Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results and Discussions
Conclusion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2016; 12(1):109-119. doi:10.7150/ijbs.12358 This issue Cite
Research Paper
The Complete Mitochondrial Genome of two Tetragnatha Spiders (Araneae: Tetragnathidae): Severe Truncation of tRNAs and Novel Gene Rearrangements in Araneae
Zheng-Liang Wang#, Chao Li#, Wen-Yuan Fang, Xiao-Ping Yu ![]()
Zhejiang Provincial Key Laboratory of Biometrology and Inspection and Quarantine, College of Life Sciences, China Jiliang University, Hangzhou, Zhejiang, 310018, People's Republic of China
# These authors contributed equally to this paper.
Received 2015-4-9; Accepted 2015-10-26; Published 2016-1-1
Abstract

Mitogenomes can provide information for phylogenetic analysis and evolutionary biology. The Araneae is one of the largest orders of Arachnida with great economic importance. In order to develop mitogenome data for this significant group, we determined the complete mitogenomes of two long jawed spiders Tetragnatha maxillosa and T. nitens and performed the comparative analysis with previously published spider mitogenomes. The circular mitogenomes are 14578 bp long with A+T content of 74.5% in T. maxillosa and 14639 bp long with A+T content of 74.3% in T. nitens, respectively. Both the mitogenomes contain a standard set of 37 genes and an A+T-rich region with the same gene orientation as the other spider mitogenomes, with the exception of the different gene order by the rearrangement of two tRNAs (trnW and trnG). Most of the tRNAs lose TΨC arm stems and have unpaired amino acid acceptor arms. As interesting features, both trnSAGN and trnSUCN lack the dihydrouracil (DHU) arm and long tandem repeat units are presented in the A+T-rich region of both the spider mitogenomes. The phylogenetic relationships of 23 spider mitogenomes based on the concatenated nucleotides sequences of 13 protein-coding genes indicated that the mitogenome sequences could be useful in resolving higher-level relationship of Araneae. The molecular information acquired from the results of this study should be very useful for future researches on mitogenomic evolution and genetic diversities in spiders.
Keywords: Mitogenome, Araneae, Tetragnatha maxillosa, Tetragnatha nitens, Phylogeny
Introduction
Generally, the arthropod mitogenome is a circular, double-stranded molecule, ranging in size from 14 kb to 20 kb, usually containing a standard set of 13 protein-coding genes (PCGs), two ribosomal RNA genes (the large and small ribosomal subunits, rRNAs), 22 transfer RNA genes (tRNAs) and an A+T-rich region in a highly variable length with essential regulatory elements for replication and transcription [1]. Due to the characteristics of small genome size, maternal inheritance, high rate of evolution and low level of intermolecular genetic recombination, the mitogenome has been widely regarded as an effective molecular marker for molecular evolution, phylogenetic studies, population genetics and phylogeography [2]. Moreover, the information of gene rearrangement, tRNA secondary structure, genetic code alteration and models of control of replication and transcription in the complete mitogenome data are also extensively exploited for deep-level phylogenetic inference at various taxonomic levels in the past decade [3,4].
Spiders (Araneae) are among the largest animal groups in the world due to their broad diversity, world-wide distribution and conspicuous synapomorphies [5]. Approximately 45,000 described species in 114 families make Araneae the seventh-largest order next to the five largest insect orders (Coleoptera, Hymenoptera, Lepidoptera, Diptera and Hemiptera) and Acari among the Arachnida [6]. Due to such high species diversity, there are many taxonomic problems about the order Araneae which remain to be unsolved [7,8]. Because of homoplasies or reduction of anatomical characters in morphological and ethological data [5], it would be desirable to examine an independent set of molecular data. Partial mitochondrial gene sequences of cytochrome oxidase subunit 1 (COI), rRNAs (16S and 12S) and tRNAs, and nuclear genes of rDNA (18S and 28S) and histone (H3) from spiders have been documented and extensively used for phylogenetic analysis [9,10]. However, these short mitochondrial or nuclear genes usually lack the phylogenetic information in resolving phylogenetic relationships among families or subfamilies within one order and sometimes resulted in controversial signal [8,11]. Hence, additional reliable data sets for phylogenetic reconstructions, such as the complete mitogenome sequences, are required.
