Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2016; 12(1):30-41. doi:10.7150/ijbs.12310 This issue Cite
Research Paper
Hepatitis B Virus X Protein Upregulates hELG1/ ATAD5 Expression through E2F1 in Hepatocellular Carcinoma
Alip Ghosh1, Suchandrima Ghosh1, Debanjali Dasgupta1, Amit Ghosh1, Somenath Datta1, Nilabja Sikdar2, Simanti Datta1, Abhijit Chowdhury3, Soma Banerjee1 ![]()
1. Centre for Liver Research, School of Digestive and Liver Diseases, Institute of Post Graduate Medical Education and Research, Kolkata, India
2. Indian Statistical Institute, Kolkata, India
3. Department of Hepatology, School of Digestive and Liver Diseases, Institute of Post Graduate Medical Education and Research, Kolkata, India
Received 2015-4-2; Accepted 2015-10-4; Published 2016-1-1
Abstract

The precise mechanism by which HBx protein of hepatitis B virus (HBV) impacts on hepato-carcinogenesis remain largely elusive despite strong evidences for its' involvement in the process. Here, we have investigated the role of HBx on expression of a novel gene hELG1/ATAD5, which is required for genome maintenance and its' importance in hepatocarcinogenesis. This study has for the first time showed that the expression of this gene was significantly higher in human cancer such as HBV-associated hepatocellular carcinoma (HCC) and in different HCC cell lines compared to normal liver. In addition, a significant elevation in ATAD5 expression was also found in HBx transfected HCC cell lines implicating HBx mediated transcriptional regulation on ATAD5. Using different deletion mutant constructs of putative promoter, the active promoter region was first identified here and subsequently the regulatory region of HBx was mapped by promoter-luciferase assay. But ChIP assay with anti-HBx antibody revealed that HBx was not physically present in ATAD5 transcription machinery whereas anti-E2F1 antibody showed the presence of E2F1 in the complex. Luciferase assay with E2F1 binding site mutant had further confirmed it. Moreover, both loss-and gain-of-function studies of ATAD5 showed that ATAD5 could enhance HBV production in transfected cells whereas knock down of ATAD5 increased the sensitivity of HCC cell line to chemotherapeutics 5-fluorouracil. Overall, this data suggests that a positive feedback loop regulation between ATAD5 and HBV contributed to both viral replication and chemo-resistance of HCC cells.
Keywords: hepatitis B virus, hepatocellular carcinoma
Introduction
Perennial and varying amount of genomic instability is a ubiquitous feature of the process of carcinogenesis. The budding yeast ELG1 (enhanced level of genomic instability 1) gene, a major subunit-I of a novel replication factor C-like complex has been described as an essential component for maintenance of chromosomal integrity [1]. Function of this gene is conserved among higher eukaryotes as depletion of hELG1/ATAD5 (located in 17q11.2 loci) in human cell line [2] or mELG1/ATAD5 (located in 11qB5 loci) in mice [3] exhibits pleiotropic chromosome instability phenotypes including abrupt chromosome rearrangements, telomere dysfunction, elevated rate of homologous recombination and cohesion defects suggesting its role in multiple cellular processes [2-4]. Two recent in vitro studies have documented its role in unloading of ubiquitinated proliferating cell nuclear antigen (PCNA) from chromatin during post replication repair (PRR) pathways in yeast [5] and in human cell line [6] where PCNA acts as a scaffold to recruit proteins on DNA. However, information related to either gain or loss of ATAD5 expression, its regulation and importance in human, particularly in carcinogenesis has not been documented yet.
Hepatocellular carcinoma (HCC) is the major type of primary liver cancer developing from repetitive cycles of liver cell injury, necro-inflammation, regeneration and repair. Chronic hepatitis B (CHB) is the leading cause of HCC globally [7]. Hepatitis B virus, the smallest member of the Hepadnaviridae family, consists of 3.2 kb partially double stranded DNA and encodes proteins namely envelop, core, polymerase and HBx. C-terminal two third (51-154 amino acids) of 154 amino acids long HBx protein contains a transactivation domain, which is responsible for multi-regulatory function of HBx [8]. As an oncoprotein, HBx regulates several cellular processes such as cell cycle progression [9], autophagy [10], apoptosis [11], cell proliferation [12], cell migration [13], proteosomal degradation [14] and expression of miRNA [15] through different transcription factors such as E2F1, NF-κβ, AP-1, AP-2, ARF/CREB and c-EBP [7,16]. Among these factors over expression of E2F1 possesses pleiotropic role in cancer. High expression of E2F1 is a risk factor for development of malignant ovarian tumor [17, 18] whereas it suppresses tumor growth in lung cancer, breast cancer and osteosarcoma [19-21].
The present study was designed to elucidate the effect of HBx on the expression of the novel gene ATAD5 and its functional importance in development of HCC. We have described the mechanism of its transcriptional regulation by HBx in HCC cell line. Our results demonstrated that HBx upregulates ATAD5 through E2F1 transcription factor and a positive feedback regulatory loop between ATAD5 expression and HBV replication provide a favorable environment for HBV persistence in cell. This study also suggests that absence of ATAD5 expression leads to enhance anticancer drug sensitivity of HCC cells.
Material and Methods
Study Subjects
A total of 11 HBV related HCC patients, who underwent either liver resection at School of Digestive and Liver Diseases, Institute of Post Graduate Medical Education and Research (IPGME&R), Kolkata, India or orthotropic liver transplantation at Center for Liver and Biliary Sciences, Indraprastha Apollo Hospital, New Delhi, India were enrolled in the study with written informed consent from each patient. Ethics Committee of IPGME&R, Kolkata has approved the study. Clinical diagnosis of HCC was achieved according to standard protocols using ultrasound, triphasic dynamic CT and serum Alfa-fetoprotein (AFP) values supplemented with histological evidence, when available. Diagnosis of cirrhosis was done by histological and/or through clinical evidences. Both Tumor and adjacent non-tumor tissues were collected freshly in RNAlater® (Life Technologies, USA) and then preserved at -800C freezer until further use.
RNA isolation from liver tissue
Frozen liver tissue of HCC patient was powdered in liquid nitrogen and homogenized in Trizol (Invitrogen, Carlsbad, CA, USA) to isolate RNA following manufacturer's instructions.
Commercially available Blue-Ribbon human total RNA (Origene, MD, USA) derived from normal human tissues of different organs was used to study ATAD5 expression.
Reverse transcription and quantitative real time PCR (qRT-PCR) for gene expression analysis
To quantify the expression of a gene, SYBR Green Real-time PCR Master Mix (Roche Diagnostics, Mannheim, Germany) was used. 5μg of total RNA was converted into cDNA using High-Capacity cDNA Reverse Transcription kit (Applied Biosystems, CA, USA) following the manufacturer's protocol. The first strand cDNA was then used to quantify the expression of gene by qRT-PCR (ABI 7500 real time PCR machine) using specific primers (Table 1). 18s ribosomal RNA or β-Actin was used as internal control for relative quantification of expression of a gene. qRT-PCR for ATAD5 mRNA expression in tissue samples were performed in triplicate.
