International Journal of Biological Sciences
ISSN: 1449-2288
10
Impact Factor
ISSN: 1449-2288
- Current Issue
- Volume 21; 2025
- Volume 20; 2024
- Volume 19; 2023
- Volume 18; 2022
- Volume 17; 2021
- Archive
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Top
Introduction
Materials and Methods
Results
Discussion
Conclusion
Supplementary Material
Acknowledgements
References
Introduction
Materials and Methods
Results
Discussion
Conclusion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2016; 12(1):42-52. doi:10.7150/ijbs.12275 This issue Cite
Research Paper
Apigenin-7-O-β-D-(-6''-p-coumaroyl)-Glucopyranoside Treatment Elicits Neuroprotective Effect against Experimental Ischemic Stroke
Min Cai1,*, Yulong Ma2,*, Wei Zhang3,*, Shiquan Wang2,*, Ying Wang1, Li Tian2, Zhengwu Peng1, Huaning Wang1, Tan Qingrong1 ![]()
1. Department of Psychiatry;
2. Department of Anesthesiology;
3. Department of Pharmacology; Xijing hospital, the Forth Military Medical School
* These authors contributed equally to this work.
Received 2015-3-29; Accepted 2015-9-21; Published 2016-1-1
Citation:
Cai M, Ma Y, Zhang W, Wang S, Wang Y, Tian L, Peng Z, Wang H, Qingrong T. Apigenin-7-O-β-D-(-6''-p-coumaroyl)-Glucopyranoside Treatment Elicits Neuroprotective Effect against Experimental Ischemic Stroke. Int J Biol Sci 2016; 12(1):42-52. doi:10.7150/ijbs.12275. https://www.ijbs.com/v12p0042.htm
Other stylesAbstract

Stroke is the major cause of permanent disability and mortality in China. Apigenin-7-O-β-D-(-6''-p-coumaroyl)-glucopyranoside (APG) is a glycoside subtype of apigenin and has the antioxidant activity; however, whether and how it plays a neuroprotective role following cerebral ischemia remains unknown. In present study, we adopted the oxygen glucose/reperfusion model in PC12 cells, bilateral common carotid artery occlusion model in C57B6 mice and middle cerebral artery occlusion model in SD rats to observe the therapeutic effects of APG on ischemic stroke. We also discussed the underlying mechanism. Treatment with 0.4 μg/ml or 0.8 μg/ml APG promoted cell viability and proliferation, reduced LDH release and apoptotic cell death levels in PC12 cells. Treatment with 50 mg/kg or 100 mg/kg APG at 30 minutes after reperfusion improved neurological outcomes in vivo, as demonstrated by elevation of neurological scores in both mice and rats. It also increased the number of survival neurons in mice and reduced infarct volume in rats. APG also increased the contents of Mn-SOD and the phosphorylation level of STAT3, elevated the antioxidant activity and reduced oxidative productions. These findings revealed a neuroprotective effect of APG, which possibly induced by the STAT3 phosphorylation-mediated Mn-SOD up-regulation.
Keywords: Apigenin-7-O-β-D-(-6''-p-coumaroyl)-glucopyranoside (APG), cerebral ischemia/reperfusion, oxidative stress, manganese-superoxide dismutase
Introduction
Stroke, especially the ischemic stroke, is still one of the leading causes of morbidity and mortality in China (1). Unfortunately, few medications or therapies have been identified to treat ischemic stroke in the clinic. The current FDA approved stroke therapeutic medication is only the tissue plasminogen activator (tPA). However, owing to its narrow therapeutic window (<4.5h) and safety concerns, only less than 5% of patients can benefit from this medication (2). Therefore, more novel maneuvers need to be revealed for stroke treatment.
It is generally accepted that multiple cellular events, such as ionic imbalance, excitotoxicity, oxidative stress, apoptosis, inflammation, and necrosis involve the pathogenesis of ischemic stroke injury (3). Accumulated evidences have shown that oxidative stress plays a significant role in the pathophysiology of cerebral ischemic/reperfusion injury. During cerebral ischemia/reperfusion, the overproduction of free radicals damages the neurons and glia cells; and interacts with cellular biomolecules, such as nucleic acid, lipids, and protein (4). Additionally, the sustained of the oxidative stress mediates the cascade amplification of impinges including mitochondrial dysfunction, inflammation response, and mitochondria/P53-dependent cell death pathway activation (5). Despite the crucial role of oxidative stress in the pathophysiological process of cerebral ischemic damage, and the promising neuroprotective results of antioxidants supplementation demonstrated in animal studies, the treatment with exogenous antioxidants failed to verify their favorable outcomes in clinical trials (6). Actually, there exerts a coordinated self-protective mechanism in brain itself, which including endogenous antioxidant capacity. Exploring a new medication or therapy that triggers at activating endogenous antioxidant system may provide a new avenue for development of neuroprotective approaches.
In searching for stroke therapeutic medications, many traditional Chinese herbs, and their active components have been demonstrated to exert neuroprotective effect against experimental ischemic stroke injury (7-10). As a member of Ranunculaceae family, Clematis tangutica (Maxim.) Korsh (Ranunculaceae) is widely distributed in southwest and northwest areas of China (11). In traditional Tibetan medicine (TTM), the whole plant was used to treat indigestion and to invigorate circulation (12). Apigenin-7-O-β-D-(-6''-p-coumaroyl)-glucopyranoside (APG) is one of the major active component in Clematis tangutica which was demonstrated by our group (13). The free radical scavenging activity of APG has been identified, and the antioxidant capacity of this glycoside flavonoe is predominantly attributed to its redox properties (14). Apigenin has been identified to induce significant neuroprotective effect against Parkinson disease(15), Alzheimer's disease (16,17) and ischemic stroke injury (18) in a variety of in vitro or in vivo experimental models. As a glycoside subtype of apigenin, whether APG confers neuroprotective effect against cerebral ischemia is still unknown. Moreover, the underlying mechanism of the neuroprotective effect induced by this component also needs to be clarified.
In the present study, we tried to determine the neuroprotective effect of APG in three different experimental stroke models in vitro and in vivo. These models include oxygen and glucose deprivation and reperfusion (OGD-R) in PC12 cell line, bilateral common carotid artery occlusion (BCCAO) in mice and middle cerebral artery occlusion (MCAO) in rats. We also examined whether the up-regulation of endogenous antioxidant enzymes mediates the neuroprotective effect induced by APG treatment.