To date, more than 130 complete mitogenomes from species of arachnids have been determined, but only 21 species in 14 families from Araneae were publicly available in GenBank. These 21 mitogenomes share a similar molecular size, ranging from 13,874 bp in Ornithoctonus huwena [12] to 14,741 bp in Wadicosa fidelis [13], but have variations in mitogenomic characteristics, such as gene content and gene arrangement. Arachnid mitogenomes, in particular spider mitogenomes, possess compact mitogenomes and their tRNAs usually lack the sequence for the T-arm and a paired aminoacyl stem of the cloverleaf structure. Such atypical tRNAs might be modified by post-transcriptional editing to be functional [14].
In the present study, we sequenced and annotated the complete mitogenome sequences of two long jawed spiders Tetragnatha maxillosa and T. nitens (Araneae: Tetragnathidae), which are the dominant arthropod predators in the agroecological system. 21 spider mitogenomes that are available in GenBank were used in the comparative analysis to reveal the gene rearrangements in two Tetragnatha species. Additionally, phylogenetic study was also performed to assess the utility of mitogenome data to understand higher phylogeny of Araneae.
Materials and methods
Sample collection and DNA extraction
Specimens of spiders, T. maxillosa and T. nitens were collected from the paddy fields in Yuyao (E121°09′, N30°03′), Zhejiang Province, China. The field collections did not involve endangered or protected species and no specific permits were required for our collecting. All collections were preserved in 95% ethanol and stored in the temperature cabinet at 4°C. Total genomic DNA was extracted from leg tissue of each spider individual of the two Tetragnatha species using the DNeasy Tissue Kit (Qiagen, Hilden, Germany) according to the manufactures protocol.
PCR amplification, cloning and sequencing
Primer sequences of long PCR amplification were designed based on the conserved sequences which were obtained by aligning the complete mitogenome sequences of four spider species that were downloaded from GenBank: Habronattus oregonnesis (NC_005942), Heptathela hangzhouensis (NC_005924), Nephila clavata (NC_008063) and Telamonia vlijmi (NC_024287). The six primer sets that were used to amplify overlapping fragments of the complete mitogenome of T. maxillosa and T. nitens are shown in Table S1.
All PCRs were performed using TaKaRa LA PCR Amplification Kit Version 2.1 (Takara Co., Dalian, China) with the following amplification condition: an initial denaturation for 5 min at 95°C, followed by 40 cycles of denaturation for 10s at 92°C, annealing for 30s at 50°C, elongation for 4 min at 68°C in the initial 20 cycles and then increase by 20s per cycle in the final 20 cycles and a final extension step for 10 min at 72°C. All PCR products were resolved by electrophoresis in 1.0% agarose gel, purified with DNA Gel Purification Kit (TianGen, China), ligated to the pGEM-T-Easy vector (Promega,USA) and sequenced using DNA Sequencer (ABI377) at Sangon Inc (Shanghai, China).
Sequencing assembling and annotation
The complete mitogenome sequences were assembled using the Staden package 1.7.0 [15]. PCGs and rRNAs were initially identified via the MITOS web server [16] and subsequently refined by hand annotation method following the procedures proposed by Cameron [17]. Gene boundaries were determined by comparing with gene regions from previously characterized spider mitogenomes: W. fidelis [13], O. huwena and H. hangzhouensis [12]. tRNAs were identified by tRNAscan-SE 1.2.1 [18] and ARWEN 1.2.3.c [19]. The putative tRNAs which were not found by the two software tools were identified based on sequence similarity to tRNAs of the other previously published spider mitogenomes. Nucleotide composition and codon usage were calculated in MEGA 4.0 [20]. Potential inverted repeats or palindromes in the A+T-rich region were determined using Tandem Repeats Finder (http://tandem.bu.edu/trf/trf.html) with the default parameters [21]. AT-skew=(A-T)/(A+T) and GC-skew=(G-C)/(G+C) were used to measure the nucleotide compositional difference between genes [22].
The complete mitogenome sequences of T. maxillosa and T. nitens have been deposited in the GenBank database under accession number KP306789 and KP306790, respectively.