Sequences of primers for cDNA and promoter of genes studied.
| Gene Names | Primer Sequences |
|---|---|
| cDNA primers | |
| ATAD5 | F: 5'-CCTTATAACAGGGCCAACAGG-3' R: 5'-CCTTGTTTGTCTACTTGATGGG-3' |
| HBx | F: 5'-TGTGCACTTCGCTTCACCTC-3' R: 5'-CAGCCTCCTAGTACAAAGAC-3' |
| HBs | F: 5'-CTCAGGCCATGCAGTGGAA-3' R: 5'-AACTGGAGCCACCAGCAG-3' |
| E2F1 | F: 5'-CATCCAGCTCATTGCCAAGAAG-3' R: 5'-GATCCCACCTACGGTCTCCTCA-3' |
| 18S | F: 5'-GTAACCCGTTGAACCCCATT-3' R: 5'CCATCCAATCGGTAGTAGCG-3' |
| β-Actin | F: 5'-GCTCGTCGTCGACAACGGCTC-3' R: 5'-CAAACATGATCTGGGTCATCTTCTC-3' |
| Promoter primers | |
| IL17β | F: 5'-GTTGTGAGGCCAGTCCATTC-3' R: 5'-TAATTGCTTCGCTCCATCCT-3' |
| ATAD5 (-703) | F: 5'-GGAGCTCCCGTCCGCGCAGCCTCCTG-3' |
| ATAD5 (-514) | F: 5'-GGAGCTCAGATGAAGCAATGACTCGGC-3' |
| ATAD5 (-410) | F: 5'-GGAGCTCTCGGATCCTGAGAGGACGT-3' |
| ATAD5 (-288) | F: 5'-GGAGCTCAAGAGGGGCTAGCCTAGGG-3' |
| ATAD5 (-178) | F: 5'-GGAGCTCTCAAATTTCCAAACTCCCG-3' |
| ATAD5 (-79) | F: 5'-GGAGCTCCCGTCCGCGCAGCCTCCTG-3' |
| ATAD5 (-158) | R: 5'-TCTCGAGCCGGGAGTTTGGAAATTTG-3' |
| ATAD5 (+103) | R: 5'-GCTCGAGTGTATGCAGGGTGCCCTC-3' |
Plasmids used for in vitro study
The plasmids ECFP-ATAD5 encoding the 5.6kb full-length cDNA of ATAD5 fused with CFP at the C-terminal end and pSV2NeoHBV2x, containing wild type HBV in tandem dimer were gifted by Dr. Kyungjae Myung (National Institute of Health, USA) and Dr. Chiaho Shih (Academia Sinica, Institute of Biomedical Sciences, Taiwan) respectively. The full-length HBx gene was amplified and cloned in pcDNA3.1/myc-His (-) version B vector (Invitrogen) where HindIII and Not1 sites were appended in primers for efficient cloning and it was referred as pcDNA3.1B/myc-His-HBx. The E2F1 over-expressing plasmid pCMVHA-E2F1 was a gift from Kristian Helin (Addgene plasmid # 24225) [22] and the HEY1 expression plasmid, pBABE-puro-HEY1, containing the full-length HEY1 cDNA was shared by Prof. Subrata Banerjee (Saha Institute of Nuclear Physics, Kolkata). The putative promoter region of ATAD5 gene [-703 to +103 sequences with respect to transcription start site (TSS)] (accession number refgene NM_024857) was cloned in Sac1 and Xho1 sites of pGL3-Basic vector containing firefly luciferase reporter gene without promoter or enhancer region (described as pGL3-FL-ATAD5). Thus luciferase activity indicated the activity of the cloned promoter sequence. Six serially deleted promoter constructs were generated by PCR amplification followed by restriction digestion with SacI/XhoI and cloned into pGL3-Basic vector. They were named as pGL3-ΔR-ATAD5 (-514 to +103), pGL3-Δ1-ATAD5 (-410 to +103), pGL3-Δ2-ATAD5 (-288 to +103), pGL3-Δ3-ATAD5 (-178 to +103), pGL3-Δ4-ATAD5 (-79 to +103) and pGL3-Δ5-ATAD5 (-703 to -158). The pGL3-E2F1mt-ATAD5 was generated by site directed mutagenesis (Stratagene, CA, USA) at 4 nucleotide positions [-23 (C to T), -21 (C to T), -19 (A to C), -17 (A to C) with respect to TSS] in pGL3-FL-ATAD5 construct.
Cell lines and Transfection
The human hepatoma cell lines HepG2, Huh7 and immortalized human kidney cell line HEK293T were gifted by Dr. Suvendra Bhattacharyya and Dr. Susanta Roychoudhury respectively (Indian Institute of Chemical Biology, Kolkata, India). Dr. Thomas Schmittgen (Ohio State University, Cleveland, USA) had gifted us SNU449, SNU387, SNU182, PLC/PRF5 and Sk-Hep-1 liver cell lines. Cells were cultured either in Dulbecco's modified Eagle's medium (DMEM) or RPMI1640 supplemented with 10% fetal bovine serum (FBS) (GIBCO, Grand Island, NY, USA) and 100 units/ml penicillin-streptomycin (Sigma Aldrich) in a 5% CO2 containing 37ºC humidified chamber. HepG2, Huh7 and HEK293T cells were transfected using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) following the manufacturer's instructions. Stable knock-down ATAD5 and vector cell lines were generated by transfecting pLK0.1-Puro-ATAD5 shRNA plasmid (Sigma Aldrich, clone number: TRCN0000134232) and pLK0.1-Puro vector respectively.
Promoter Assay
100ng of each luciferase-reporter construct was co-transfected with 500ng of pcDNA3.1B/myc-His-HBx plasmid in HepG2 and Huh7 cells. After 36h of transfection, luciferase activity was determined using Dual-Luciferase® Reporter Assay System (Promega, Madison, WI, USA) in Luminometer. The firefly luciferase activity was normalized with total protein concentration in each lysate. Each assay was done in triplicate and repeated at least three times. HepG2 cell line was preferably used for in vitro experiments as data was significant and reproducible.
Chromatin immuno-precipitation Assay (ChIP)
ChIP assay was done with anti-HBx antibody (MA1-081, Pierce, Thermo Scientific, Rockford, IL, USA), anti-E2F1 antibody (sc-22820, Santa Cruz, CA, USA) and control human IgG antibody (MA5-14728, Pierce, Thermo Scientific, Rockford, IL, USA). HepG2 cells growing on 10cm tissue culture dish (90-95% confluent) were cross-linked using 1% formaldehyde for 15 minutes at room temperature and quenched with 125mM of fresh glycine. Then cells were harvested in SDS lysis buffer, sheared by sonication to generate 300-800bp fragments of DNA. Immunoprecipitation was performed with 4μg of specific antibody at 4ºC for overnight in a rotor. It was further incubated with 25μl of Protein A sepharose resin (Sigma Aldrich) for 2h to capture the protein-DNA-antibody complexes and DNA fragments were eluted in TE buffer by heating at 900C for 10 minutes and analyzed by semi-quantitative PCR using ELG1 promoter specific primers (Table 1) (-79 to +103bp sequences with respect to TSS). IL17β gene was used as a positive control for ChIP experiment and amplified from anti-HBx antibody pooled DNA [23] using specific primers (Table 1) of IL17β (114 bp from IL17β promoter).