Materials and Methods
Materials
APG was isolated from C. tangutica, and the molecular formula was established as C30H26O12 (molecular weight: 578). The chemical structure of this compound is shown in Supplemental Fig. 1. Purified APG (Purity > 98%) used in current study was supplied by the Pharmacy Department of Xijing Hospital, the Fourth Military Medical University, Xi'an, China.
Male Sprague Dawley (SD) rats aged 8-12 weeks and weighting 250-280g were purchased from The Experimental Animal Centre of the Forth Military Medical University. Male C57Bl mice aged 8-12 weeks and weighting 20-25g were also obtained from this Centre. All experimental procedures were carried out according to protocols approved by the Ethics Committee for Animal Experimentation of the Fourth Military Medical University (Xi'an, Shaanxi, China) and in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Antibodies used in the current study were: anti-phosphorylated STAT3 (705Y) rabbit monoclonal antibody (Cell signal technology; 1:1000); anti-total STAT3 rabbit monoclonal antibody (Cell signal technology; 1:1000); anti-Mn-SOD rabbit polyclonal antibody (Millipore; 1:500); and GAPDH monoclonal antibody (Abcam; 1:2000), β-tubulin monoclonal antibody (Abcam; 1:1000).
In vitro Oxygen and Glucose deprivation and re-oxygenation (OGD-R) model and APG treatment
The PC12 cells were plated at a density of 3×105 cells per well in 6-well multi well or ×104 cells per well in 96-well multi well at 37℃ in 5% CO2 and 95% air in DMEM administrated with 10% fetal bovine serum, streptomycin (100 μg/ml) and penicillin (100 units/mL). OGD-R model was established as described previously (19). Briefly, PC12 cells were switched to d-Hanks buffer (OGD medium) and then to a modular incubator chamber subsequently. The chamber was flushed with a gas mixture of 95% N2 /5% CO2 for 30 minutes at room temperature at the rate of 3 L/min. After flushing, the chamber was sealed and placed in a 37℃ thermostat container. OGD was carried out for 4h. Following OGD, the cells were incubated with dulbecco's modified eagle medium (DMEM, without fetal bovine serum) with the vehicle or different concentrations of APG for additional 20 h reperfusion under normal conditions. The concentrations of APG were 0.2 μg/ml, 0.4 μg/ml, and 0.8 μg/ml and dimethyl sulfoxide (DMSO) was used as vehicle in this experiment.
In vitro cell viability assay
The cell viability was determined by 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) cell viability examination. Briefly, cells were cultured at 15×103 cells per well in 96-well tissue plates. At the end of the culture period, cells were washed with phosphate-buffered saline (PBS) and MTT was added to each well for incubation at 37 ℃ for 4 h according to the manufacturer's instructions. Then, the medium was replaced with 150 μl of DMSO. The optical density (OD) was recorded on a Universal Microplate Reader (Elx 800, Bio-TEK Instruments Inc., USA) at 490 nm. Cell viability was shown as percentage changes in the control value.
Lactate Dehydrogenase (LDH) release assessment
Cell cytotoxicity was quantitatively assessed by evaluating the activity of LDH that was released from the injured cells into the culture medium after OGD-R. Then the cells were treated with 0.5 % Triton X-100, and the media containing detached cells were collected and centrifuged subsequently to yield supernatant for the assay of LDH activity. The LDH activity was measured using an assay kit according to the manufacturer's recommendations. LDH leakage was expressed as the percentage of the total LDH activity (LDH in the both medium and cells), according to the equation, LDH released (%) = (LDH activity in the medium/total LDH activity) × 100%. Cultures without OGD-R treatment (sham group) were represented as basal LDH release.
Flow cytometric analysis
Cell apoptosis was analyzed by flow cytometry (20). Briefly, the cells were washed with 1 × annexin V-FITC binding buffer and then stained with annexin V-FITC and PI for 15 min at room temperature in a dark room. Subsequently, the stained cells were analyzed by flow cytometric method. Apoptotic and necrotic cells were quantitated by annexin V binding and PI uptake. The cell populations of annexin V-FITC+/PI- were calculated to represent apoptotic cells.
Cell cycle analysis
Cell cycle was evaluated by flow cytometry as previously described (21). The cells were collected by trypsinization and centrifuged by PBS twice, then fixed in pre-cooling 70% ethanol at -20 ℃ and subsequently stained with PI solution. DNA content was measured by flow cytometry using Cell Quest Software. Totally, 10,000 events were counted for each sample (FACSC alibur, Becton-Dickinson). The percentage of cells in any particular cell cycle stage was calculated using the ModFit software (Becton-Dickinson, USA).
Global cerebral ischemia/reperfusion model establishment in mice
In the current study, global cerebral ischemia/reperfusion was induced by BCCAO operation. Mice were inhaled with 3 % isoflurane for anesthetic induction, and the anesthetic was maintained with 1.5 % isoflurane inhalation. After a midline incision between the neck and sternum had been made, both common carotid arteries were located lateral to the sternocleidomastoid. The surrounding tissues and vagus nerve were separated carefully. Global cerebral ischemia was induced by clamping the common carotid arteries with two miniature artery clips. After 20 min of ischemia, the clips were removed to allow the blood reflow. During the surgical procedure, the pericranial temperature was maintained at 37.0 ± 0.5 ℃ with a heating pad. Additionally, regional cerebral blood flow (rCBF) was monitored by a laser Doppler flowmeter (PeriFlux System 5000; Perimed, Stock- holm, Sweden). The measurement started from the time of anesthetic induction till 5 minutes after reperfusion. In this experiment, only mice whose cortical rCBF was suppressed to <15% of baseline during ischemia and was recovered to 85% during reperfusion process were included for data analysis. Mice were treated with the vehicle or different concentrations of APG via intraperitoneal injection at 30 minutes after reperfusion. The concentrations of APG were 25 mg/ml, 50 mg/ml, and 100mg/ml and 10% DMSO was used as vehicle in this experiment.