Phylogenetic analysis
A total 21 spider mitogenomes available in the GenBank were included in the phylogenetic analysis (Table 1). Limulus polyphemus (Xiphosura) was selected as an outgroup [23]. To overcome of base compositional heterogeneity and explore the effect of method choice on phylogenetic reconstruction, three datasets were assembled: (1) a concatenated nucleotide sequence alignment of 13 PCGs (PCG123); (2) a concatenated nucleotide sequence alignment of the first and second codons of 13 PCGs (PCG12); (2) a concatenated nucleotide sequence alignment of the first and second codons of 13 PCGs and two rRNAs (PCR12R). The gene order of 13 PCGs in three concatenated datasets is as follows: ATP6, ATP8, COI, COII, COIII, Cytb, ND1, ND2, ND3, ND4, ND4L ND5 and ND6. Both Maximum Likelihood (ML) and Bayesian Inference (BI) were used to infer phylogenetic trees. The GTR+I+G was selected as the best model for three nucleotide datasets under the Akaike information criterion by ModelTest 3.7 [24]. ML analysis was performed by a PHYML online web server [25] with default parameters and the node support values were evaluated via a bootstrap test with 100 replicates. BI analysis was conducted by MrBayes version 3.1.2 [26]. Two set of four chains (one cold chain and 3 hot chains) were allowed to run simultaneously for 1,000,000 generations and trees were sampled every 1,000 generations, with the first 25% discarded as burn-in. Stationarity was considered to be reached when the average standard deviation of split frequencies was less than 0.01.
Nucleotide composition bias of all spider mitogenomes available from GenBank and an outgroup species used in phylogenetic analysis.
| Order | Family | Species | Length (bp) | AT-skew | GC-skew | Accession number |
|---|---|---|---|---|---|---|
| Araneae | Tetragnathidae | Tetragnatha maxillosa | 14578 | -0.087 | 0.246 | KP306789 |
| Tetragnathidae | Tetragnatha nitens | 14639 | -0.055 | 0.200 | KP306790 | |
| Araneidae | Argiope amoena | 14121 | -0.060 | 0.241 | NC_024281* | |
| Araneidae | Argiope bruennichi | 14063 | -0.035 | 0.263 | NC_010596* | |
| Araneidae | Araneus ventricosus | 14617 | -0.048 | 0.243 | NC_025634* | |
| Araneidae | Neoscona theisi | 14156 | -0.059 | 0.252 | NC_026290* | |
| Salticidae | Habronattus oregonensis | 14381 | -0.112 | 0.301 | NC_005942* | |
| Salticidae | Telamonia vlijmi | 14601 | -0.081 | 0.235 | NC_024287* | |
| Salticidae | Plexippus paykull | 14316 | -0.105 | 0.318 | NC_024877* | |
| Lycosidae | Pirata subpiraticus | 14528 | -0.111 | 0.295 | NC_025523* | |
| Lycosidae | Pardosa laura | 14513 | -0.109 | 0.285 | NC_025223* | |
| Lycosidae | Wadicosa fidelis | 14741 | -0.130 | 0.312 | NC_026123* | |
| Pholcidae | Pholcus sp. | 14279 | -0.188 | 0.372 | NC_010775* | |
| Pholcidae | Pholcus phalangioides | 14459 | -0.191 | 0.371 | NC_020324* | |
| Dipluridae | Phyxioschema suthepium | 13931 | -0.040 | 0.472 | NC_020322* | |
| Hypochilidae | Hypochilus thorelli | 13991 | -0.140 | 0.266 | NC_010777* | |
| Nemesiidae | Calisoga longitarsis | 14070 | -0.146 | 0.365 | NC_010780* | |
| Nephilidae | Nephila clavata | 14436 | -0.053 | 0.242 | NC_008063* | |
| Theraphosidae | Ornithoctonus huwena | 13874 | -0.083 | 0.344 | NC_005925* | |
| Selenopidae | Selenops bursarius | 14272 | -0.123 | 0.321 | NC_024878* | |
| Oxyopidae | Oxyopes sertatus | 14442 | -0.130 | 0.321 | NC_025224* | |
| Liphistiidae | Liphistius erawan | 14197 | 0.024 | -0.361 | NC_020323* | |
| Heptathelidae | Heptathela hangzhouensis | 14215 | -0.023 | -0.235 | NC_005924* | |
| Xiphosura | Limulidae | Limulus polyphemus | 14985 | 0.114 | -0.399 | NC_003057* |
* refers to the mitogenomes that have been downloaded from GenBank.
