertaken for the study described here (IACUC Protocol No: 110689 and 110648). The mouse strains used in this study were housed, maintained and provided by the A*STAR Biopolis Biological Resource Center, or by the School of Biological Sciences (SBS), Nanyang Technological University (IACUC Protocol No: ARF SBS/NIE-A009). The mouse lines described here will be made available to the research community upon acceptance of the manuscript.
Generation of Ttc9a Knockout mouse
The Ttc9a gene is comprised of three exons. Exon 1 is targeted for deletion because it codes for over 70% of the TTC9A protein. The Ttc9a targeting vector was generated by replacing the Ttc9a exon 1 with a loxp flanked neomycin cassette (Figure 1A). The homologous arms were cloned from the Bacterial Artificial Chromosome (BAC) RP24-382B7 clone using the Red/Et BAC recombineering approach [19-21]. The Ttc9a targeting vector was linearized by digestion with EcoRV and electroporated into R1 mouse embryonic stem (ES) cells. Transfected ES cells were selected for G418-resistance (200-400 μg/ml). The individual G418 resistant ES colonies were screened by Southern hybridization for targeted homologous recombination at the Ttc9a exon 1 locus with the 5' probe as indicated in Figure 1. The Ttc9a heterozygous ES clones were microinjected into 8 cell stage mouse embryos isolated from C57BL/6J [22]. The Ttc9a chimeras were crossed to wild type C57BL6/6J mice to generate Ttc9a heterozygous mice. These Ttc9a heterozygous mice were crossed to obtain the Ttc9a homozygous mice.
Genotyping by Southern blotting analysis and PCR
The genomic DNA from ES cells or tail biopsies was digested with Sca I, separated on a 0.8% agarose gel and blotted onto a positively charged nylon membrane overnight. These membranes were then hybridized with the digoxigenin (DIG)-labelled 5' probe overnight, washed, then blocked with blocking buffer and finally probed with anti-DIG-alkaline phosphatase antibody at a 1:5000 dilution. These nylon membranes with digoxigenin-labelled nucleic acids were detected using chemiluminescent immunodetection methods. PCR was also used for routine mouse genotyping and the following PCR conditions and primer pairs were used for this purpose: 94°C (30 sec), 60°C (30 sec), 72°C (1 min) for 30 cycles and a final extension step at 72°C (7 min). Ttc9a allele primers: Ttc9a-FP: 5'-CACACGAGTTCAAGAGCCAAGGG-3'; Ttc9a-RP: 5'-GCTTCAACCGTCTTGCTCTG-3'. Neomycin primers: neo-FP: 5'-ACTGGGCACAACAGACGATCGG-3'; neo-RP: 5'-GGAAGCGGTCAGCCCATTCG-3'.
RT-PCR analysis
Mammary gland tissue was grinded in liquid nitrogen with mortar and pestle. Approximately 20 mg of ground tissue powder was weighed; homogenised and total RNA isolated using the TRIzol reagent (Invitrogen) as per manufacturer's instructions. From this 5 μg of total RNA was reverse transcribed using Superscript II reverse transcriptase (Invitrogen, Carlsbad, CA). Real-time PCR was performed with SYBRGreen master mix (Kapa Biosystems, US) on an ABI Prism 7000 sequence detection system (PE Applied Biosystems, Foster City, CA).
The real time PCR primer sequences used for the analysis of PR, Areg, Greb1, Muc1, 36B4 and 18sRNA expressions were: PR forward 5'-CGCCATCTACCAGCCGCTC-3', PR reverse 5'-TGAATCCGGCCTCAGGTAGTT-3'; Areg forward 5'-CACAGCGAGGATGACAAGGA-3', Areg reverse 5'-GAGGATGATGGCAGAGACAAAGA-3'; Greb1 forward 5'-CGGGCTTTGTGAGGAGTCA-3', Greb1 reverse 5'-CCGGAAGCACAAAGAAGACAGA-3'; Muc1 forward 5'-CCACCAGAGCTCCCGAAGAC-3', Muc1 reverse 5'-TAGAGGGCTGGTGGCTGGAG-3', 36B4 forward 5'-GATCGGGTACCCAACTGTTGCC-3', 36B4 reverse 5'-CAGGGGCAGCAGCCGCAAATGC-3', 18sRNA forward 5'-GTAACCCGTTGAACCCCATT-3', 18sRNA reverse 5'-CCATCCAATCGGTAGTAGCG-3'. Fold changes were calculated from the Ct values and the expression levels of PR, Areg, Greb1 and Muc1 were determined by normalizing their Ct values against 36B4 and 18sRNA Ct values. PCR primers were designed from exon junctions to prevent any amplification from contaminating genomic DNA within the RNA samples.
Western Blotting analysis
Mouse tissues were dissected, snap frozen in liquid nitrogen, and grinded to powder with a mortar and pestle. To prepare total protein lysates, approximately 20 mg of the ground tissue powder was homogenised in cold lysis buffer containing 100 mM NaF, 50 mM HEPES (pH 7.5), 150 mM NaCl, 1% Triton X-100, 1 mM PMSF and the cocktail of proteinase inhibitors (5 μg/ml pepstatin A, 5 μg/ml leupeptin, 2 μg/ml aprotinin and 1mM Na3VO4). Protein lysates were cleared by centrifuging at 13,800 rpm for 20 min at 4oC. Protein concentrations were determined using the Bicinchoninic Acid (BCA) protein assay protocol from Pierce (Rockford, Illinois).
Protein lysates (25 µg) were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose membrane (Amersham Biosciences, UK). Membranes were briefly stained with Ponceau S (containing Ponceau S, trichloroacetic acid, and sulfosalicylic acid) to check equal protein loading. Membranes were then blocked with 5% milk for 1 hr and the TTC9A protein was detected using an anti-mouse Ttc9a polyclonal antibody which was raised in-house [16]. On the following day the blots were washed and incubated with horseradish peroxidase-conjugated secondary antibody (anti-mouse IgG) diluted in 5% milk at 1:2000 for 2-3 hr. Western blots were visualized using the enhanced chemiluminescence, ECL plus system (Amersham Biosciences Inc). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as internal control. GAPDH was detected using a mouse monoclonal antibody (Ambion, Austin, TX, USA).
Whole-mount analysis of mammary glands
Number 4 inguinal mammary glands were dissected from mice, spread onto glass slides, fixed in 6:3:1 mixture of ethanol:chloroform:glacial acetic acid fixative, hydrated, and stained overnight in 0.2% carmine (Sigma-Aldrich, St. Louis, MO, USA) and 0.5% aluminium potassium sulphate. After overnight staining the slides were dehydrated in graded ethanol series, cleared in Xylene and mounted with DPX mounting media (Merck, Germany).
Mammary gland morphometric analysis
The number of mammary ductal branch points, ductal outgrowth length and number of terminal end buds (TEBs) were determined from mammary whole mount images taken under bright field microscope using SteREOLumar, a V12 microscope with Axiocam Zeiss MRc camera and AxioVision Release 4.6.3 software (Carl Zeiss AG, Germany). Morphometric analysis was performed using Image J 1.44 software. To determine the number of branch points, grids were overlaid on images and all resolvable branch points were counted at the leading edge of advancing ducts. The number of branch points was then averaged to yield the mean number in each grid. Mammary ductal length was measured as the length from the primary duct to the leading edge of the duct. The number of TEBs was determined by counting the number of TEBs with areas ≥ 0.03 mm2 at the leading edge of advancing ducts. Only TEBs with areas ≥ 0.03 mm2 and having distinct bulbous feature were counted as TEB structures. This is to differentiate TEBs from terminal end ducts [23].