Neurobehavioral evaluation in mice
At 72 h after reperfusion, mice were subjected to modify neurological measurements designed to detect motor deficits by an observer unaware of the grouping. Tree tests were performed in this experiment. In the first test, a 10×20 cm screen with 0.2cm×0.2cm grid size, which could be rotated from horizontal to vertical, was used. During the test, the mouse was placed on the horizontal screen, and then the screen was rotated into a vertical plane. The duration time that the mouse held onto the screen was recorded for a maximum of 15 s. Next, the mouse was put in the middle of a horizontal wooden rod with a diameter of 1.5 cm. The time that the mouse was able to remain balanced on the rod was recorded for a maximum of 30 seconds. Finally, a prehensile traction test was performed. The time that the mouse could cling to a horizontal rope was recorded for a maximum of 5 seconds. Every test was allowed a total of 3 points. From 3 tests, a total motor score was computed for 9 possible points.
Histological evaluation of neuronal damage in mice
The evaluation of histological damage was performed 72 hours after reperfusion when the neurological test had finished. After perfused with freshly ice-cold paraformaldehyde in 0.01 M phosphate buffered saline (PBS), brains were removed, and coronal sectioned. Then sections that included the dorsal hippocampus were stained with hematoxylin-eosin, thus the pyramidal neurons in CA1 region could be stained. The total number of viable neurons in the CA1 region was counted in a blind manner by light microscopy at ×400 magnification in 3 different fields for each section. Data from 6 mice in each group were averaged.
Transient focal cerebral ischemia/reperfusion model in rats
Middle cerebral artery occlusion model was used to induce transient focal cerebral ischemia reperfusion injury as described previously (22). For the preparation of surgery, the rats were fasted overnight with free access to tap water. Rats were anesthetized with 10% chloral hydrate (350 mg/kg). To obstruct blood flow in MCA, a 3-0 nylon suture with blunted head was inserted into internal carotid artery through the right common carotid artery until a middle resistance was felt. The occlusion was maintained in position for 2 h and subsequently removed to allow blood reperfusion. The administration of APG was confirmed according to the paradigm used in mice, as described above.
Neurological score and infarct volume evaluation in rats
The neurological outcome was evaluated at 72 h after reperfusion by a blinded observer according to Garcia's methods.
After neurological outcome examination, rats' brains were removed and sectioned. The infarct volume was evaluated as described previously. Briefly, the slices were stained with 2% solution of 2,3,5-triphenyltetrazolium chloride at 37 ℃ and then transferred to 4% paraformaldehyde for 24 h. The brain slices were photographed using a digital camera (Canon Ixus 220HS, Japan). The infarct volume was calculated according to the following equation: relative infarct size = (contralateral area - ipsilateral non-infarct area) / contralateral area.
Western blot assessment
Tissue from MCAO models was used to investigate the mechanism of APG induced neuroprotective effect. The penumbra tissue of the anesthetized rats from another independent groups was promptly removed at 2 h after reperfusion (n = 5 per group). Tissue was homogenized in ice-cold RIPA lysis buffer (Beyotime, Nantong, China) mixed with 1% phenylmethanesulfonyl fluoride (PMSF). Equal amount of protein (40 μg) was loaded into each lane of a polyacrylamide-SDS gel and electrophoresed. The resolved proteins were transferred to a PVDF membrane. Membranes were blocked with 5% bovine serum albumin (BSA) and incubated with the appropriate primary antibodies overnight at 4 °C. The membranes were each washed three times in Tris-Buffered Saline Tween-20 (TBST) for 5 min. Then the membranes were incubated for 1h at room temperature with the appropriate secondary antibodies. The immune-blots were immersed in enhanced chemiluminescent (ECL; Millipore) reagent to analyze specific protein. Protein expression or activation changes were presented with the ratio of β-tubulin or GAPDH.
ELISA measurement of antioxidants and oxidative productions
Penumbra samples from the ischemic hemisphere were harvested and homogenized in ice-cold saline with a 1:10 weight-to volume value. Then the samples were centrifuged at 3000 rpm for 15 min at 4 °C and the supernatant was collected for ELSIA assessment. The contents of antioxidant enzymes, total superoxide dismutase (T-SOD, Beyotime), catalase (CAT, Beyotime) and total antioxidant capacity (T-AOC, Beyotime); the oxidative productions, malondialdehyde (MDA, Beyotime), protein nitrotyrosine (Millipore) and reactive oxygen species (ROS, West-tone) were measured by aviable ELISA kits, according to their instructions.
Statistical Analysis
Nonparametric values such as neurological scores were analyzed by a Kruska-Wallis test followed by the Mann-Whitney U statistic with Bonferroni correction. Other values were presented as mean ± SD and were analyzed by one-way analysis of variance, followed by Bonferroni correction for post-hoc T-test. Values of P<0.05 were considered statistically significant.
Results
Protective effect of APG against OGD-R injury
As shown in Figure 1, OGD-re-oxygenation reduced cell viability to 64±5.13% of the sham group and APG at administration of 0.2 ug/ml showed no significant effect on increasing the cell viability. Treatment with APG at 0.4 ug/ml and 0.8 ug/ml restored the cell viability (Fig. 1A, P<0.05 vs. the control group, respectively). However, no dose-dependent effect was detected in these two groups.
Compared to the sham group, the LDH level in the cells of OGD-R groups was increased significantly. Treatment with APG at 0.2 ug/ml did not reduce the elevated LDH level induced by OGD-R injury. APG treatment at 0.4 ug/ml and 0.8 ug/ml showed significant protective effect on attenuation of the incresed LDH level induced by OGD-R, as compared to OGD group (Fig. 1B, 53.4% reduction in 0.4 ug/ml group, P<0.01; 45.3% reduction in 0.8 ug/ml group; P<0.05).
To test the effect of APG treatment on cell apoptosis, flow cytometric analysis was also conducted in this study (Fig. 2A-E). The apoptotic index of the control and vehicle group were increased in comparison with the sham group (Fig. 2K). Both treatment with 0.4 ug/ml and 0.8 ug/ml APG attenuated OGD-R-induced cell apoptosis significantly as compared with that in OGD-R group (63.8% reduction in 0.4 ug/ml group; 59.2% reduction in 0.8 ug/ml group; P<0.01, respectively). Conversely, treatment with 0.2 ug/ml APG had no protective effect on cell apoptosis.