Results and Discussions
Genome organization, structure and composition
The complete mitogenome sequences of T. maxillosa and T. nitens are 14,578 bp and 14,639 bp in size, respectively (Fig. 1). The relatively small mitogenome sizes (<15 kb) are well within the observed range of other spider mitogenomes available from GenBank [13]. Both mitogenomes share the same 37 typical metazoan gene set (13 PCGs, 22 tRNAs and two rRNAs) and an A+T-rich region [1]. Among these 37 genes, twenty-three are coded on the major strand (J-strand) while the rest are coded on the minor strand (N-strand). Gene orientation of T. maxillosa and T. nitens mitogenomes is identical to that of all previously determined araneoid mitogenomes [27,28]. With the exception of eight translocated tRNAs, mitogenome gene order of the two species are similar to that of L. polyphemus, which is considered to represent the putative ground pattern of the arthropods [23].
Map of mitogenome of T. maxillosa (A) and T. nitens (B). Transfer RNAs are designated by the IUPAC-IUB single letter amino acid codes (L1: trnLCUN; L2: trnLUUR; S1: trnLAGN; L1: trnLUCN) and those encoded by major- and minor-strand are shown outside and inside of the circular mitogenome map, respectively. Gene names not underlined indicate a clockwise direction of transcription, and those with underline indicate a counterclockwise direction transcription.
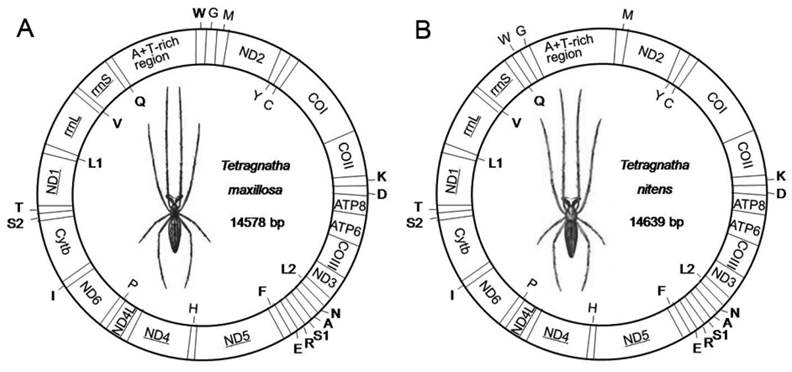
As in most of the arthropod mitogenomes, the nucleotide composition of the J-strands shows a highly A+T content which accounts for 74.5% in T. maxillosa and 74.3% in T. nitens, respectively. Metazoan mitogenomes usually present a clear strand bias in nucleotide composition [29], and arthropod mitogenomes tend to have a positive AT-skew and a negative GC-skew on the J-strand [30]. However, the strength of the skew in both T. maxillosa and T. nitens mitogenomes shows the opposite pattern, as the AT-skew and GC-skew in the J-strands are -0.087 and 0.246 in T. maxillosa, -0.055 and 0.200 in T. nitens, respectively (Table 1). The similar patterns of nucleotide skew are also commonly found in mitogenomes of other chelicerata taxa, such as in those of scorpions [31]. Through comparison of the nucleotide skews among all the sequenced spider mitogenomes, the divergence between the two suborders was detected. All the Opisthothelae spider mitogenomes exhibit a negative AT-skew but a positive GC-skew, while mitogenomes of two species from Meaothelae are characterized by a positive AT-skew and negative GC-skew. Similar phenomenon was also observed in pseudoscorpion mitogenomes [32]. These results indicate that their ancestors underwent a reversal bias in nucleotide composition on the J-strand between the two suborders. The nucleotide composition and skewness in different gene regions of T. maxillosa and T. nitens mitogenomes are shown in Table S2.