Western blot analysis
Western blot analysis was performed following standard protocol. Primary antibodies used for detection of proteins were anti-E2F1, anti-α-Tubulin (T6074, Sigma Aldrich) and Rabbit polyclonal anti-ATAD5, which was a kind gift from Dr. Kyungjae Myung. HRP conjugated Goat anti-Rabbit and Goat anti-Mouse was used as secondary antibody (Thermo Fisher Scientific, MA, USA).
Cell viability assay after treatment with 5-fluorouracil (5-FU) and Methyl methanesulfonate (MMS)
ATAD5 knock down and control cells (2.5x103) were seeded on 96 well plates and treated with different doses of 5-fluorouracil (5-FU) for 24h and Methyl methanesulfonate (MMS) for 1h. Cells were washed thrice with 1X PBS at the end of treatment and cultured in fresh media for 24h. Cell viability was then quantitated using WST1 reagent (Roche Diagnostics, Mannheim, Germany) following manufacturer's instruction.
HBV Core particle Isolation and quantification of viral DNA
The pSV2NeoHBV2x plasmid was digested with EcoR1 restriction enzyme to isolate linear full-length monomer of HBV DNA [24], so that after transfection viral genes are expressed using its own promoters. 1μg of monomer HBV DNA was then co-transfected with either 1.5μg of ECFP-ATAD5 plasmid or ATAD5-shRNA in HepG2 cells in a 6 well plate. After 72h of transfection core particle associated viral DNA was isolated as previously described [25] and quantified using HBV surface (HBs) specific primers (Table 1).
Statistical analysis
GraphPad Prism 5 (GraphPad Software, La Jolla, California, USA) was used for statistical analysis. Data between two different groups were compared by Student's t-test. All the in vitro experiments were performed at least three times and mean value with standard deviation was presented. p<0.05 was considered as statistically significant unless mentioned otherwise.
Results
Expression pattern of ATAD5 in different normal tissues
As there is no available published data about human ATAD5 gene expression in different healthy organs, the tissue specific mRNA expression of ATAD5 was first determined using Blue Ribbon human total RNA from 21 different organs by qRT-PCR. Maximum expression of ATAD5 was observed in breast tissue followed by thymus, kidney, placenta and thyroid (Fig. 1A). In normal adult liver the expression of ATAD5 was comparatively lower than other organs except in bladder and esophagus. Similar lower expression of ATAD5 in liver compared to other organs was found in different transcriptome databases such as (i) UCSC genome browser (https://genome.ucsc.edu) and (ii) Genecards (http://www.genecards.org).
mRNA expression pattern of hELG1/ATAD5 (A) in tissues from 21 different human organs, (B) in 7 hepatocellular carcinoma (HCC) cell lines and (C) in 11 pairs of HCC liver tissues. Relative expression of ATAD5 was determined by qRT-PCR. NL in (B) represents RNA of normal liver tissue from commercially available Blue-Ribbon human total RNA. (** and * denote p value <0.005 and <0.05 respectively. NS indicates non-significant value.)
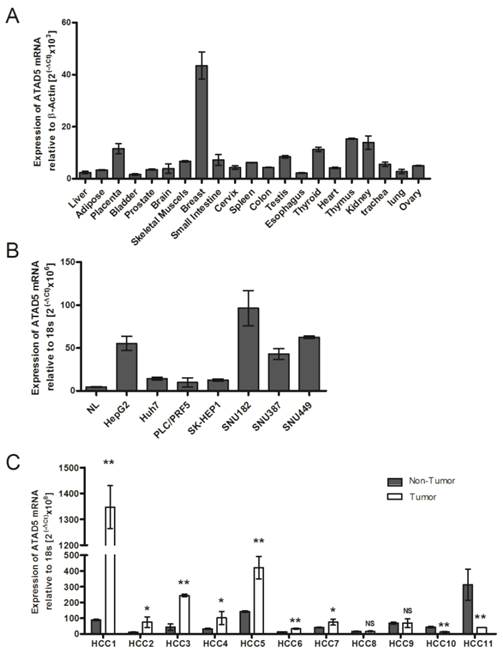
Quantitative analysis of ATAD5 expression in different liver cancer cell lines and in HCC tissues
We have investigated the expression level of ATAD5 mRNA in 7 different liver cancer cell lines by qRT-PCR and a significant increase in expression of ATAD5 was observed in all the cell lines i.e. HepG2, Huh7, PLC/PRF5, SK-HEP1, SNU182, SNU387 and SNU449 compared to normal liver (NL) tissue (p<0.05) (Fig. 1B). The expression pattern of ATAD5 was also investigated in HBV infected HCC tissues. As shown in Fig.1C, the ATAD5 gene expression was found significantly higher in 64% of HBV infected HCC (7/11) tissues compared to adjacent non-tumor tissues (p<0.05).
To understand the underlying mechanism and importance of over-expression of ATAD5 in HCC in presence of HBV, transcriptional regulation on ATAD5 by HBV trans-activator protein HBx [8] was explored.
HBx regulates ATAD5 expression in hepatoma cell lines
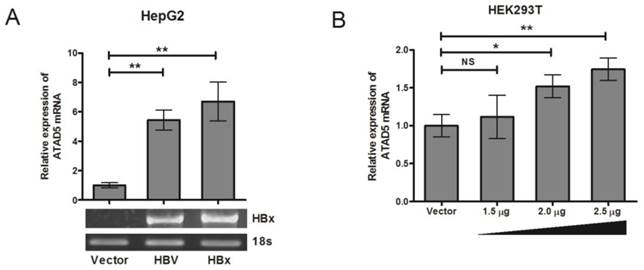
In vitro transfection of HepG2 cell line (HBV negative HCC cell line) with either pSV2NeoHBV2x (carrying two times of full length HBV DNA) or pCDNA3.1B/myc-His-HBx plasmid (HBx expressing plasmid) showed that after 48h of transfection, the expression of ATAD5 was increased significantly (p<0.05) in presence of both HBV (5.5 fold) and HBx (6.8 fold) compared to the vector transfected cells (Fig. 2A) suggesting HBx protein of HBV had primarily regulated the expression of ATAD5. The transactivation property of HBx on ATAD5 was further confirmed by transfecting increasing doses of pCDNA3.1B/myc-His-HBx plasmid in a non-liver cell line HEK293T (Fig.2B) and an increasing trend in ATAD5 expression was observed (p<0.05).
Regulation of hELG1/ATAD5 mRNA expression by HBx protein. (A) Control vector and plasmids pSV2NeoHBV2x (Full length HBV) and pCDNA3.1B/myc-His-HBx (HBx expressing plasmid) were transfected in HepG2 cell line. (B) pCDNA3.1B/myc-His-HBx was transfected with different doses in HEK293T cell line. The relative expression of ATAD5 was determined by qRT-PCR. Ectopic expression of HBx gene was verified by semi-quantitative PCR. (** and * denote p value <0.001 and <0.01 respectively. NS indicates non-significant value.)