Figure 1
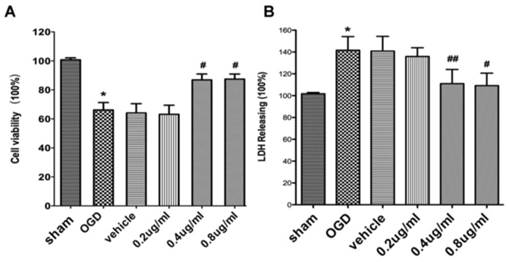
The protective effect of APG against OGD-R injury in PC12 cell line. Effect of APG treatment on the cell viability (A) and plasma lactate dehydrogenase (LDH) releasing level (B) in the PC12 cell culture after OGD-R. A, cell viability analysis; B. LDH releasing level evaluation. APG treatment significantly increased cell viability and decreased the LDH releasing level compared with OGD group. * P < 0.05 vs. sham group; #, ## P < 0.05, P < 0.01 vs. OGD group. n = 5 per group.
Figure 2
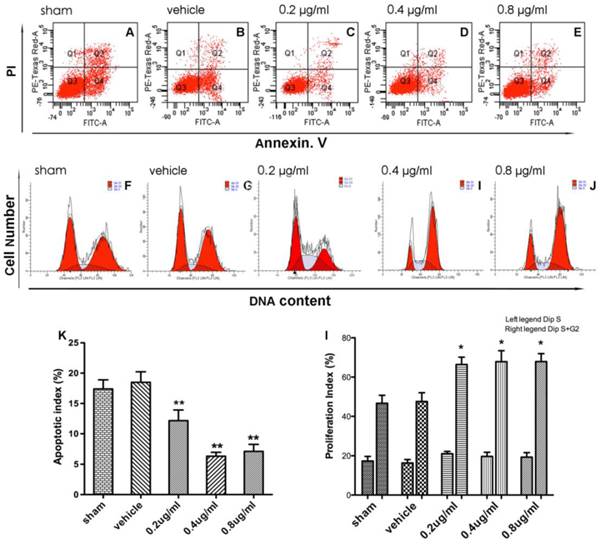
Effect of APG therapy on the apoptotic level and cell proliferation in PC12 cells. A-E. Representative dot plots showing flow cytometric analysis of PC12 cells treated with different concentrations of APG for the dedicated time periods and then stained with FITC-conjugated Annexin V and propidium iodide in OGD (A), vehicle (B) and 0.2 ug/ml (C), 0.4 ug/ml (D), 0.8 ug/ml (E) APG treatment groups, respectively. F-J. Flow cytometric analysis of cell cycle phase distribution of PC12 cells after treatment with different concentrations of APG or vehicle (F, control; G, vehicle; H, 0.2 ug/ml; I, 0.4 ug/ml; J, 0.8 ug/ml). K. Evaluation of apoptotic index in each group (n = 5). I. Analysis of proliferation index in each group (n = 5). All concentrations of APG administration increased the percentage of S+G2 cells but not altered the percentage of S cells (the left legend is the percentage of S cells, the left legend is the percentage of S+G2 cells). * P < 0.05, ** P < 0.01 vs. sham group.
The effect of APG treatment on cell proliferation was also measured in the present study (Fig. 2F-J). Treatment of PC12 cells with different doses of APG increased the percentage of the S and G2 phase cells as compared with sham group, however no dose-dependent changes were found (Fig. 2I, 38.2%, 50.9%, 45.5% improvement in 0.2 ug/ml group, 0.4 ug/ml group, 0.8 ug/ml group; P<0.01, respectively).
Neuroprotective effect of APG treatment in a global cerebral ischemia/perfusion mice model
As shown in Fig. 3A, there was no significant difference in rCBF changes among all groups at the corresponding time points (Fig. 3B). No mice died until the neurological assessment at 72 h after reperfusion. In the BCCAO and vehicle groups, the total motor scores were reduced in global ischemia/reperfusion group, as compared with the one in sham groups. Both treatment with 50mg/kg (7.67[7, 9]) and 100 mg/kg (7.67[7-9]) APG attenuated the total motor scores deficiency evidently (Fig. 3B, in 100mg/kg versus 5.9[5-7] in BCCAO group). However, no statistical difference was detected between these two different dose groups. No protective effect was found in 25 mg/kg APG treatment group (6.8 [6-8]).
Figure 3
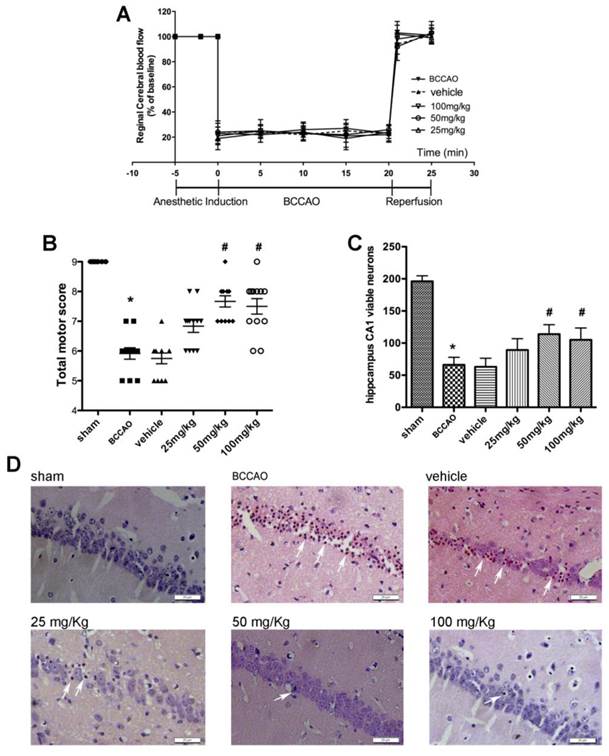
APG treatment improved the total motor scores (B) and protected hippocampal CA1 neurons (C) against global ischemic/reperfusion injury. A. Changes in regional cerebral blood flow (rCBF) in C57BL/6 mice during BCCAO operation. Data are expressed as mean ± SD. B. Triangle, square and circle symbols represent mice in each group (n = 12), respectively. C. Counting of viable neuronal cells in hippocampal area in each group (n = 12). D. Representative microphotographs of hematoxylin eosin staining in sham, BCCAO, vehicle, 25 mg/kg, 50 mg/kg and 100 mg/kg APG treatment groups. * P < 0.05 vs. sham group; # P < 0.05, ## P < 0.01 vs. BCCAO group. BCCAO = bilateral common carotid artery occlusion. Bar = 20 μm
The histopathological results are shown in Fig. 3C-D. Viable neurons in hippocampal CA1 region were significantly decreased at 72 h after ischemia/reperfusion compared with which in the sham group. Treatment with 50 mg/kg or 100 mg/kg APG increased the number of reduced viable neurons induced by ischemia/reperfusion injury as compared with the BCCAO group. However, administration with 25 mg/kg APG had no protective effect on viable neurons.