Steroid hormone treatment of mice
Three-week-old female Ttc9a+/+ and Ttc9a-/- mice were ovariectomized following anesthesia using a combination of Ketamine and Xylazine. Animals were allowed to rest for one week after surgery before treatment. The mice were then divided into 3 groups. Group one mice were injected with sesame oil, group two injected with 17-β-Estradiol benzoate at 10 µg/kg dosage (E2) and group three injected with 17-β-Estradiol benzoate (10 µg/kg) plus progesterone 10 mg/kg (E2P) (Sigma Aldrich, USA). All injections were given subcutaneously on alternate days. After 16 days, the treated mice were sacrificed; mammary glands collected and snap frozen in liquid nitrogen for protein and RNA study. Additionally fresh tissues of the inguinal no. 4 mammary glands were collected for whole-mount analysis to study mammary ductal development.
Cell culture and luciferase reporter assay
MCF-7 cells and COS7 cells were routinely maintained in phenol red-containing Dulbecco's modified Eagle's medium (DMEM) (PAA Laboratories Ltd., Somerset, UK) supplemented with 7.5% fetal bovine serum (FBS) (Sigma-Aldrich) and 2 mM L-glutamine (PAA Laboratories Ltd). Cells were plated in phenol red-free DMEM supplemented with 5% dextran-coated charcoal-treated FBS (DCC-FBS) for 48 hr before hormone treatment to remove the residual effect of hormones from serum.
For luciferase reporter assay, MCF-7 cells in 6-well plates were transfected with 0.5 μg of ERE-luciferase, 0.3 ng of Renillar-luciferase, 25 ng of FLAG-TTC9A plasmids or 50 ng of FLAG-FKBP plasmids using Polyethylenimine (PEI) (Polysciences, Warrington, PA, USA) according to the CELLnTEC advanced cell systems transfection protocol for PEI. Twenty four hour after transfection, cells were treated with 10 nM 17β-estradiol (E2) for 24 hr. To test the effect of TTC9A knockdown on estrogen-induced ERα activity, MCF-7 cells were transfected with control or TTC9A specific siRNA (Ambion) with Lipofectamine 2000 (Invitrogen) for 24 hr before transfected with 0.5 μg of ERE-luciferase, 0.3 ng of Renilla luciferase vector for an additional 24 hr, followed by hormone treatment for 24 hr. Lysate was collected and analyzed using Promega Luciferase assay kit (Promega, Madison, WI, USA). Firefly-luciferase signals were detected using GloMax®-Multi Luminescence Detection System (Promega) and normalized against Renilla signal.
Co-immunoprecipitation (Co-IP)
For Co-IP of GFP-TTC9A with FLAG-FKBP38 and FLAG-FKBP51, COS7 cells were transfected with GFP-TTC9A with or without FLAG-FKBP38 (or FLAG-FKBP51) for 48 hr before total cell lysate were isolated using cold lysis buffer at 4oC. Similarly, ERα and FLAG-FKBP38 (or FLAG-FKBP51) were co-transfected in COS-7 cells to test their interaction. For Co-IP of FLAG-TTC9A with endogenous FKBP51, HeLa cells were transfected with FLAG-TTC9A and then treated with 10 nM dexamethasone for 24 hr to induce the expression of FKBP51. 500 μg of total cell lysates were incubated with 1 μg of anti-FLAG antibody plus protein A/G agarose beads (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Immunoprecipitated proteins were analyzed by Western blotting. Antibodies used in the Co-IP are anti-FLAG (F1804, Sigma-Aldrich), anti-GFP (G1544, Sigma-Aldrich). Anti-ERα (sc-002), anti-Hsp70 (sc-24) and anti-Hsp90 (sc-13119) were purchased from Santa Cruz Biotechnology. The in-house anti-FKBP51 has been described previously [24].
Statistical analysis
Statistical analysis of the animal studies of two experimental groups was evaluated using the Mann Whitney non-parametric analysis. For analysing luciferase results students T-test was used. The analysis was conducted using SPSS software. Results are expressed as mean ± standard error of the mean (SEM) and p < 0.05 was assigned as significant.
Results
Generation and characterization of Ttc9a gene deletion
To produce a functional deletion of the Ttc9a gene by homologous recombination, exon 1 of the Ttc9a gene was replaced by neomycin-resistant (neo) gene that is flanked by a 2 kb 5' homologous arm 1 (HR1) and a 14 kb 3' homologous arm 2 (HR2) as shown in Figure 1A. The neo cassette is flanked by loxp sites for cre-recombinase, but it is not excised for studies reported in this article. Southern blotting analysis of Sca I digested DNA with a 5' probe of 919 bp DNA identified ES cells with heterologous integration of the target vector at the desired genetic locus. The wild type DNA showed an 11 Kb DNA fragment whereas the DNA with neo cassette replacing exon 1 showed a 4 Kb fragment with the 5' probe. The insertion of the neo cassette introduced a new Sca I site. This new Sca I site leads to a smaller Sca I restriction fragment when analysed using the 5' probe by Southern blotting (Figure 1B, left panel). These mouse ES cells heterozygous for exon 1 deletion were used to generate chimeric males which carried the exon 1 deletion through the germ line. Mice heterozygous for the Ttc9a knockout were bred to produce mice of genotypes Ttc9a+/+ (Wild), Ttc9a+/- (Het) and Ttc9a-/- (Homo), respectively (Figure 1B, right panel). The genotypes were also confirmed by PCR screening (Figure 1C) and by Western blot analysis (Figure 1D) of the TTC9A protein expression in different tissues (brain, mammary gland, lung and uterus). It is to be noted that the mammary gland does express significant amount of TTC9A as was previously reported [15] although the band in this blot of Ttc9a+/+ sample is faint. In summary, Ttc9a exon 1 deletion successfully disrupted the Ttc9a gene, resulting in the loss of TTC9A protein expression (Figure 1D).
Ttc9a knockout mice appear healthy and fertile
We next determined the consequence of Ttc9a deficiency. Both Ttc9a+/- (Het) and Ttc9a-/- (Homo) mice were viable and reproductively normal. The genotype frequency of Ttc9a+/+, Ttc9a+/- and Ttc9a-/- followed the Mendelian inheritance ratio of 1:2:1 (Supplementary Material: Table S1), suggesting the absence of embryonic lethality. Although there seems to be greater male-female ratio in Ttc9a-/- mice than the Ttc9a+/+ mice, this difference however is not significant (Supplementary Material: Table S2). The body weights of 2 day old mice seem to be greater in Ttc9a-/- mice than Ttc9a+/+, in both the male (p=0.01) and female (p<0.05) group (Figure 2A). However, this difference is not observed in adult male mice of 6 - 12 weeks old (Figure 2B). On the other hand, the body weights of female Ttc9a-/- mice were 5 - 10% higher than Ttc9a+/+, mice at all stages analyzed albeit only significantly higher at 6-week and 10-week old mice (Figure 2C). The data therefore suggests that Ttc9a knockout affected the energy homeostasis in females but had little effect in males. It is plausible that Ttc9a is involved in the action of female steroid hormones estrogen and progesterone. This notion is supported by the observation that the new-born Ttc9a-/- mice (2-day old) of both sexes were heavier than Ttc9a+/+ mice as the new-born mice have been exposed to high levels of estrogen and progesterone while in mother's womb.