HBx protein up-regulates the promoter activity of ATAD5
The enigmatic HBx protein transactivates promoter-enhancer structures of several genes which are comprised of different regulatory regions. It has no specific interactive DNA sequence or interactive protein partner [26]. To investigate whether HBx protein could alter the promoter activity of ATAD5, we first identified the putative promoter region of ATAD5 considering the 1000 base pairs (bp) upstream to 100bps downstream sequences from the TSS retrieved from UCSC genome browser. Five deletion constructs from the ATAD5 putative promoter region were generated (ΔR and Δ1-Δ4) in pGL3-luciferase vector which were differed by near about 120bps (Fig. 3A). Each construct was transfected in two different cell lines HepG2 and Huh7 and luciferase activity was determined. The constructs with -1000bps and -703bps upstream sequences of ATAD5 showed similar luciferase activities (data not shown) and thus the region between -703 to +103bps was considered as full-length promoter (pGL3-FL-ATAD5). To our surprise, the construct comprised of -514 to +103bps (pGL3-ΔR-ATAD5) exhibited extremely low promoter activity compared to pGL3-Δ1-ATAD5 (Fig. 3B). Therefore, we had tested our hypothesis using five constructs pGL3-FL-ATAD5 and pGL3-Δ1-ATAD5 to pGL3-Δ4-ATAD5 spanning -410 to +103 sequences with respect to TSS as described in methods. The construct having -288bps upstream region from the TSS (pGL3-Δ2-ATAD5) was recognized as basal promoter region of ATAD5 gene (Fig. 3B). Next, to decipher the effect of HBx on ATAD5 promoter, luciferase activity was determined in both HepG2 and Huh7 cell lines co-transfected with pGL3-FL-ATAD5 and with either pcDNA3.1B/myc-His-HBx plasmid or control vector pcDNA3.1B/myc-His. The luciferase activity of ATAD5 was increased significantly (>1.6 fold and p<0.05) in presence of HBx compared to control vector suggested that ATAD5 was upregulated at the transcriptional level by HBx (Fig. 3C).
Transcriptional regulation of ATAD5 by HBx. (A) Six deletion mutant constructs of ATAD5 promoter were generated in pGL3-Basic vector. Arrow indicates the putative transcription start site (TSS), which was deduced from the extreme 5' nucleotide of the longest expressed mRNA sequence identified in NCBI database. Number indicates the position of deletion point with respect to the TSS. HepG2 and Huh7 cells were transfected (B) with each promoter construct independently and (C) with pGL3-FL-ATAD5 and either control vector or pcDNA3.1B/myc-His-HBx. HepG2 cells were co-transfected with (D) different deletion mutant constructs and either with control vector or pcDNA3.1B/myc-His-HBx separately. The average relative luciferase activity (RLU) was presented in bar diagram. (** and * denote p value <0.005 and<0.05 respectively. NS indicates non-significant value).
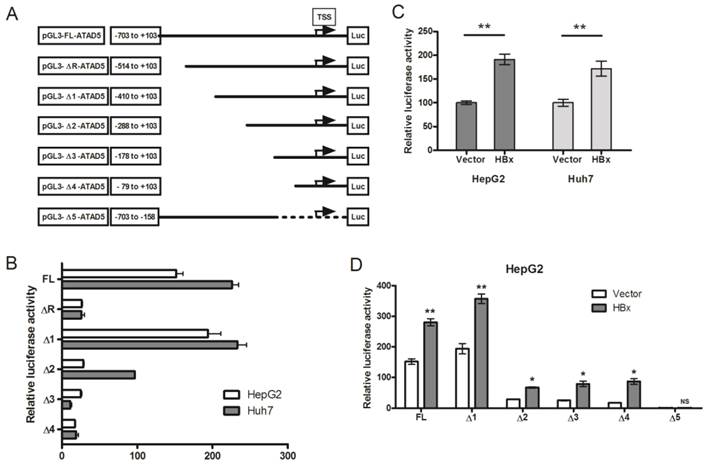
Having observed the transcriptional regulation of ATAD5 by HBx, we identified the exact regulatory region on the ATAD5 promoter sequence by deletion mapping. Each deletion construct was co-transfected with pcDNA3.1B/myc-His-HBx plasmid in HepG2 cells and luciferase activity was determined. The increased luciferase activities of all deletion mutant constructs in presence of HBx (Fig. 3D) indicated the presence of HBx regulatory region in close proximity (from -79 to +103) of the TSS in the ATAD5 promoter. To further confirm this, the luciferase activity of pGL3-Δ5-ATAD5 plasmid where -157 to +103 region was deleted, was evaluated and as expected HBx was found unable to increase the luciferase activity of this promoter construct (Fig. 3D).
HBx protein is not physically associated to the ATAD5 promoter but functions via E2F1 transcription factor
Next we examined the presence of HBx protein in the transcription complex on the ATAD5 promoter. HBx protein does not have DNA binding domain but it interacts to several transcription factors and trans-regulates transcriptional activity of several genes [26, 23]. ChIP assay was performed with anti-HBx antibody to investigate the presence of HBx on ATAD5 promoter but no PCR product was obtained from pooled DNA after repeated ChIP experiments (Fig. 4A). Whereas a positive interaction between HBx and ILl7β promoter, a known target protein of HBx [23], had confirmed that HBx indirectly regulated the expression of ATAD5 and it was presumably through another transcription factor which was upregulated in presence of HBx and in turn activated ATAD5 expression (Fig. 4A). Therefore we scanned the promoter region (-79 to +103) of ATAD5 to identify the putative binding sites of the transcription factors which are deregulated by HBx protein using bioinformatics tools such as Alibaba2.0, MATINSPECTOR and the ChIPSeq data from ENCODE study (UCSC genome browser) (Fig.4B). Binding sites of the transcription factor E2F1, which is a direct target of HBx [27] and another factor HEY1, which is regulated by E2F1 [28] were identified in the promoter region (-79 to +103) of ATAD5.
Transcriptional regulation of ATAD5 by HBx through E2F1 transcription factor. (A) Chromatin immune-precipitation (ChIP) assay was performed with anti-HBx antibody using cell extract from pcDNA3.1B/myc-His-HBx plasmid transfected HepG2 cells. IgG antibody was used as negative control. Semi-quantitative PCR with ATAD5 and IL17β specific primers were performed using ChIP DNA and a fraction of input was used as positive control. NC is the PCR negative control. (B) Schematic diagram of putative transcription factors binding sites in ATAD5 promoter sequence. Luciferase activity was determined in HepG2 cells (C) co-transfected with three different promoter constructs (pGL3-FL-ATAD5 (-703 to +103), pGL3-Δ3-ATAD5 (-178 to +103) and pGL3-Δ5-ATAD5 (-703 to -158) independently and either pCMVHA-E2F1 or pBABE-puro-HEY1 plasmid, (D) co-transfected with either plasmid pGL3-FL-ATAD5 (wild type E2F1) or pGL3-mtE2F1-ATAD5 (mutant E2F1) and pcDNA3.1B/myc-His-HBx or pCMVHA-E2F1 or both. (E) The relative mRNA expression of E2F1 was quantified after transfection with pcDNA3.1B/myc-His-HBx plasmid in HepG2 cells. Empty vector was used as control. (F) Semi-quantitative PCR was performed with ATAD5 specific primers from ChIP DNA with anti-E2F1 antibody, non-specific IgG antibody and a fraction of input. (G) ATAD5 and E2F1 protein levels were altered by HBx. Western blot analysis was performed with extract from HepG2 cells transfected with pcDNA3.1B/myc-His-HBx using anti-ATAD5 and anti-E2F1 antibody. Anti-α-Tubulin antibody was taken as loading control. (H) Pearson's correlation analysis between the log (fold change) value of the expression of ATAD5 and E2F1 in tumor tissue compared to non-tumor tissue of HCC patients. (** and * denote p value <0.005 and<0.05 respectively. NS indicates non-significant value).