Neuroprotective effect of APG in a rat focal cerebral ischemia/perfusion model
No significant difference in rCBF changes was detected among all groups at the corresponding time points (Fig. 4A). The neurological score assessment is presented in Fig. 4B. The MCAO group revealed a significant neurological deficit score when compared to the sham group. Treatment with APG significantly improved the neurological scores, in 50mg/kg (13.63[10-16]) or 100mg/kg (12.38[9, 14]) respectively, as compared with that in the MCAO group (8.0[6-12]). However, no statistical difference was detected in neurological score between the MCAO group and 25 mg/kg APG treatment group (9.62[7-13]).
As shown in Fig. 4C-D, cerebral I/R injury induced a 41.12% infarction volume in the MCAO group while the sham group showed no infarction area. Compared with the control group, the cerebral infarct volumes had a 30.0% deduction in 50mg/kg APG group (P < 0.01) and a 23.3% deduction in 100mg/kg APG group (P<0.05). No difference was detected between the latter two groups.
APG treatment promoted STAT3 phosphorylation
As shown in Fig. 5A, the difference of STAT3 phosphorylation level between MCAO and vehicle group was not statistically significant. APG treatment increased the phosphorylation of STAT3 at 705Y as compared to the MCAO group at 2 h after reperfusion (P<0.05). However, no significant change was detected in overall expression of total STAT3 among all the groups.
Figure 4
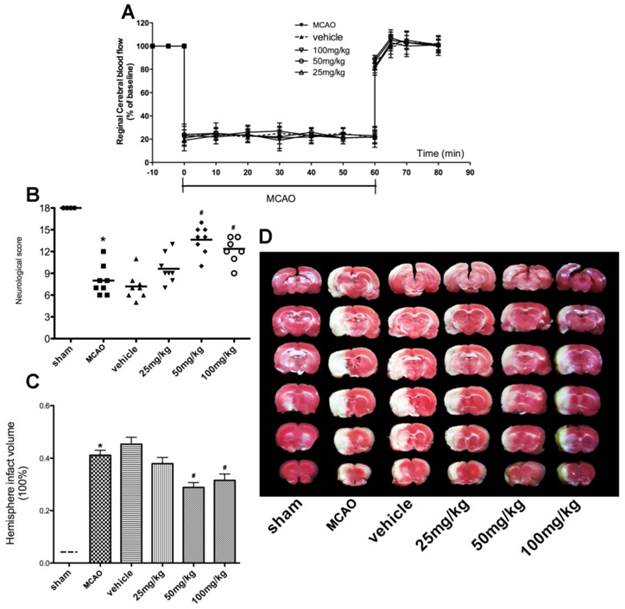
Neuroprotective effect of APG therapy against focal cerebral ischemic/reperfusion injury in rats. A. Regional cerebral blood flow in ischemic hemispheres of rats during MCAO operation. Data are expressed as means ± SD. B. Neurological scores; C. infarct volumes at 72 h after reperfusion; D. representative photographs of TTC staining. APG treatment significantly improved the neurological scores and attenuated infarct volume as compared with the MCAO group (n = 8). * P < 0.05 vs. sham group; # P<0.05 vs. MCAO group. MCAO = middle cerebral artery occlusion.
Figure 5
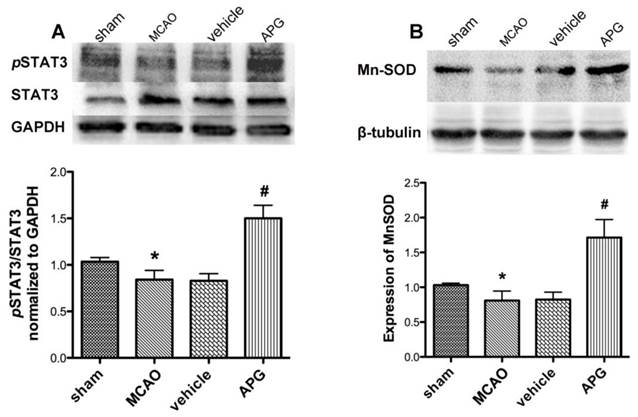
Effect of APG therapy on the phosphorylation of STAT3 at 705Y (A) and the expression of Mn-SOD (B). A. The upper part is the photograph of pSTAT3 (705Y), total STAT3, and the corresponding GAPDH bands, respectively. The lower part is the result of western-blot statistical analysis in the sham, control, vehicle and APG treatment groups (n = 5). APG treatment increased STAT3 phosphorylation as compared with the MCAO group. B. The upper part is the photograph of Mn-SOD and its corresponding β-tubulin bands. The lower part is the result of western-blot statistical analysis in the sham, MCAO, vehicle and APG treatment groups (n = 5). APG treatment increased Mn-SOD2 expression as compared with the control group. * P < 0.05 vs. sham group; # P < 0.05 vs. MCAO group. MCAO = middle cerebral artery occlusion.
APG treatment upregulated expression of Mn-SOD protein
As shown in Fig. 5B, ischemic/reperfusion injury attenuated the content of SOD2 protein as compared with that in the sham group. There is no statistical significant difference in Mn-SOD protein expression between MCAO and vehicle group subjected to MCAO injury. APG treatment increased the expression of Mn-SOD as compared with the control group at 2 h after reperfusion (P<0.05).
APG therapy increased protective antioxidant effect
The impact of APG treatment on antioxidant enzymes expression was compared with the MCAO groups at 24 h after reperfusion. Some antioxidant enzymes, CAT, T-AOC, and T-SOD were activated by APG treatment as measured by ELISA kits (Fig. 6A-C; P<0.05, respectively).