Figure 1
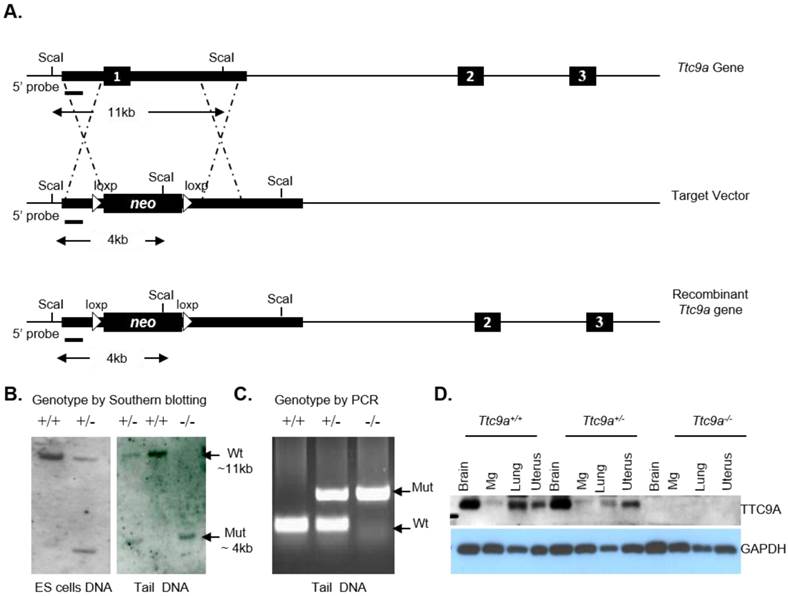
Ttc9a transgene construct and screening strategy. (A) Strategy for targeted disruption of Ttc9a gene. Ttc9a exon1 was replaced by neomycin cassette by homologous recombination as indicated by the dotted lines in the mouse ES cells. Exons are indicated by filled boxes; homology arms are indicated by thick lines; introns are indicated by thin lines; and loxp sites are indicated by triangles. (B) Southern blot analysis of Sca I-digested ES cell and tail genomic DNA from wild (+/+), heterozygote (+/-) and homozygous (-/-) Ttc9a KO mice. 5' probe detect a ~11 kb wild-type (Wt) and a ~4 kb mutant (Mut) band. (C) Genotyping tail DNA by PCR screening. The wild-type allele yielded a 200 bp band size and the mutant allele gave 654 bp. (D) TTC9A protein expression is abolished in Ttc9a-/- mice. Total protein extract from mice tissues (brain, mammary gland (Mg), lung and uterus) were analysed by immunoblotting with TTC9A specific antibody. GAPDH was used as a loading control. The blot shows complete absence of TTC9A protein in the 4 tissues assayed from Ttc9a-/- mice.
Figure 2
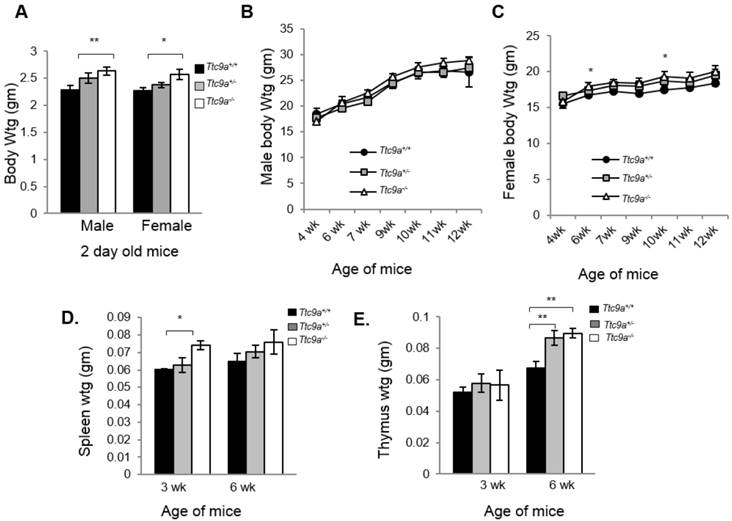
Ttc9a KO mice show greater body, spleen and thymus mass than wild-type mice. (A) Both male and female Ttc9a-/- mice at postnatal day 2 showed greater body weight than Ttc9a+/+ mice. (B) Body weights of male mice from 4-12 week old were similar across Ttc9a+/+, Ttc9a+/- and Ttc9a-/- mice. (C) The body weights of Ttc9a-/- female mice was significantly higher than Ttc9a+/+ mice at 6- and 10-week of age. (D) The spleen and (E) thymus mass in the Ttc9a-/-mice were significantly greater compared to Ttc9a+/+ mice. Animal and organ weight data are expressed as means ± SEM; *p value < 0.05; **p value < 0.01. Supplement data indicates the number of animals used in each analysis.
Significant increase in spleen and thymus weights in Ttc9a null mice
Since Ttc9a is a downstream target of both estrogen and progesterone and Ttc9a knockout resulted in greater body weight in female mice, we next focused on characterizing the effect of Ttc9a knockout in female mice. First, we evaluated the organ weights of the sexually immature 3-week old and then the mature 6-week old mice (Supplementary Material: Table S3). There is no difference in the weight of most organs among different Ttc9a genotypes except for the spleen and thymus. Spleen weight is greater in Ttc9a-/- mice than Ttc9a+/+, and Ttc9a+/- mice at both 3-week and 6-week old but the difference is statistically significant (p<0.05) only for 3-week old mice (Figure 2D). We also observed a significant increase of the thymus weight in Ttc9a-/- mice compared to wild-type mice at 6-week old (p <0.01). Furthermore we observed the increment in thymus mass from 3-week to 6-week was greater in the Ttc9a-/-and Ttc9a+/-mice (58.5% and 49.7%, respectively) compared to Ttc9a+/+ mice (29.62%). This implies that the lack of TTC9A enabled better pubertal thymus development. Since TTC9A protein is expressed at high levels in these two organs in adult mice [15], bigger spleen and thymus in Ttc9a-/- mice suggest that Ttc9a plays a role in regulating these organs' homeostasis.
Increased mammary ductal branching in Ttc9a null mice
Mammary TTC9A expression is up-regulated by estrogen and progesterone [15], both of which are critical for mammary development [25]. To determine the contribution of Ttc9a in mammary gland development, we analyzed the effect of Ttc9a gene inactivation on the mammary development in the pre-pubertal (3-week old) and post-pubertal (6-week old) mice. There was no difference in the rudimentary ductal tree at 3 weeks among the Ttc9a+/+, Ttc9a+/-, and Ttc9a-/- mice (data not shown). At 6 weeks old, however, mammary gland from Ttc9a-/- mice appears to have more ductal side branching and terminal end buds (Figure 3A and 3B). Quantitative analysis revealed that there is significantly more (p<0.05) mammary ductal branching in 6-week old Ttc9a-/- mice compared with Ttc9a+/+ mice (Figure 3C). There is also more mammary terminal end buds (TEB) in both the Ttc9a-/- and Ttc9a-+/- than Ttc9a+/+ mice but the difference is not significant (0.1>p>0.05) (Figure 3E). Unexpectedly, there was no significant difference in the mammary ductal lengths among mice with different Ttc9a genotypes (Figure 3D). We speculate that the lack of effect of Ttc9a knockdown on mammary ductal growth in 6-weeks old mice may be the result of compensatory effect over time by other factors such as the members of the Ttc9a family, Ttc9b and Ttc9c.
Ttc9a-/- mice is more sensitive to ovarian steroid hormone treatment
The observation that the Ttc9a-/- mice exhibit greater mammary side branching and a trend for more TEB formation in the sexually mature 6-week mice suggests that the mammary gland in Ttc9a-/- mice could be more sensitive to estrogen and progesterone for their stimulation on mammary side branching and TEB formation [26, 27]. We tested this postulation in ovariectomized mice treated with control vehicle, estradiol benzoate (E2), or E2 and progesterone (E2P) for 16 days. Interestingly, Ttc9a-/- mammary gland responded to E2P treatment, but not E2 alone, with greater mammary ductal branching than Ttc9a+/+ gland (Figure 4A, 4B, 4C and 4D) (p<0.05). This is consistent with the essential roles of P in mammary ductal branching in the presence of E2, which is responsible for the induction of PR expression [28, 29]. On the other hand, there was no significant difference in ductal branching between Ttc9a+/+ and Ttc9a-/- gland in untreated controls (Figure 4D). These mice were ovariectomized. Since the expression of progesterone receptor is largely estrogen-dependent, the lack of estrogen due to ovariectomy would mean the lack of progesterone receptor to stimulate ductal side branching in both Ttc9a+/+ mice and Ttc9a-/- mice. It is useful to note at this point that Ttc9a knockout did not affect PR gene expression in the absence of E2 (Figure 4G). Hence there would not be a difference in unliganded PR activity between Ttc9a+/+ and Ttc9a-/- mammary gland.