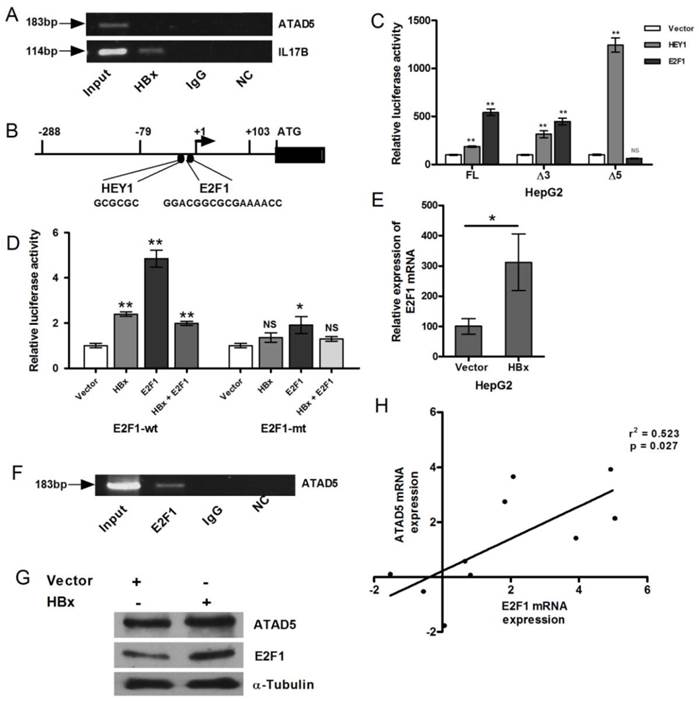
The putative consensus binding sites of HEY1 and E2F1 transcription factors are located in the close proximity to TSS (HEY1 from -36 to -31 and E2F1 from -29 to -14 respectively from TSS), the effect of these two transcription factors on the expression of ATAD5 was studied (Fig. 4B). Luciferase promoter assay was performed to determine the direct effect of E2F1 and HEY1 protein on ATAD5 promoter. HepG2 cells were co-transfected with each of the three ATAD5 promoter constructs (pGL3-FL-ATAD5, pGL3-Δ3-ATAD5 and pGL3-Δ5-ATAD5) and either pCMVHA-E2F1 or pBABE-puro-HEY1 plasmids (Fig. 4C). Although pGL3-Δ5-ATAD5 construct did not possess HBx regulatory region but in presence of HEY1 the enhanced luciferase activities (>2 fold, p<0.05) with all the three constructs implicated that HEY1 might not play the primary role in HBx mediated upregulation of ATAD5 promoter activity. Again, increased luciferase activities of pGL3-FL-ATAD5 and pGL3-Δ3-ATAD5 promoter constructs in presence of E2F1 (p<0.005) and its reduction in pGL3-Δ5-ATAD5 construct suggested that E2F1 is the primary player in HBx mediated upregulation of ATAD5 promoter activity. To further confirm this, the consensus E2F1 binding site was mutated by site directed mutagenesis in the pGL3-FL-ATAD5 promoter construct and the luciferase activity of pGL3-E2F1mt-ATAD5 construct was compared with that of the wild type pGL3-FL-ATAD5 construct by co-transfecting them with pCMVHA-E2F1 in HepG2 cells. The luciferase activity of the pGL3-E2F1mt-ATAD5 promoter construct was significantly reduced (about 50%) compared to that of wild type construct (p<0.005) (Fig. 4D).
Similar result was obtained when HBx was co-transfected with wild type and mutant ATAD5 promoter constructs. But HBx did not cooperate with E2F1 when they were co-transfected (1:1) in HepG2 cell line. The luciferase activity of the pGL3-FL-ATAD5 construct was declined in this setting compared to that of E2F1 alone and the level was almost similar to HBx (Fig. 4D). The enhanced expression of E2F1 mRNA in presence of HBx was verified in HepG2 cells (p<0.05) (Fig. 4E).
Now to further confirm the recruitment of E2F1 to the HBx regulatory promoter region of ATAD5, ChIP assay was performed with anti-E2F1 antibody. Antibody bound protein-DNA complexes were pooled down in HepG2 cells and the -79 to +103 region of ATAD5 promoter was detected by semi-quantitative PCR (Fig. 4F) from the E2F1 pooled genomic DNA. Human IgG antibody was used as negative control in ChIP experiment. Input DNA was used as loading control.
Consistent with HBx-E2F1 mediated transcriptional control on ATAD5, effect of HBx regulation was also observed at the protein level. In HBx transfected HepG2 cell line both E2F1 and ATAD5 proteins were increased (Fig. 4G). In concordance with this in vitro data, a positive correlation between the mRNA expression of ATAD5 and E2F1 in HBV related HCC liver tissues were also observed (Fig.4H).
ATAD5 induces HBV replication
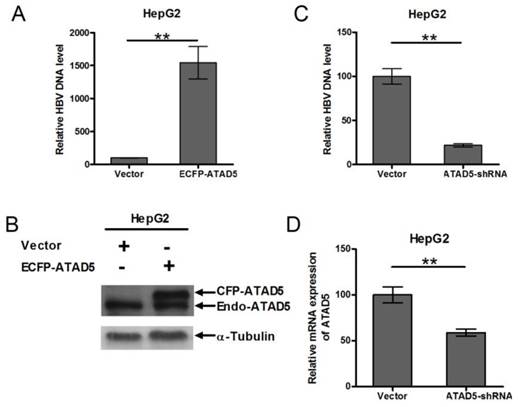
To determine role of ATAD5 on HBV replication, HepG2 cell line was co-transfected with pSV2NeoHBV2x and either ECFP-ATAD5 or control vector. After 3 days of transfection, the intracellular core particle associated viral DNA was quantified by qRT-PCR and observed a significantly higher number of HBV DNA copies within the intracellular core particle in ATAD5 over expressed cells compared to that of the vector using HBV surface specific primers (Fig. 5A). The level of ATAD5 protein was confirmed by western blot analysis using anti-ATAD5 antibody, which detected both endogenous and ectopically expressed ATAD5 (Fig. 5B). Conversely, co-transfection of pSV2NeoHBV2x with either ATAD5-shRNA or control vector revealed that HBV DNA copies within viral core was significantly reduced (p<0.005) (Fig. 5C) when ATAD5 mRNA expression was dropped by ATAD5-shRNA (Fig. 5D). Thus both gain- and loss-of-function of ATAD5 showed pronounced effect on HBV replication efficiency.
Effect of ATAD5 on HBV replication. HepG2 cells were co-transfected with HBV DNA and with either (A, B) ECFP-ATAD5 or (C, D) ATAD5-shRNA. Intracellular HBV DNA level was quantified by qRT-PCR with HBV surface specific primers. The relative change in HBV DNA copy number was represented in bar diagram (A,C). The ectopic over expression of CFP-ATAD5 (B) and down regulation of endogenous ATAD5 by shRNA (D) were confirmed by western blot analysis with anti-ATAD5 antibody and qRT-PCR respectively. [** p value <0.005].