Some oxidative productions were also measured in the current study. As shown in Fig. 6D-E, the content of MDA, nitrotyrosine and ROS increased in the MCAO group compared to the sham group. APG treatment attenuated the expression of MDA and ROS compared to the MCAO group (P<0.05, respectively).
Discussion
In present study, we demonstrated for the first time as we known that APG exhibited neuroprotective effects against cerebral ischemic/reperfusion injury. APG markedly increased cell proliferation ratio, reduced the number of apoptotic cells; improved cell viability and attenuated LDH releasing level against OGD-R as demonstrated in vitro. Also, APG treatment significantly ameliorated neurological behavior, elevated survival neurons and reduced infarct volume in both global cerebral ischemic and focal cerebral ischemic models in vivo. Furthermore, the results of the current study showed that APG treatment dramatically reduced levels of MDA, protein nitrotyrosine, ROS, as well as improved the content of antioxidant enzymes. In addition, APG treatment increased Mn-SOD protein expression and the phosphorylation level of STAT3 at 705Y. These results indicate that the antioxidant activity induced by APG treatment was mediated by Mn-SOD up-regulation via STAT3 phosphorylation dependent mechanism, which may be one of the probable mechanisms of the neuroprotection induced by APG treatment.
Accumulating studies suggest that some bioactive compounds discovered from traditional Chinese medicines such as apigenin can attenuate cerebral ischemic/reperfusion injury in the animal models (23). As a glycoside flavonoid, APG is one of the major compounds isolated from C. tangutica. The molecular weight of APG is small than 3,000, and it is a liposoluble constituent. Therefore, APG is believed to easily and rapidly cross the blood-brain barrier and exert biological activities in the brain.
Figure 6
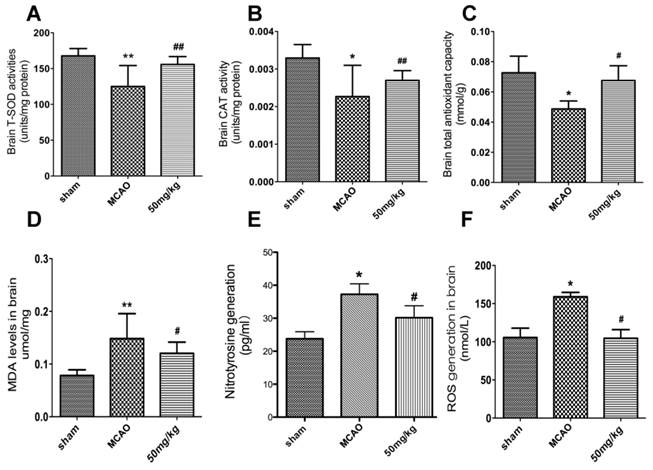
Effect of APG therapy on the antioxidant enzymes activity and oxidative productions generation. A-C. The comparisons of T-SOD (A), CAT (B) and TAOC (C) level in the sham, MCAO, vehicle and APG treatment groups (n = 6 for each group). D-E. The comparisons of MDA (D), nitrotyrosine (E) and ROS (F) level in the sham, MCAO, vehicle and APG treatment groups (n = 6 for each group). APG treatment increased the antioxidants activity and reduced the contents of oxidative productions. * P < 0.05 vs. sham group; # P < 0.05 vs. MCAO group. T-SOD = Total superoxide dismutase; CAT = Catalase; TAOC = Total antioxidant capacity; MDA = Malondialdehyde; ROS = Reactive oxygen species. MCAO = middle cerebral artery occlusion.
Currently, we have demonstrated that APG can confer neuroprotection in three considerable ischemic/reperfusion injury models, respectively. In PC12 cell line, APG improved cell viability, increased cell proliferation ratio, attenuated LDH releasing level and reduced the number of apoptotic cells. Additionally, we have also shown that APG can induce neuroprotective effect in global cerebral ischemic/reperfusion model of mice, as evidenced by improved neurological function and increased survival neurons. Moreover, APG also ameliorated neurological function and attenuated infarct volume measurement in a transient rat MCAO model. The neuroprotective dose of APG at 0.4 μg/ml in vitro or 50 mg/kg in vivo was more potent than those at 0.2 μg/ml in vitro or 25 mg/kg in vivo. However, the higher dose of APG (0.8 μg/ml in vitro or 100 mg/kg in vivo) did not show any more protective potency, indicating that the middle dose (0.4 mg/ml in vitro or 50 mg/kg in vivo) of APG might be an appropriate dose for the further studies.
It is well established that the overproduction of ROS is a crucial injurious mechanism in the context of I/R injury (24). During I/R process, suddenly accumulated ROS cannot be efficiently scavenged by the endogenous antioxidant enzymes, which subsequently interacts with subcellular biomolecules, causing the cascade amplification of injury pathogenesis such as mitochondrial dysfunction, apoptosis (24,25). Therefore, raising the expression of endogenous antioxidant enzymes can efficaciously reduce cerebral I/R injury. The prominent free radical scavenging activity of APG has been identified (26, 27). In the present study, we have demonstrated that APG increased the expression of antioxidant enzymes including SOD, CAT, and T-AOC. These enzymes constitute the first-line cellular defense system against oxidative injury, thus the activation of antioxidant enzymes induced by APG treatment could induce neuroprotective effect.
The half-life of ROS such as O2.- was very short. Thus, it is very difficult to real-time monitor them in vivo. The breaking out of ROS induced by I/R also induces the damage of biomolecules, such as lipid and protein. So the assessment of oxidative biomolecules was constantly used to evaluate the oxidative injury. In the current study, we demonstrated that APG reduced the nitrotyrosine protein, MDA and ROS levels in brain tissue. This result indicates the involvement of the antioxidant effect in the neuroprotection induced by APG. However, the underlying mechanism of the antioxidant effect induced by APG is still unknown.