In accordance with the effect of E2 on mammary ductal lengthening [30], Ttc9a-/-mammary gland also exhibited significantly (p<0.01) greater ductal lengthening than Ttc9a+/+ mammary gland in response to E2 treatment (Figure 4E). The enhanced response of Ttc9a-/- mammary gland to estrogen was also associated with increase of E2-induced gene expression (Figure 4). Estrogen up-regulated the expression of its target genes progesterone receptor (PR) and amphiregulin (Areg) in Ttc9a-/- mammary gland to a significantly (p<0.05) greater extent than the Ttc9a+/+ mammary gland (Figure 4G, 4H). E2 treatment also enhanced greater increase in the expression of other two ER target genes Greb1 and Muc1 in Ttc9a-/- mammary gland but the difference was not statistically significant. Taken together, the data support the postulation that Ttc9a-/- mammary gland is more sensitive to the effect of E2 and P. Accordingly, Ttc9a may negatively regulate mammary response to estrogen.
Figure 3
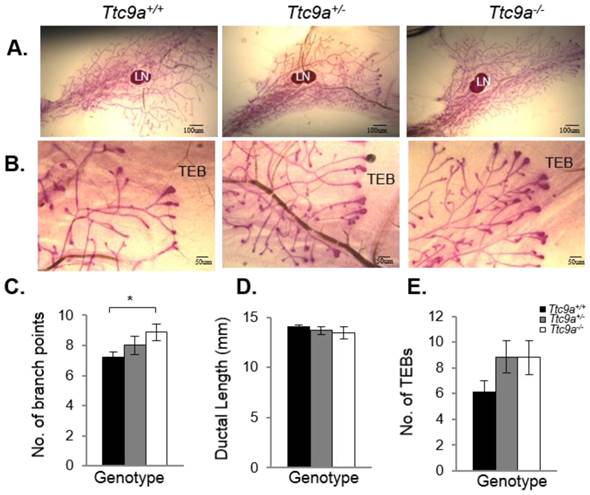
Ttc9a KO mice exhibit increased mammary ductal branching. Whole-mount images of inguinal mammary gland from Ttc9a+/+, Ttc9a+/- and Ttc9a-/- mice at 6 week old at (A) 4X magnification and (B) 10X magnification. Morphometric analysis using Image J quantitated the ductal development in the three genotypes. There was significant increase in the (C) ductal branch points in Ttc9a-/- mice compared to Ttc9a+/+, but no difference in the (D) ductal length. (E) Ttc9a-/- and Ttc9a+/-mammary gland showed an increase of TEBs compared to Ttc9a+/ but this difference was not statistically significant. LN - lymph node; TEB - terminal end bud. Data are expressed as means ± SEM; * p < 0.05.Supplement data indicates the number of animals used in each analysis.
Figure 4
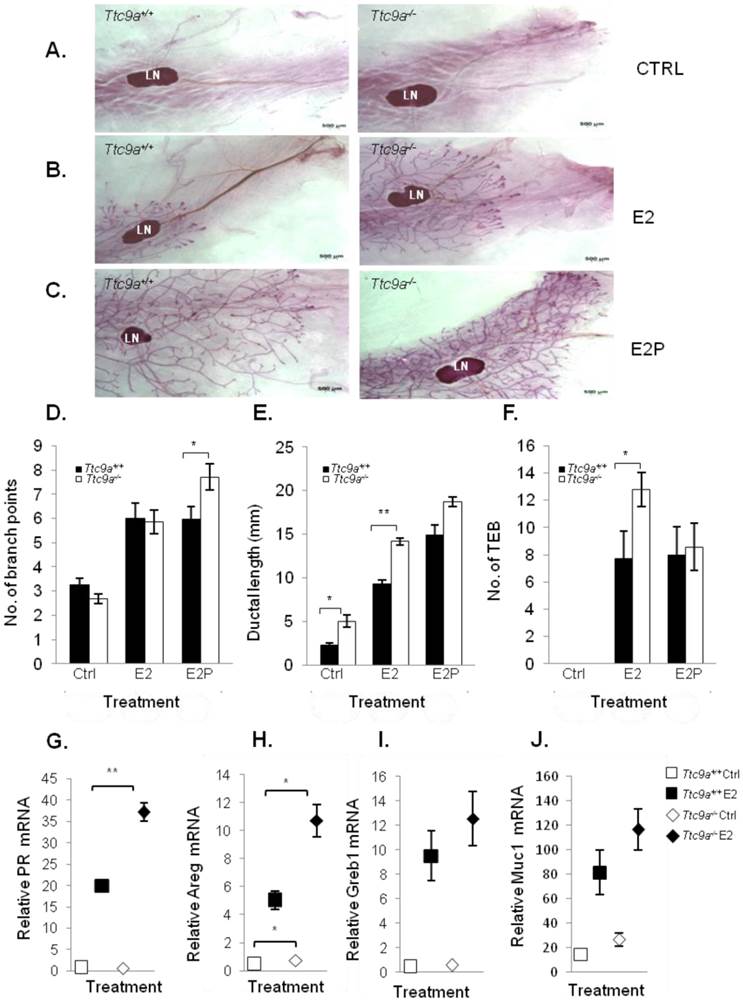
The mammary glands from Ttc9a KO mice are more responsive to estradiol. Representative whole mount images of the 4th inguinal mammary gland of (A) control (ctrl) (B) 17β estradiol benzoate (E2) and (C) 17β estradiol benzoate and progesterone (E2P) treated Ttc9a+/+and Ttc9a-/- mice. The mammary ductal morphology between the Ttc9a+/+ and Ttc9a-/- gland was quantified using the Image J software. The histograms illustrated significant increases in (D) ductal branch points and (E) ductal length in Ttc9a-/- mice compared to Ttc9a+/+ on E2P and E2 treatment respectively. Also there was a significant increase in the (F) TEB count in Ttc9a-/- gland on E2 treatment compared to Ttc9a+/+. This increased ductal morphology were associated with greater induction of E2 target genes (PR, Areg, Greb1 and Muc1) in Ttc9a-/- mice than Ttc9a+/+ mice as analyzed by real time PCR (G-J). Data are expressed as means ± SEM; for each sample set. *p<0.05, **p< 0.01.Supplement data indicates the number of animals used in each analysis.
Intriguingly, Ttc9a-/- mice treated with control vehicle (Figure 4E) also had greater ductal lengthening than the corresponding Ttc9a+/+ controls (p<0.01). This is associated with increases of the gene expression of Areg (p=0.046) and Muc1 (p=0.075) in Ttc9a-/- mice (Figure 4H, 4J), both of which are important for mammary growth [31] [32]. Since these are ovariectomized mice without ovarian steroid hormone, the observation suggests that TTC9A also negatively regulates mammary ductal lengthening via an estrogen-independent pathway.
TTC9A down-regulates the transcriptional activity of ERα
Since mouse studies showed that Ttc9a may be a negative regulator of ERα, we examined the effect of TTC9A on the transcriptional activity of ERα in vitro using human breast cancer cells MCF-7. In agreement with the in vivo observation, TTC9A over-expression suppressed the transcriptional activity of ERα in an E2-dependent manner (Figure 5A). While the basal activity remained similar with and without TTC9A over-expression, TTC9A over-expression caused a small (11%) but significant (p<0.01) reduction in ligand induced ERα transcriptional activity. Western blotting analysis shows that endogenous ERα protein level was not affected by TTC9A over-expression (Figure 5A), confirming that the observed change was due to TTC9A over-expression. The negative effect of TTC9A on ERα activity is further supported by TTC9A gene silencing study. Figure 5B shows that transfection with TTC9A siRNA1 and siRNA2 significantly up-regulated E2-induced transcriptional activity. In addition, TTC9A siRNA also increased E2-independent activity but this effect is only statistically significant (p<0.05) in one of the two siRNA transfected cells.