Knock down of ATAD5 enhances chemo-sensitivity of HCC cell line
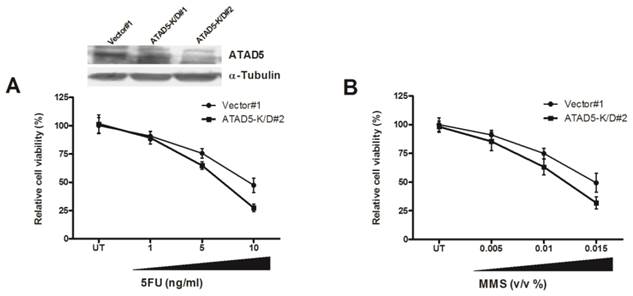
To further understand the role of enhanced ATAD5 in HCC cell line in presence of HBx, ATAD5 knock down HepG2 cells were treated with different doses of chemotherapeutics 5-fluorouracil (5-FU). In anticipation a sharp decrease in cell viability was observed in knock down cells compared to control cells (Fig. 6A). Similar result was noted after treatment of ATAD5 knock down cells with different doses of MMS, a DNA damaging agent (Methyl methanesulfonate) (Fig. 6B) indicating ATAD5 protected cells from apoptosis induced by different stresses and thus ATAD5 over expressed HCC cells achieved chemo-resistance.
Chemo-sensitivity of ATAD5 knock down HCC cell line. Stable ATAD5 knock down (K/D) and control cells were treated with indicated doses of (A) 5-fluorouracil (5-FU) and (B) Methyl methanesulfonate (MMS). Cell viability was quantified using WST1 reagent as described in methods. Data was presented as percentage of viable cells at each dose considering viability of the untreated (UT) vector cells 100%. Each experiment was repeated at least twice in triplicate. The down regulation of ATAD5 in two K/D clones was confirmed by western blot analysis as shown in upper portion of (A).
Therefore, activation of E2F1, a key regulator of cell cycle progression, plays the pivotal role in upregulation of ATAD5 gene which promotes HBV replication and safeguards tumor cells against anticancer drug.
Discussion
The contribution of HBx protein of HBV in the development of HCC is well documented, although the exact function and molecular mechanism is not clearly understood. We have demonstrated here the role of HBx in regulation of a novel gene ATAD5 which is required for genome maintenance and its impact on hepatocarcinogenesis. This study has for the first time mapped the active promoter region of the gene and also showed the over expression of this novel gene in HBV associated HCC liver tissues. As a trans-activator, HBx protein of HBV was found to regulate the expression of this gene in in vitro transfection assay through transcription factor E2F1. Thus this study provides a new insight into the regulatory network of this novel gene. Both gain- and loss-of-function of ATAD5 in HCC cell line suggested that over expression of ATAD5 by HBx might contribute to the viral replication. Most importantly, enhanced chemo-sensitivity of ATAD5 knock down HCC cells provided a basis for considering ATAD5 as a therapeutic target in HBV infection.
Over expression of this gene was observed in significant number of HBV associated HCC tissues compared to adjacent non-tumor tissues and also in several HCC cell lines compared to normal liver. The high expression of ATAD5 in HCC tissues could be a consequence of amplification of chromosome 17q arm in which this gene (17q.11.2 loci) is located. But a meta-analysis study consisting of 31 CGH arrays with 785 HCC samples showed highest frequency of chromosomal imbalances exist specifically at 17q23-q25 locus [29], suggesting alternative mechanisms of regulation attributed to over-expression of ATAD5 gene in HCC. One of the key mechanisms of HBV related hepatocarcinogenesis include transcriptional regulation by HBx protein of HBV. HBx transcriptionally regulates several host genes either by directly binding to the proteins of transcription machinery [30-35] or indirectly by upregulating the expression of several transcription factors [36-38]. The role of this viral oncogenic protein in hepatocarcinogenesis has been investigated extensively in several in vitro studies [12-15]. To evaluate the significance of ATAD5 in HBx mediated HCC, we examined the role of HBx in regulating the expression of ATAD5 in HCC cell line. Our in vitro gene expression analysis in presence of HBx indicated that the overexpression of ATAD5 was HBx dependent. Next we tried to understand the underlying mechanism of up regulation of ATAD5 by HBx. To decipher this about 1kb putative promoter region of this novel gene was considered and the basal promoter region, which was -288 to +103 with respect to TSS was mapped from promoter luciferase assay using series of deletion mutant constructs of promoter. Subsequently, HBx regulatory region which was located in the region of -79 to +103 was identified.
But the viral oncogene HBx is not a DNA binding protein [23] and the ChIP assay with anti-HBx antibody negated the hypothesis of direct recruitment of HBx protein to the transcription machinery of the ATAD5 promoter. Searching for binding of other transcription factors using two different prediction tools and Encode ChIPSeq data suggested that E2F1 might be the probable transcriptional regulator of ATAD5. Deregulation of RB-E2F1 pathway is a hallmark of cancer and Swarnalatha et al. had showed recently that over expression of HBx stabilizes E2F1 by blocking its degradation and then activated E2F1 upregulates many proteins involve in G1-S transition and replication [27]. Now, E2F1-HBx interaction has been shown to function synergistically in the promoter of the several tumor suppressor genes including p53 and pRB [39, 40]. HBx augments DNA binding activity of E2F1 by direct interaction with its RB interacting domain [39]. In this study, we have observed that the promoter activity of ATAD5 was increased by both E2F1 and HBx independently, although the effect of E2F1 was more pronounced than HBx. However, HBx and E2F1 co-transfection experiment revealed that HBx failed to show any synergistic effect with E2F1 (1:1) in HepG2 cell line, rather the promoter activity of ATAD5 was decreased. To our surprise, the expression of ATAD5 was found increased with enhanced dose of E2F1 (1:2) in similar co-transfection setting, which was again reduced by over dose of HBx (data not shown). Thus, it suggests that although ATAD5 is regulated mainly by E2F1 but a feedback loop regulation between E2F1 and HBx persist in co-transfection setting, mechanism of which is not clear yet.
Now to understand the physiological importance of the overexpression of ATAD5 in HBV associated HCC, intracellular HBV copy number was measured in HCC cell line after gain or loss of expression of ATAD5. Although there were differences in the level of gain or loss of ATAD5 expression, but the overall mutual relation of ATAD5 expression and intracellular HBV DNA copy number suggested that ATAD5 supported HBV replication. Further studies are needed to understand the mechanism of regulation on HBV replication by ATAD5.
HCC is highly chemo-resistant to conventional chemotherapeutic drugs but the underlying mechanism is not clearly understood. In 2009 Sikdar et al has reported that ATAD5 is stabilized upon treatment of different DNA damaging agents [2]. While a preliminary study in our laboratory with ATAD5 over expressed Hela cell (cervical cancer cell line) treated with DNA damaging agents (5FU or MMS) revealed accumulation of low level of replication stress induced DNA damages as indicated by reduced level of expression of γH2A.X protein compared to that of control cells. Moreover, existence of γH2A.X protein in ATAD5 over expressed cells for shorter period of time after DNA damage in a time course experiment suggested that DNA damages were repaired and cells were survived (Ghosh et al, 2015, unpublished data). This data was supported by the high sensitivity of ATAD5 knock down HCC cells after treatment with either chemo-therapeutic agent 5-FU or DNA damaging agent MMS which again indirectly implicated that restoration of ATAD5 enhanced the chemo-resistance of HCC cells.