Mn-SOD primarily mediated the mitochondrial mechanism to eliminate or maintain the homeostatic level of O2-, to process ROS into fewer toxic substances (28, 29). It is also an important particle of endogenous protective mechanism against cerebral ischemic damage. In this study, we have shown that the Mn-SOD protein expression was suppressed in the penumbra at 2 h after reperfusion, which was in consistent with previous studies (30). In addition, in the APG-treated brain, the content of Mn-SOD protein was much more pronounced than that in mice subjected to only cerebral I/R injury. This result indicated the involvement of Mn-SOD in the neuroprotective effect induced by APG. The up-regulation of Mn-SOD can undergo transcriptional, translational and post-translational regulation mechanism, which induced protective effect against oxidative injury. However, it is still unclear which upstream signal pathway transduced the activation of Mn-SOD induced by APG. As a signaling transducer, the activation of STAT3 mediated much subcellular protective mechanism, also including the antioxidant effect (31, 32). It has been identified that STAT3 was a novel transcription factor of the Mn-SOD gene (30), in present study, we have demonstrated that APG increases the STAT3 phosphorylation level at 705Y, indicating that APG induces the Mn-SOD up-regulation via STAT3 phosphorylation dependent- mechanism and then attenuates the oxidative stress, resulting in neuroprotective effect.
Limitations
Some limitations should be pointed out in this study. Firstly, more experimental evidences are needed to determine the correlation of Mn-SOD up-regulation and the neuroprotection induced by APG. Moreover, as a transcriptional factor, the phosphorylation of STAT3 could be increased by APG, but more investigations are needed to determine the upstream pathway that transduced the phosphorylation of STAT3 induced by APG.
Conclusion
Our study provided both in vitro and in vivo evidence that APG exerted neuroprotective effects against cerebral ischemic/reperfusion injury. Although much more studies are still needed to explain the involvement of transduction signal pathways between APG and antioxidant system fully, the present study demonstrates that APG treatment can provide ischemic tolerance to cerebral ischemic oxidative injury. This was mediated by activation of Mn-SOD signaling pathway via STAT3 phosphorylation dependent- mechanism. Thus, these results indicate that APG may be a potent therapeutic drug for the treatment of ischemic stroke.
Supplementary Material
Supplementary Figure 1.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 81371478 for QingRong Tan) and Discipline supporting program funding from Xijing Hospital (XJZT14M13 for Min Cai). The authors appreciate Dr. Bairen Wang (Department of Anesthesiology, Xijing Hospital) for carefully revising the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Yang G, Wang Y, Zeng Y, Gao GF, Liang X, Zhou M. et al. Rapid health transition in China, 1990-2010: findings from the Global Burden of Disease Study 2010. Lancet. 2013;381:1987-2015
2. Fonarow GC, Smith EE, Saver JL, Reeves MJ, Bhatt DL, Grau-Sepulveda MV. et al. Timeliness of tissue-type plasminogen activator therapy in acute ischemic stroke: patient characteristics, hospital factors, and outcomes associated with door-to-needle times within 60 minutes. Circulation. 2011;123:750-8
3. Chan PH. Reactive oxygen radicals in signaling and damage in the ischemic brain. J Cerebr Blood F Met. 2001;21:2-14
4. Niizuma K, Endo H, Chan PH. Oxidative stress and mitochondrial dysfunction as determinants of ischemic neuronal death and survival. J Neurochem. 2009;109(Suppl 1):133-8
5. Crack PJ, Taylor JM. Reactive oxygen species and the modulation of stroke. Free Radical Bio Med. 2005;38:1433-44
6. Shuaib A, Lees KR, Lyden P, Grotta J, Davalos A, Davis SM. et al. NXY-059 for the treatment of acute ischemic stroke. New Engl J Med. 2007;357:562-71
7. Lin Y, Chen F, Zhang J, Wang T, Wei X, Wu J. et al. Neuroprotective effect of resveratrol on ischemia/reperfusion injury in rats through TRPC6/CREB pathways. J Mol Neurosci. 2013;50:504-13
8. Ha SK, Lee P, Park JA, Oh HR, Lee SY, Park JH. et al. Apigenin inhibits the production of NO and PGE2 in microglia and inhibits neuronal cell death in a middle cerebral artery occlusion-induced focal ischemia mice model. Neurochem Int. 2008;52:878-86
9. Wang S, Wei H, Cai M, Lu Y, Hou W, Yang Q. et al. Genistein attenuates brain damage induced by transient cerebral ischemia through up-regulation of ERK activity in ovariectomized mice. Int J Biol Sci. 2014;10:457-65
10. Sun L, Jin Y, Dong L, Sumi R, Jahan R, Li Z. The neuroprotective effects of Coccomyxa gloeobotrydiformis on the ischemic stroke in a rat model. Int J Biol Sci. 2013;9:811-7
11. Zhong HM, Chen CX, Tian X, Chui YX, Chen YZ. Triterpenoid saponins from Clematis tangutica. Planta Med. 2001;67:484-8
12. Du Z, Zhu N, Ze-Ren-Wang-Mu N, Shen Y. Two new antifungal saponins from the Tibetan herbal medicine Clematis tangutica. Planta Med. 2003;69:547-51
13. Zhang W, Lu YY, Li Y, Hai WL, LI Y, Tang HF. Flavonoid glycosides constituents from Clematis tangutica. Centr South Pharma. 2014;12(5):1-4
14. Delazar A, Celik S, Gokturk RS, Unal O, Nahar L, Sarker SD. Two acylated flavonoid glycosides from Stachys bombycina, and their free radical scavenging activity. Die Pharmazie. 2005;60:878-80