TTC9A contains 3 TPR domains and shares high level of homology with co-chaperones FKBP38 and FKBP51 [16, 33]. We investigated if TTC9A exerts its inhibitory effect on ERα as a cochaperone protein. As there is little information on the involvement of FKBP38 and FKBP51 in ERα activity, we first tested the effect of FKBP38 and FKBP51 over-expression on ERα activity in ERE-Luc assay. Figure 5C shows that FKBP38 over-expression in MCF-7 cells significantly inhibited both E2-induced and E2-independent ERα activity. In contrast, FKBP51 over-expression promoted ERα activity in the presence and absence of E2, which is consistent with the previous report [34].
We next tested if TTC9A may regulate ERα activity as a part of ER-chaperone complex. Interestingly, both FKBP38 and FKBP51 interacted with ERα and Hsp90 (Figure 5D), although they exert opposite effect on ERα activity. This suggests that FKBP38 and FKBP51 are part of ERα-chaperone complex to modulate ERα activity. We next investigated if TTC9A exerts its inhibitory effect through physical interaction with this complex. Although TTC9A did not interact with ERα or Hsp90 (Figure 5E), it binds to FKBP38 and FKBP51 strongly as both FKBP38 and FKBP51 could pull-down TTC9A very well (Figure 5F). At the same time FKBP38 and FKBP51 also pulled-down endogenous Hsp90. TTC9A interaction with FKBP51 is further supported by FLAG-TTC9A co-immunoprecipitation with endogenous FKBP51 (Figure 5G). In this experiment, Hela cells were treated with dexamethasone to induce the endogenous FKBP51 [35, 36]. It is clear that FLAG-TTC9A pulled-down endogenous FKBP51 following dexamethasone induction. In contrast, the control samples (cells transfected with control vector, or cells transfected with FLAG-TTC9A but without dexamethasone treatment) showed no or little FKBP51 pull-down. It is plausible therefore that TTC9A could affect ERα-chaperone complex assembly or maturation through interacting with co-chaperone proteins FKBP51 and FKBP38, both of which are associated with ERα and Hsp90 complex. Since FKBP38 and FKBP51 have opposite effect on ERα activity in ERE-Luc assay, interaction of TTC9A with these two proteins could balance their influence on ERα with a net inhibitory outcome.
Discussion
TTC9A is expressed in embryonic stem cells [15]. It is also ubiquitously expressed in all animal organs after birth except for the liver which expresses distinctively low levels of TTC9A [15, 16]. It is surprising that a complete ablation of TTC9A expression in mice did not result in any significant developmental abnormality or dysfunction. Ttc9a-/- mice exhibit normal growth and reproductive function. In fact, Ttc9a-/- female mice shows better development with greater body weight, bigger spleen and thymus. Importantly, the study indicates that the absence of Ttc9a permits better mammary development and greater mammary sensitivity to estrogen. Ttc9a-/- mice of 6-week old, but not of the sexually immature 3-week old mice, have greater number of mammary ductal branching and TEBs. Furthermore, Ttc9a-/- mice responded to estrogen treatment with a greater ductal lengthening, and to estrogen plus progesterone with an increase in ductal branching than Ttc9a+/+ mice. This heightened hormone responsiveness of Ttc9a-/- mice were further illustrated at the gene expression level where estrogen up-regulated the expression of its target genes Areg and PR to a greater extent in Ttc9a-/- mammary gland. Areg is the major paracrine mediator of estrogen-stimulated ductal development [37, 38]. Greater up-regulation of Areg could favor the greater mammary ductal elongation observed in the Ttc9a-/- mammary gland. Similarly, greater stimulation of PR expression by estrogen would sensitize mammary gland to the effect of progesterone, resulting in enhanced mammary ductal side-branching that was observed in Ttc9a-/- mice. These data supports the view that Ttc9a is negatively involved in the regulation of ERα activity in the mammary gland.
Figure 5
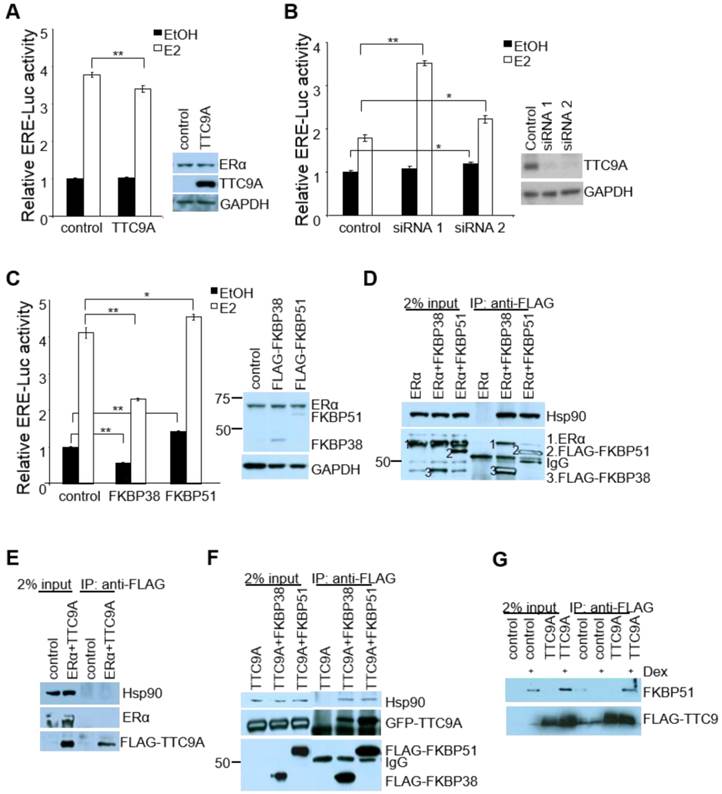
TTC9A down-regulates ERα transcriptional activity in breast cancer cells. (A, B) ERE-luciferase assay showing a significant decrease in ERα transcriptional activity by TTC9A (25ng) and up-regulation in ERα transcriptional activity with TTC9A siRNA knockdown in MCF-7. Western blot showing that endogenous ERα expression level was not affected by TTC9A over-expression, and TTC9A protein level was drastically reduced by knockdown. Result shown (mean±SEM (n = 3) is representative of two independent experiments. The p values were obtained by Student's t-test. **p<0.01. (C) ERE-luciferase assay showing the effects of FKBP38 and FKBP51 (50ng) on ERα's transcriptional activity in MCF-7 cells. Western blot showing that endogenous ERα expression level was not affected by FKBP over-expression. Luciferase reporter assay result (mean±SEM (n = 3) shown is representative of two independent experiments *p<0.05, **p<0.01. (D) FKBP38 and FKBP51 interact with ERα and Hsp90. COS7 cells were transfected with ERα alone or together with FLAG-FKBP38 or FLAG-FKBP51. Anti-FLAG antibody pulled down both FKBPs and co-immunoprecipitated both ERα and Hsp90. ERα and Hsp90 were detected using anti-ERα and anti-Hsp90 antibodies respectively. FKBP38 and 51 were detected by anti-FLAG antibody. (E) TTC9A does not interact with ERα and Hsp90. COS7 cells were transfected with pXJ-FLAG control vector with ERα or FLAG-TTC9 with ERα. Anti-FLAG antibody pulled down FLAG-TTC9, but no ERα or endogenous Hsp90 proteins were co-immunoprecipitated. (F) TTC9A interacts with FKBP38 and FKBP51. COS7 cells were transfected with GFP-TTC9A and FLAG-FKBP38 or FLAG-FKBP51. Anti-FLAG antibody pulled down both FKBPs and co-immunoprecipitated TTC9A and Hsp90 at the same time. (G) TTC9A interacts with endogenous FKBP51. HeLa cells were transfected with FLAG-TTC9A or control vector and treated with 10 nM dexamethasone to induce the endogenous FKBP51. Anti-FLAG antibody was used to pull-down FLAG-TTC9A in the whole cell lysates and the co-immunoprecipitated FKBP51 was detected by anti-FKBP51 antibody.