Over-expression of several tumor suppressor genes such as retinoblastoma (RB) [41], SMAD4/DPC4 [42], S100A1 [43] in cancer have been reported and this phenotype was correlated with reduction of tumor growth. Further study is needed to understand the detailed molecular basis of over expression of ATAD5 in HCC. Maintenance and progression of several types of cancers are associated with the presence of cancer stem cells and HBx has the ability to promote “stemness” in HCC cells [44], overexpression of ATAD5 might be involved in this process.
Thus, this study provides a new insight on the transcriptional regulation of the novel gene ATAD5 in human cancer by HBx protein of HBV through E2F1 and enhanced ATAD5 promotes chemo-resistance in cancer cells. In addition, positive feedback regulation between HBV and ATAD5 may be advantageous to viral replication. Thus HBV-HBx-E2F1-ATAD5 circuit would act in a novel way to promote HBV infection and liver disease progression to HCC. Despite improvements in surgical techniques and identification of new chemotherapeutics for treatment of HCC, the possible mechanism of anticancer drug resistance due to overexpression of this novel gene in HCC may provide useful information to guide future design of therapeutic intervention in HBV associated HCC.
Abbreviations
HCC: Hepatocellular carcinoma; HBV: Hepatitis B virus; PCNA: proliferating cell nuclear antigen; PRR: post replication repair; qRT-PCR: quantitative real time polymerase chain reaction; TSS: Transcription start site.
Acknowledgements
This work was supported by Council of Scientific and Industrial Research (CSIR), New Delhi, India, partially through project no: 60(0089)/08/EMR-II and 27(0234)/10/EMR-II. Alip Ghosh, Somenath Datta, Amit Ghosh and Debanjali Dasgupta have registered at the West Bengal University of Health Sciences for their PhD degrees. Amit Ghosh and Suchandrima Ghosh are supported by CSIR and University Grant Commission (UGC) respectively. We thank to Professor Nitai P Bhattacharya (Saha Institute of Nuclear Physics, Kolkata), Dr. Susanta Roychoudhury and Dr. Subash Gupta (Apollo hospital, New Delhi) for their constant support.
Author Contributions
AG(1) performed most of the in vitro experiments and prepared the manuscript. SG and DDG did qRT-PCR of ATAD5 in liver tissues and mutagenesis respectively. qRT-PCR with multi-organ tissue panel was performed by NS. SD(1), AG(2) collected tissue samples. SD(2) and AC read the manuscript carefully. SB conceived, designed experiments and prepared the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Smith S, Hwang JY, Banerjee S, Majeed A, Gupta A, Myung K. Mutator genes for suppression of gross chromosomal rearrangements identified by a genome-wide screening in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 2004Jun15;101(24):9039-44
2. Sikdar N, Banerjee S, Lee KY, Wincovitch S, Pak E, Nakanishi K. et al. DNA damage responses by human ELG1 in S phase are important to maintain genomic integrity. Cell Cycle. 2009;8:3199-207
3. Bell DW, Sikdar N, Lee K-y, Price JC, Chatterjee R, Park HD. et al. Predisposition to Cancer Caused by Genetic and Functional Defects of Mammalian Atad5. PLoS Genet. 2011;7(8):e1002245
4. Banerjee S, Myung K. Increased genome instability and telomere length in the elg1-deficient Saccharomyces cerevisiae mutant are regulated by S-phase checkpoints. Eukaryot Cell. 2004;3:1557-66
5. Kubota T, Nishimura K, Kanemaki MT, Donaldson AD. The Elg1 replication factor C-like complex functions in PCNA unloading during DNA replication. Mol Cell. 2013Apr25;50(2):273-80
6. Lee KY, Yang K, Cohn MA, Sikdar N, D'Andrea AD, Myung K. Human ELG1 regulates the level of ubiquitinated proliferating cell nuclear antigen (PCNA) through Its interactions with PCNA and USP1. J Biol Chem. 2010Apr2;285(14):10362-9
7. Gomaa AI, Khan SA, Toledano MB, Waked I, Taylor-Robinson SD. Hepatocellular carcinoma: epidemiology, risk factors and pathogenesis. World J Gastroenterol. 2008;14:4300-4308
8. Bouchard MJ, Schneider RJ. The enigmatic X gene of hepatitis B virus. J Virol. 2004;78:12725-12734
9. Quétier I, Brezillon N, Duriez M, Massinet H, Giang E, Ahodantin J. et al. Hepatitis B virus HBx protein impairs liver regeneration through enhanced expression of IL-6 in transgenic mice. J Hepatol. 2013Aug;59(2):285-91
10. Liu B, Fang M, Hu Y, Huang B, Li N, Chang C. et al. Hepatitis B virus X protein inhibits autophagic degradation by impairing lysosomal maturation. Autophagy. 2014Mar;10(3):416-30
11. Rawat S, Clippinger AJ, Bouchard MJ. Modulation of apoptotic signaling by the hepatitis B virus X protein. Viruses. 2012Nov8;4(11):2945-72
12. Tralhao JG, Roudier J, Morosan S, Giannini C, Tu H, Goulenok C. et al. Paracrine in vivo inhibitory effects of hepatitis B virus X protein (HBx) on liver cell proliferation: an alternative mechanism of HBx-related pathogenesis. Proc Natl Acad Sci U S A. 2002May14;99(10):6991-6
13. Liu H, Xu L, He H, Zhu Y, Liu J, Wang S. et al. Hepatitis B virus X protein promotes hepatoma cell invasion and metastasis by stabilizing Snail protein. Cancer Sci. 2012Dec;103(12):2072-81
14. Jung JK, Kwun HJ, Lee JO, Arora P, Jang KL. Hepatitis B virus X protein differentially affects the ubiquitin-mediated proteasomal degradation of beta-catenin depending on the status of cellular p53. J Gen Virol. 2007;88:2144-54
15. Tian Y, Yang W, Song J, Wu Y, Ni B. Hepatitis B virus X protein-induced aberrant epigenetic modifications contributing to human hepatocellular carcinoma pathogenesis. Mol Cell Biol. 2013Aug;33(15):2810-6
16. Kekulé AS, Lauer U, Weiss L, Luber B, Hofschneider PH. Hepatitis B virus transactivator HBx uses a tumour promoter signalling pathway. Nature. 1993Feb25;361(6414):742-5
17. Martín-Pérez D, Artiga MJ, Menárguez J, Flemington EK, Piris MA, Campanero MR. E2F1 expression is deregulated and plays an oncogenic role in sporadic Burkitt's lymphoma. Cancer Res. 2009;69:4052-4058
18. Gala S, Marreiros A, Stewart GJ, Williamson P. Overexpression of E2F-1 leads to cytokine-independent proliferation and survival in the hematopoietic cell line BaF-B03. Blood. 2001;97:227-234