15. Patil SP, Jain PD, Sancheti JS, Ghumatkar PJ, Tambe R, Sathaye S. Neuroprotective and neurotrophic effects of Apigenin and Luteolin in MPTP induced parkinsonism in mice. Neuropharmacology. 2014;86:192-202
16. Liu R, Zhang T, Yang H, Lan X, Ying J, Du G. The flavonoid apigenin protects brain neurovascular coupling against amyloid-beta(2)(5)(-)(3)(5)-induced toxicity in mice. J Alzheimers Dis. 2011;24:85-100
17. Zhao L, Wang JL, Liu R, Li XX, Li JF, Zhang L. Neuroprotective, anti-amyloidogenic and neurotrophic effects of apigenin in an Alzheimer's disease mouse model. Molecules. 2013;18:9949-65
18. Yamagata K, Kitazawa T, Shinoda M, Tagawa C, Chino M, Matsufuji H. Stroke status evoked adhesion molecule genetic alterations in astrocytes isolated from stroke-prone spontaneously hypertensive rats and the apigenin inhibition of their expression. Stroke. 2010. 2010
19. Ma YL, Qin P, Li Y, Shen L, Wang SQ, Dong HL. et al. The effects of different doses of estradiol (E2) on cerebral ischemia in an in vitro model of oxygen and glucose deprivation and reperfusion and in a rat model of middle carotid artery occlusion. BMC Neurosci. 2013;14:118
20. Lin H, Zhang X, Cheng G, Tang HF, Zhang W, Zhen HN. et al. Apoptosis induced by ardipusilloside III through BAD dephosphorylation and cleavage in human glioblastoma U251MG cells. Apoptosis. 2008;13:247-57
21. Majewski L, Sobczak M, Wasik A, Skowronek K, Redowicz MJ. Myosin VI in PC12 cells plays important roles in cell migration and proliferation but not in catecholamine secretion. J Muscle Res Cell M. 2011;32:291-302
22. Dong H, Fan YH, Zhang W, Wang Q, Yang QZ, Xiong LZ. Repeated electroacupuncture preconditioning attenuates matrix metalloproteinase-9 expression and activity after focal cerebral ischemia in rats. Neurolo Res. 2009;31:853-8
23. Choi AY, Choi JH, Lee JY, Yoon KS, Choe W, Ha J. et al. Apigenin protects HT22 murine hippocampal neuronal cells against endoplasmic reticulum stress-induced apoptosis. Neurochem Int. 2010;57:143-52
24. Chen SD, Yang DI, Lin TK, Shaw FZ, Liou CW, Chuang YC. Roles of oxidative stress, apoptosis, PGC-1alpha and mitochondrial biogenesis in cerebral ischemia. Int J Mol Sci. 2011;12:7199-215
25. Saito A, Maier CM, Narasimhan P, Nishi T, Song YS, Yu F. et al. Oxidative stress and neuronal death/survival signaling in cerebral ischemia. Mol Neurobiol. 2005;31:105-16
26. Heijnen CG, Haenen GR, van Acker FA, van der Vijgh WJ, Bast A. Flavonoids as peroxynitrite scavengers: the role of the hydroxyl groups. Toxicol In Vitro. 2001;15:3-6
27. Murota K, Shimizu S, Miyamoto S, Izumi T, Obata A, Kikuchi M. et al. Unique uptake and transport of isoflavone aglycones by human intestinal caco-2 cells: comparison of isoflavonoids and flavonoids. J Nutr. 2002;132:1956-61
28. Wang Q, Li L, Li CY, Pei Z, Zhou M, Li N. SIRT3 protects cells from hypoxia via PGC-1alpha- and MnSOD-dependent pathways. Neuroscience. 2015;286:109-21
29. Li F, Wang H, Li L, Huang C, Lin J, Zhu G. et al. Superoxide plays critical roles in electrotaxis of fibrosarcoma cells via activation of ERK and reorganization of the cytoskeleton. Free Radical Bio Med. 2012;52:1888-96
30. Jung JE, Kim GS, Narasimhan P, Song YS, Chan PH. Regulation of Mn-superoxide dismutase activity and neuroprotection by STAT3 in mice after cerebral ischemia. J Neurosci. 2009;29:7003-14
31. Yang Y, Duan W, Jin Z, Yi W, Yan J, Zhang S. et al. JAK2/STAT3 activation by melatonin attenuates the mitochondrial oxidative damage induced by myocardial ischemia/reperfusion injury. J Pineal Reas. 2013;55:275-86
32. Dixit D, Sharma V, Ghosh S, Koul N, Mishra PK, Sen E. Manumycin inhibits STAT3, telomerase activity, and growth of glioma cells by elevating intracellular reactive oxygen species generation. Free Radical Bio Med. 2009;47:364-74
Author contact
![]() Corresponding author: Dr. Tang Qingrong, MD. PhD., Department of Psychiatry, Xijing Hospital, the Fourth Military Medical University, Xi'an 710032, Shaanxi Province, China. Tel: +86-29-89661593; Fax: +86-29-89661593; E-mail: tanqingredu.cn
Corresponding author: Dr. Tang Qingrong, MD. PhD., Department of Psychiatry, Xijing Hospital, the Fourth Military Medical University, Xi'an 710032, Shaanxi Province, China. Tel: +86-29-89661593; Fax: +86-29-89661593; E-mail: tanqingredu.cn
Citation styles
APA
Cai, M., Ma, Y., Zhang, W., Wang, S., Wang, Y., Tian, L., Peng, Z., Wang, H., Qingrong, T. (2016). Apigenin-7-O-β-D-(-6''-p-coumaroyl)-Glucopyranoside Treatment Elicits Neuroprotective Effect against Experimental Ischemic Stroke. International Journal of Biological Sciences, 12(1), 42-52. https://doi.org/10.7150/ijbs.12275.
ACS
Cai, M.; Ma, Y.; Zhang, W.; Wang, S.; Wang, Y.; Tian, L.; Peng, Z.; Wang, H.; Qingrong, T. Apigenin-7-O-β-D-(-6''-p-coumaroyl)-Glucopyranoside Treatment Elicits Neuroprotective Effect against Experimental Ischemic Stroke. Int. J. Biol. Sci. 2016, 12 (1), 42-52. DOI: 10.7150/ijbs.12275.
NLM
Cai M, Ma Y, Zhang W, Wang S, Wang Y, Tian L, Peng Z, Wang H, Qingrong T. Apigenin-7-O-β-D-(-6''-p-coumaroyl)-Glucopyranoside Treatment Elicits Neuroprotective Effect against Experimental Ischemic Stroke. Int J Biol Sci 2016; 12(1):42-52. doi:10.7150/ijbs.12275. https://www.ijbs.com/v12p0042.htm
CSE
Cai M, Ma Y, Zhang W, Wang S, Wang Y, Tian L, Peng Z, Wang H, Qingrong T. 2016. Apigenin-7-O-β-D-(-6''-p-coumaroyl)-Glucopyranoside Treatment Elicits Neuroprotective Effect against Experimental Ischemic Stroke. Int J Biol Sci. 12(1):42-52.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.