The ovariectomized Ttc9a-/- mice treated with control vehicle also had significantly more mammary ductal lengthening (Figure 4E), suggesting that TTC9A negatively regulates mammary ductal growth in an estrogen-independent manner. Since Areg is critical for mammary ductal growth, the associated up-regulation of Areg in Ttc9a-/- mice (Figure 4H) may be involved in the estrogen-independent mammary ductal growth. It has been reported that unliganded ERα promotes Areg gene expression in ERα-positive breast cancer cells MCF-7 and [39]. It is plausible that TTC9A also negatively regulates the activity of unliganded ERα on induction of Areg in mouse mammary gland. This notion is supported by the evidence that TTC9A knockdown by siRNA in MCF-7 cells increased ERE-Luc activities in the absence of E2 (Figure 5B).
The notion that Ttc9a negatively regulates ERα activity is also supported by the observation that Ttc9a gene deletion permits better puberty-associated thymus development in female mice (Figure 2E). Estrogen is required for thymus development and ERα knockout mice displayed thymus atrophy [40-42]. The increase in thymus weight observed in the Ttc9a knockout mice may also be related to greater ERα activity.
How Ttc9a is potentially involved in mammary sensitivity to hormonal stimulation is at present a matter of speculation. TTC9A is a TPR-containing protein. In addition to its homology with TTC9B and TTC9C, TTC9A shares significant homology at its TPR1 or TPR2 domains to the TPR domain containing proteins cyclophillin-40, FKBP38, FKBP52, WAF1/CIP1 stabilizing protein 39 (WISp39) and FKBP51 in order of significance in homology. All except for WISp39 are known to be co-chaperone proteins for steroid hormone receptors and compete for binding of a common site on Hsp90 [5, 11]. These co-chaperones participate in hormone-receptor complex assembly, maturation and hence play a critical role in regulating cellular response to hormones. It is also known that steroid hormone receptors have an order of preference to different co-chaperones for their complex assembly and activation [5]. Some of the chaperones facilitate hormone receptor activation whereas others confine their activities. For example, FKBP51 is inhibitory whereas FKBP52 is stimulatory on the activity of glucocorticoid receptor; together the chaperones fine-tune the glucocorticoid response [43-45]. Another example is protein phosphate 5 (PP5), a well-known TPR-domain containing protein which negatively regulates ERα-mediated gene activation and ERα phosphorylation at S118 [46]. We have provided evidence to suggest that TTC9A function to regulate ERα activity through interacting with ERα co-chaperone proteins FKBP38 and FKBP51. Although TTC9A does not appear to interact with ERα-Hsp90 complex directly, it binds very well to both FKBP38 and FKBP51, both of which are associated with ERα and Hsp90. Thus, TTC9A may modulate the binding of FKBP38 and FKBP51 with ERα complex to regulate ERα activity. The means of this modulation may be cell/tissue-context dependent. In MCF-7 cells, TTC9A over-expression exerted a small but significant inhibitory effect on ERα activity. In contrast, FKBP38 inhibited markedly ERα activity, whereas FKBP51 promoted it. It is conceivable that the outcome of this regulation under physiological conditions would depend on the relative expression of various chaperones and co-chaperones. In mouse mammary gland, TTC9A appears to exert negative control over ERα activity since TTC9A gene knockout led to increased ERα activity and better mammary development.
To our knowledge, this is the first observation that TPR-containing proteins interact with each other in mammalian cells. TTC9A was pulled-down by both FKBP38 and FKBP51 in large amount although it is not clear if the binding involves TPR domains. The interaction is likely direct because TTC9A does not interact with Hsp90, which was reported to use its C-terminal EEVD motif to bind co-chaperones harboring a TPR domain [47, 48].
Since TTC9A interacted well with FKBP38 or FKBP51, why was it not present in ERα complex with FKBP38 or FKBP51? We can offer the following explanations. First, TTC9A is in the ERα-Hsp90 complex via interaction with FKBP38 or FKBP51 but TTC9A in the complex, in relation to ERα is tertiary or quaternary and hence the interaction is weak and cannot survive the assay condition. Second, the formation of TTC9A-Hsp90-FKBPs complex could precede its association with ERα and the presence of TTC9A may influence the dynamics of ERα-Hsp90-FKBPs complex formation and hence ERα activity.
Estrogen is known to stimulate the development of breast cancer. The negative regulation of estrogen activity by TTC9A in mice would suggest that TTC9A could inhibit estrogen stimulation of breast cancer development. However, the regulation of some aspects of TTC9A function may be disrupted in breast cancer. For example, estrogen induced TTC9A protein levels in mice mammary gland but suppressed it in breast cancer cells MCF-7 [15, 16]. Furthermore, TTC9A was found to be over-expressed in breast cancer but this over-expression was not associated with ERα status or response to endocrine therapy [16]. It is plausible that breast cancer cells may have developed a mechanism in estrogen action pathway that could evade some aspects of the inhibitory effect of TTC9A.
It is also important to note that the changes in mammary development or in thymus and spleen weight in Ttc9a knockdown mice are generally modest. It is possible that other members (Ttc9b and Ttc9c) of the TTC9 family have functional overlap with Ttc9a such that the loss of Ttc9a function is offset by Ttc9b or Ttc9c. To our knowledge, there has been no report on the study of Ttc9b and Ttc9c function, except for a recent report on the association between Ttc9b gene hypermethylation and postpartum depression in women [49]. However, since the protein sequence of the TTC9 family is conserved at the regions corresponding to the 3 predicted TPR domains of Ttc9a (Figure 6), it is reasonable to speculate that the TTC9 proteins may have functional overlap at the level of protein interaction. Whether the loss of Ttc9a function in the Ttc9a knockout mice is partly compensated by Ttc9b or Ttc9c requires further investigation using single or double gene knockout approach.
In summary, this study reports for the first time the generation of Ttc9a knockout mice. Ttc9a deficiency is associated with greater body weight, bigger thymus and better mammary development in post-pubertal female mice. Ttc9a-deficient mammary gland was also more responsive to estrogen with greater mammary ductal lengthening, ductal branching and estrogen target gene induction. Since Ttc9a is induced by estrogen in estrogen target tissues [15], these results suggest that Ttc9a is a negative regulator of estrogen function through a negative feedback mechanism. In vitro studies also showed that TTC9A over-expression suppresses the transcriptional activity of ERα in an E2 dependent manner. In addition, TTC9A interacts with FKBP38 and FKBP51 that are associated with ERα-Hsp90 complex and modulate ERα transcriptional activity. We propose that TTC9A may regulate ERα-chaperone complex formation and dynamics through interaction with FKBP proteins. In light of the pivotal involvement of estrogen in breast cancer etiology, further understanding of the mechanisms by which TTC9A interact with estrogen signaling in breast cancer may have important therapeutic implications.
Figure 6
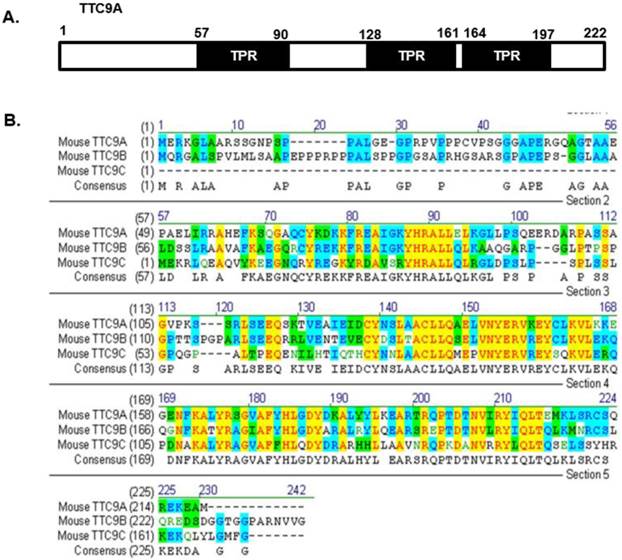
The protein sequence of the Ttc9 family is conserved at the regions corresponding to the TPR domains of Ttc9a. (A) The TTC9A protein comprises of three TPR domains. (B) Alignment of the mouse TTC9A (222 aa), TTC9B (239 aa) and TTC9C (171 aa) amino acids using Vector NTI software. Yellow - Identical amino acid residues; Green - residues of similar properties; Blue - conserved residues across species.