19. Peng B, Cao J, Yi S, Wang C, Zheng G, He Z. Inhibition of proliferation and induction of G1-phase cell-cycle arrest by dFMGEN, a novel genistein derivative, in lung carcinoma A549 cells. Drug Chem Toxicol. 2013;36:196-204
20. Shen WH, Jackson ST, Broussard SR, McCusker RH, Strle K, Freund GG. et al. IL-1beta suppresses prolonged Akt activation and expression of E2F-1 and cyclin A in breast cancer cells. J Immunol. 2004;172:7272-7281
21. Liontos M, Niforou K, Velimezi G, Vougas K, Evangelou K, Apostolopoulou K. et al. Modulation of the E2F1-driven cancer cell fate by the DNA damage response machinery and potential novel E2F1 targets in osteosarcomas. Am J Pathol. 2009;175:376-39
22. Lukas J, Petersen BO, Holm K, Bartek J & Helin K. Deregulated expression of E2F family members induces S-phase entry and overcomes p16INK4A-mediated growth suppression. Mol Cell Biol. 1996;16(3):1047-57
23. Sung WK, Lu Y, Lee CW, Zhang D, Ronaghi M, Lee CG. Deregulated direct targets of the hepatitis B virus (HBV) protein, HBx, identified through chromatin immunoprecipitation and expression microarray profiling. J Biol Chem. 2009Aug14;284(33):21941-54
24. Günther S, Sommer G, Von Breunig F, Iwanska A, Kalinina T, Sterneck M & Will H. Amplification of full-length hepatitis B virus genomes from samples from patients with low levels of viremia: frequency and functional consequences of PCR-introduced mutations. J Clin Microbiol. 1998;36(2):531-8
25. Yuan TT, Sahu GK, Whitehead WE, Greenberg R, & Shih C. The mechanism of an immature secretion phenotype of a highly frequent naturally occurring missense mutation at codon 97 of human hepatitis B virus core antigen. J Virol. 1999;73(7):5731-5740
26. Feitelson MA, Reis HM, Tufan NL, Sun B, Pan J, Lian Z. Putative roles of hepatitis B x antigen in the pathogenesis of chronic liver disease. Cancer Lett. 2009Dec1;286(1):69-79
27. Swarnalatha M, Singh AK, Kumar V. Promoter occupancy of MLL1 histone methyltransferase seems to specify the proliferative and apoptotic functions of E2F1 in a tumour microenvironment. J Cell Sci. 2013Oct15;126(Pt 20):4636-46
28. Iwanaga R, Komori H, Ishida S, Okamura N, Nakayama K, Nakayama KI, Ohtani K. Identification of novel E2F1 target genes regulated in cell cycle-dependent and independent manners. Oncogene. 2006;25:1786-1798
29. Moinzadeh P, Breuhahn K, Stützer H, Schirmacher P. Chromosome alterations in human hepatocellular carcinomas correlate with aetiology and histological grade-results of an explorative CGH meta-analysis. Br J Cancer. 2005Mar14;92(5):935-41
30. Maguire HF, Hoeffler JP, Siddiqui A. HBV X protein alters the DNA binding specificity of CREB and ATF-2 by protein-protein interactions. Science. 1991May10;252(5007):842-4
31. Williams JS, Andrisani OM. The hepatitis B virus X protein targets the basic region-leucine zipper domain of CREB. Proc Natl Acad Sci U S A. 1995Apr25;92(9):3819-23
32. Qadri I, Maguire HF, Siddiqui A. Hepatitis B virus transactivator protein X interacts with the TATA-binding protein. Proc Natl Acad Sci U S A. 1995Feb14;92(4):1003-7
33. Lin Y, Nomura T, Cheong J, Dorjsuren D, Iida K, Murakami S. Hepatitis B virus X protein is a transcriptional modulator that communicates with transcription factor IIB and the RNA polymerase II subunit 5. J Biol Chem. 1997Mar14;272(11):7132-9
34. Cheong JH, Yi M, Lin Y, Murakami S. Human RPB5, a subunit shared by eukaryotic nuclear RNA polymerases, binds human hepatitis B virus X protein and may play a role in X transactivation. EMBO J. 1995Jan3;14(1):143-50
35. Kong GY, Zhang JP, Zhang S, Shan CL, Ye LH, Zhang XD. Hepatitis B virus X protein promotes hepatoma cell proliferation via upregulation of MEKK2. Acta Pharmacol Sin. 2011Sep;32(9):1173-80
36. Avantaggiati ML, Balsano C, Natoli G, De Marzio E, Will H, Elfassi E, Levrero M. The hepatitis B virus X protein transactivation of c-fos and c-myc proto-oncogenes is mediated by multiple transcription factors. Arch Virol Suppl. 1992;4:57-61
37. Qu J, Li J, Chen K, Qin D, Li K, Sheng Y. et al. Hepatitis B virus regulation of Raf1 promoter activity through activation of transcription factor AP-2α. Arch Virol. 2013Apr;158(4):887-94
38. Qadri I, Conaway JW, Conaway RC, Schaack J, Siddiqui A. Hepatitis B virus transactivator protein, HBx, associates with the components of TFIIH and stimulates the DNA helicase activity of TFIIH. Proc Natl Acad Sci U S A. 1996Oct1;93(20):10578-83
39. Choi M, Lee H, Rho HM. E2F1 activates the human p53 promoter and overcomes the repressive effect of hepatitis B viral X protein (Hbx) on the p53 promoter. IUBMB Life. 2002Jun;53(6):309-17
40. Choi BH, Choi M, Jeon HY, Rho HM. Hepatitis B Viral X Protein Overcomes Inhibition of E2F1 Activity by pRb on the Human Rb Gene Promoter. DNA Cell Biol. 2001;20(2):75-80
41. Huynh H. Overexpression of tumour suppressor retinoblastoma 2 protein (pRb2/p130) in hepatocellular carcinoma. Carcinogenesis. 2004Aug;25(8):1485-94
42. Hunt KK, Fleming JB, Abramian A, Zhang L, Evans DB, Chiao PJ. Overexpression of the Tumor Suppressor Gene Smad4/DPC4 Induces p21waf1 Expression and Growth Inhibition in Human Carcinoma Cells. Cancer Res. 1998;58(24):5656-5661
43. Ohuchida K, Mizumoto K, Ohhashi S, Yamaguchi H, Konomi H, Nagai E. et al. S100A11, A Putative Tumor Suppressor Gene, Is Overexpressed in Pancreatic Carcinogenesis. Clin Cancer Res. 2006Sep15;12(18):5417-22
44. Wang C, Yang W, Yan HX, Luo T, Zhang J, Tang L. et al. Hepatitis B virus X (HBx) induces tumorigenicity of hepatic progenitor cells in 3,5-diethoxycarbonyl-1,4-dihydrocollidine-treated HBx transgenic mice. Hepatology. 2012Jan;55(1):108-20
Author contact
![]() Corresponding author: Dr. Soma Banerjee, Associate Professor, Centre for Liver Research, School of Digestive and Liver Diseases, Institute of Post Graduate Medical Education and Research, 244, Acharya Jagadish Chandra Bose Road, Kolkata-700020, India. TeleFax: (91)-033- 2223 5435; E.mail: somabanerjee70com
Corresponding author: Dr. Soma Banerjee, Associate Professor, Centre for Liver Research, School of Digestive and Liver Diseases, Institute of Post Graduate Medical Education and Research, 244, Acharya Jagadish Chandra Bose Road, Kolkata-700020, India. TeleFax: (91)-033- 2223 5435; E.mail: somabanerjee70com