Supplementary Material
Tables S1 - S4.
Acknowledgements
This study was supported by a grant (BMRC 06/1/22/19/455) from the Biomedical research Council of Singapore. MBC has research grant support from NIH/NIMHD/RCMI grant (G12MD007592) and State of Texas Cancer Prevention and Research Institute grant (RP110444-P2). We would like to thank Ms Geraldine Leong and Ms Sivakamasundari V at the Genome Institute of Singapore for their assistance in the generation of Ttc9a knockout mice. We would also like to thank Dr. Chung Hwa Hwa from the School of Biological Sciences, Nanyang Technological University for helping with the surgical procedures.
Author Contributions
VL, SS, TL, SY and MBC conceived and designed the experiments; SS, SY, PK, FT, YAG, NG, and PR performed the experiments; VL, SS, SY, FT, MCB, YAG, NG, and PR analyzed the data; VL, SS, SY and TL wrote the paper. All authors have read and approved the final manuscript.
Competing interests
All authors declare that they have no competing financial, professional or personal interests that might have influenced the performance or presentation of the work described in this paper.
References
1. Nagase T, Seki N, Ishikawa K, Ohira M, Kawarabayasi Y, Ohara O. et al. Prediction of the coding sequences of unidentified human genes. VI. The coding sequences of 80 new genes (KIAA0201-KIAA0280) deduced by analysis of cDNA clones from cell line KG-1 and brain. DNA Res. 1996;3:321-9 41-54
2. Strausberg RL FE, Grouse LH, Derge JG, Klausner RD, Collins FS, Wagner L. et al. Generation and initial analysis of more than 15,000 full-length human and mouse cDNA sequences. Proc Natl Acad Sci U S A. 2002;99:16899-903
3. Allan RK, Ratajczak T. Versatile TPR domains accommodate different modes of target protein recognition and function. Cell stress & chaperones. 2011;16:353-67 doi:10.1007/s12192-010-0248-0
4. Storer CL, Dickey CA, Galigniana MD, Rein T, Cox MB. FKBP51 and FKBP52 in signaling and disease. Trends in endocrinology and metabolism: TEM. 2011;22:481-90 doi:10.1016/j.tem.2011.08.001
5. Smith DF, Toft DO. Minireview: the intersection of steroid receptors with molecular chaperones: observations and questions. Molecular endocrinology. 2008;22:2229-40 doi:10.1210/me.2008-0089
6. Harst A, Lin H, Obermann WM. Aha1 competes with Hop, p50 and p23 for binding to the molecular chaperone Hsp90 and contributes to kinase and hormone receptor activation. Biochem J. 2005;387:789-96
7. Riggs DL, Roberts PJ, Chirillo SC, Cheung-Flynn J, Prapapanich V, Ratajczak T. et al. The Hsp90-binding peptidylprolyl isomerase FKBP52 potentiates glucocorticoid signaling in vivo. EMBO J. 2003;22:1158-67 doi:10.1093/emboj/cdg108
8. Cao S, Ho GH, Lin VC. Tetratricopeptide repeat domain 9A is an interacting protein for tropomyosin Tm5NM-1. BMC Cancer. 2008;8:231. doi:1471-2407-8-231 [pii] 10.1186/1471-2407-8-231
9. Blatch GL, Lassle M. The tetratricopeptide repeat: a structural motif mediating protein-protein interactions. Bioessays. 1999;21:932-9
10. Lamb JR, Tugendreich S, Hieter P. Tetratrico peptide repeat interactions: to TPR or not to TPR? Trends Biochem Sci. 1995;20:257-9
11. Smith DF. Tetratricopeptide repeat cochaperones in steroid receptor complexes. Cell stress & chaperones. 2004;9:109-21
12. Hirano T, Kinoshita N, Morikawa K, Yanagida M. Snap helix with knob and hole: essential repeats in S. pombe nuclear protein nuc2+. Cell. 1990;60:319-28
13. Das AK, Cohen PW, Barford D. The structure of the tetratricopeptide repeats of protein phosphatase 5: implications for TPR-mediated protein-protein interactions. The EMBO journal. 1998;17:1192-9
14. Scheufler C, Brinker A, Bourenkov G, Pegoraro S, Moroder L, Bartunik H. et al. Structure of TPR domain-peptide complexes: critical elements in the assembly of the Hsp70-Hsp90 multichaperone machine. Cell. 2000;101:199-210
15. Shrestha S, Cao S, Lin VC. The local microenvironment instigates the regulation of mammary tetratricopeptide repeat domain 9A during lactation and involution through local regulation of the activity of estrogen receptor alpha. Biochemical and biophysical research communications. 2012;426:65-70 doi:10.1016/j.bbrc.2012.08.036
16. Cao S, Iyer JK, Lin V. Identification of tetratricopeptide repeat domain 9, a hormonally regulated protein. Biochemical and biophysical research communications. 2006;345:310-7
17. Lin VC, Ng EH, Aw SE, Tan MG, Chan VS, Ho GH. Progestins inhibit the growth of MDA-MB-231 cells transfected with progesterone receptor complementary DNA. Clin Cancer Res. 1999;5:395-403
18. Lin VC, Ng EH, Aw SE, Tan MG, Bay BH. Progesterone induces focal adhesion in breast cancer cells MDA-MB-231 transfected with progesterone receptor complementary DNA. Mol Endocrinol. 2000;14:348-58
19. Osoegawa K, Tateno M, Woon PY, Frengen E, Mammoser AG, Catanese JJ. et al. Bacterial artificial chromosome libraries for mouse sequencing and functional analysis. Genome Res. 2000;10:116-28
20. Angrand PO, Daigle N, van der Hoeven F, Scholer HR, Stewart AF. Simplified generation of targeting constructs using ET recombination. Nucleic Acids Res. 1999;27:e16. doi:gnc016 [pii]
21. Muyrers JP, Zhang Y, Testa G, Stewart AF. Rapid modification of bacterial artificial chromosomes by ET-recombination. Nucleic Acids Res. 1999;27:1555-7 doi:gkc282 [pii]
22. Kraus P, Leong G, Tan V, Xing X, Goh JW, Yap SP. et al. A more cost effective and rapid high percentage germ-line transmitting chimeric mouse generation procedure via microinjection of 2-cell, 4-cell, and 8-cell embryos with ES and iPS cells. Genesis. 2010;48:394-9 doi:10.1002/dvg.20627
23. Bonnette SG, Hadsell DL. Targeted disruption of the IGF-I receptor gene decreases cellular proliferation in mammary terminal end buds. Endocrinology. 2001;142:4937-45
24. Nair SC, Rimerman RA, Toran EJ, Chen S, Prapapanich V, Butts RN. et al. Molecular cloning of human FKBP51 and comparisons of immunophilin interactions with Hsp90 and progesterone receptor. Mol Cell Biol. 1997;17:594-603
25. Hennighausen L, Robinson GW. Think globally, act locally: the making of a mouse mammary gland. Genes Dev. 1998;12:449-55
26. Feng Y, Manka D, Wagner KU, Khan SA. Estrogen receptor-alpha expression in the mammary epithelium is required for ductal and alveolar morphogenesis in mice. Proc Natl Acad Sci U S A. 2007;104:14718-23 doi:0706933104 [pii] 10.1073/pnas.0706933104
27. Beleut M, Rajaram RD, Caikovski M, Ayyanan A, Germano D, Choi Y. et al. Two distinct mechanisms underlie progesterone-induced proliferation in the mammary gland. Proc Natl Acad Sci U S A. 2010;107:2989-94 doi:10.1073/pnas.0915148107
28. Fernandez-Valdivia R, Mukherjee A, Mulac-Jericevic B, Conneely OM, DeMayo FJ, Amato P. et al. Revealing progesterone's role in uterine and mammary gland biology: insights from the mouse. Semin Reprod Med. 2005;23:22-37 doi:10.1055/s-2005-864031
29. Fernandez-Valdivia R, Mukherjee A, Creighton CJ, Buser AC, DeMayo FJ, Edwards DP. et al. Transcriptional response of the murine mammary gland to acute progesterone exposure. Endocrinology. 2008;149:6236-50 doi:10.1210/en.2008-0768
30. Bocchinfuso WP, Korach KS. Mammary gland development and tumorigenesis in estrogen receptor knockout mice. Journal of mammary gland biology and neoplasia. 1997;2:323-34
31. Schroeder JA, Thompson MC, Gardner MM, Gendler SJ. Transgenic MUC1 interacts with epidermal growth factor receptor and correlates with mitogen-activated protein kinase activation in the mouse mammary gland. J Biol Chem. 2001;276:13057-64 doi:10.1074/jbc.M011248200
32. Nautiyal J, Steel JH, Mane MR, Oduwole O, Poliandri A, Alexi X. et al. The transcriptional co-factor RIP140 regulates mammary gland development by promoting the generation of key mitogenic signals. Development. 2013;140:1079-89 doi:10.1242/dev.085720
33. Pratt WB, Toft DO. Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr Rev. 1997;18:306-60
34. Ni L, Yang CS, Gioeli D, Frierson H, Toft DO, Paschal BM. FKBP51 promotes assembly of the Hsp90 chaperone complex and regulates androgen receptor signaling in prostate cancer cells. Molecular and cellular biology. 2010;30:1243-53 doi:10.1128/MCB.01891-08
35. Cox MB SD. Functions of the Hsp90-Binding FKBP Immunophilins. 2000.
36. Blatch GL. The Networking of Chaperones by Co-chaperones. 2007.
37. LaMarca HL, Rosen JM. Estrogen regulation of mammary gland development and breast cancer: amphiregulin takes center stage. Breast Cancer Res. 2007;9:304. doi:bcr1740 [pii] 10.1186/bcr1740
38. Ciarloni L, Mallepell S, Brisken C. Amphiregulin is an essential mediator of estrogen receptor alpha function in mammary gland development. Proc Natl Acad Sci U S A. 2007;104:5455-60 doi:10.1073/pnas.0611647104
39. Zhang X, Diaz MR, Yee D. Fulvestrant regulates epidermal growth factor (EGF) family ligands to activate EGF receptor (EGFR) signaling in breast cancer cells. Breast Cancer Res Treat. 2013;139:351-60 doi:10.1007/s10549-013-2541-y
40. Seiki K, Sakabe K. Sex hormones and the thymus in relation to thymocyte proliferation and maturation. Arch Histol Cytol. 1997;60:29-38
41. Staples JE, Gasiewicz TA, Fiore NC, Lubahn DB, Korach KS, Silverstone AE. Estrogen receptor alpha is necessary in thymic development and estradiol-induced thymic alterations. J Immunol. 1999;163:4168-74 doi:ji_v163n8p4168 [pii]
42. Erlandsson MC, Ohlsson C, Gustafsson JA, Carlsten H. Role of oestrogen receptors alpha and beta in immune organ development and in oestrogen-mediated effects on thymus. Immunology. 2001;103:17-25 doi:imm1212 [pii]
43. Reynolds PD, Ruan Y, Smith DF, Scammell JG. Glucocorticoid resistance in the squirrel monkey is associated with overexpression of the immunophilin FKBP51. J Clin Endocrinol Metab. 1999;84:663-9
44. Denny WB, Valentine DL, Reynolds PD, Smith DF, Scammell JG. Squirrel monkey immunophilin FKBP51 is a potent inhibitor of glucocorticoid receptor binding. Endocrinology. 2000;141:4107-13
45. Cheung-Flynn J, Roberts PJ, Riggs DL, Smith DF. C-terminal sequences outside the tetratricopeptide repeat domain of FKBP51 and FKBP52 cause differential binding to Hsp90. J Biol Chem. 2003;278:17388-94 doi:10.1074/jbc.M300955200 [pii]
46. Ikeda K, Ogawa S, Tsukui T, Horie-Inoue K, Ouchi Y, Kato S. et al. Protein phosphatase 5 is a negative regulator of estrogen receptor-mediated transcription. Molecular endocrinology. 2004;18:1131-43 doi:10.1210/me.2003-0308
47. Brinker A, Scheufler C, Von Der Mulbe F, Fleckenstein B, Herrmann C, Jung G. et al. Ligand discrimination by TPR domains. Relevance and selectivity of EEVD-recognition in Hsp70 x Hop x Hsp90 complexes. J Biol Chem. 2002;277:19265-75 doi:10.1074/jbc.M109002200 [pii]
48. Young JC, Moarefi I, Hartl FU. Hsp90: a specialized but essential protein-folding tool. J Cell Biol. 2001;154:267-73
49. Guintivano J, Arad M, Gould TD, Payne JL, Kaminsky ZA. Antenatal prediction of postpartum depression with blood DNA methylation biomarkers. Mol Psychiatry. 2014;19:560-7 doi:10.1038/mp.2013.62
Author contact
![]() Corresponding author: Dr Valerie Lin Chun Ling, Associate Professor, Division of Molecular Genetics & Cell Biology, School of Biological Sciences, Nanyang Technological University, Singapore637551. Email: cllinedu.sg Office phone: 65 - 6316 2843.
Corresponding author: Dr Valerie Lin Chun Ling, Associate Professor, Division of Molecular Genetics & Cell Biology, School of Biological Sciences, Nanyang Technological University, Singapore637551. Email: cllinedu.sg Office phone: 65 - 6316 2843.
Citation styles
APA
Shrestha, S., Sun, Y., Lufkin, T., Kraus, P., Or, Y., Garcia, Y.A., Guy, N., Ramos, P., Cox, M.B., Tay, F., Lin, V.CL. (2015). Tetratricopeptide Repeat Domain 9A Negatively Regulates Estrogen Receptor Alpha Activity. International Journal of Biological Sciences, 11(4), 434-447. https://doi.org/10.7150/ijbs.9311.
ACS
Shrestha, S.; Sun, Y.; Lufkin, T.; Kraus, P.; Or, Y.; Garcia, Y.A.; Guy, N.; Ramos, P.; Cox, M.B.; Tay, F.; Lin, V.CL. Tetratricopeptide Repeat Domain 9A Negatively Regulates Estrogen Receptor Alpha Activity. Int. J. Biol. Sci. 2015, 11 (4), 434-447. DOI: 10.7150/ijbs.9311.
NLM
Shrestha S, Sun Y, Lufkin T, Kraus P, Or Y, Garcia YA, Guy N, Ramos P, Cox MB, Tay F, Lin VCL. Tetratricopeptide Repeat Domain 9A Negatively Regulates Estrogen Receptor Alpha Activity. Int J Biol Sci 2015; 11(4):434-447. doi:10.7150/ijbs.9311. https://www.ijbs.com/v11p0434.htm
CSE
Shrestha S, Sun Y, Lufkin T, Kraus P, Or Y, Garcia YA, Guy N, Ramos P, Cox MB, Tay F, Lin VCL. 2015. Tetratricopeptide Repeat Domain 9A Negatively Regulates Estrogen Receptor Alpha Activity. Int J Biol Sci. 11(4):434-447.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.