In the T. maxillosa mitogenome, there are 10 intergenic spacers with a total of 135 bp long varying from 1 bp to 41 bp. The three long intergenic spacers are located between trnN and trnA (41 bp), COIII and ND3 (35 bp), and trnG and trnM (30 bp), respectively. The T. nitens mitogenome has 12 intergenic spacers with a total of 139 bp with various lengths of 2-38 bp. The longest intergenic spacer is situated between COIII and ND3. Gene overlaps in the mitogenome of T. maxillosa are found in 20 locations and their total size is 166 bp. Size of each overlapped region varies from 2 to 22 bp. 17 overlapped regions are found in the mitogenome of T. nitens and their total size is 143 bp. Size of each overlapped region ranges from 1 to 23 bp. Overlapping tRNAs have long been known throughout metazoans [33]. There are 7 and 8 overlaps which are located between two adjacent tRNAs in T. maxillosa and T. nitens mitogenomes, respectively, and the longest overlap occurs between trnE and trnF in both of the spider mitogenomes.
Protein-coding genes and codon usage
Among 13 mitochondrial PCGs of the two Tetragnatha species, only four genes (ND5, ND4, ND4L and ND1) are coded on the N-strand, while the other genes are all coded on the J-strand (Table 2). Start codons follow the ATN rule in all 13 PCGs, except of three genes (COI, COII and COIII). The start codon for COI is TTT in T. maxillosa and TTA in T. nitens, respectively. Both COII and COIII use TTG as an initial codon in T. maxillosa and T. nitens. The start codons of TTT or TTA for COI and TTG for COII and COIII are also presented in mitogenomes of many species, especially for spiders [12,13]. The standard stop codons (TAA and TAG) and one incomplete stop codon (T) are utilized in all 13 PCGs. In T. maxillosa, three genes (COII, Cytb and ND1) terminate with TAG, two genes (ND2 and ND4L) use T and the rest genes are all stop with TAA. The stop codons are identical between the two spider mitogenomes with the exception of TAA as stop codon for COII and ND1 in T. nitens. The incomplete termination codon was commonly found in metazoan mitogenomes, which is presumed to be completed via post-transcriptional polyadenylation [34]. In accordance with other arthropod mitogenomes, overlapping regions between two adjacent PCGs are present in both T. maxillosa and T. nitens mitogenomes [35]. A 4 bp overlap exists in the junction between ATP8 and ATP6 in both spider mitogenomes. Generally, hairpin structures forming at the 3' end of the upstream protein's mRNA may act as a signal for the cleavage of the polycistronic primary transcript [36]. Either incomplete stop codons or overlaps between genes may be a product of the selective pressure to reduce genome size noted in mitochondria [37].
Mitochondrial genome organization of Tetragnatha maxillosa and T. nitens*.
| Feature | Strand | T. maxillosa | T. nitens | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Position | Spacer (+)/Overlap (-) | Start/Stop codon | Position | Spacer (+)/Overlap (-) | Start/Stop codon | ||||
| tRNAMet | J | 1-62 | +30 | 1-61 | 0 | ||||
| ND2 | J | 53-986 | -10 | ATT/T | 48-981 | -14 | ATT/T | ||
| tRNATyr | N | 989-1043 | +2 | 985-1040 | +3 | ||||
| tRNACys | N | 1032-1094 | -12 | 1030-1095 | -11 | ||||
| COI | J | 1093-2628 | -2 | TTT/TAA | 1092-2627 | -4 | TTA/TAA | ||
| COII | J | 2632-3291 | +3 | TTG/TAG | 2631-3290 | +3 | TTG/TAA | ||
| tRNALys | J | 3288-3342 | -4 | 3293-3344 | +2 | ||||
| tRNAAsp | J | 3334-3393 | -9 | 3338-3398 | -7 | ||||
| ATP8 | J | 3385-3540 | -9 | ATT/TAA | 3399-3542 | 0 | ATA/TAA | ||
| ATP6 | J | 3537-4199 | -4 | ATA/TAA | 3539-4201 | -4 | ATA/TAA | ||
| COIII | J | 4203-4988 | +3 | TTG/TAA | 4205-4990 | +3 | TTG/TAA | ||
| ND3 | J | 5024-5344 | +35 | ATA/TAA | 5029-5343 | +38 | ATT/TAA | ||
| tRNALeu(UUR) | N | 5331-5385 | -14 | 5332-5387 | -12 | ||||
| tRNAAsn | J | 5381-5439 | -5 | 5385-5441 | -3 | ||||
| tRNAAla | J | 5481-5546 | +41 | 5474-5535 | +32 | ||||
| tRNASer(AGN) | J | 5544-5598 | -3 | 5534-5584 | -2 | ||||
| tRNAArg | J | 5600-5650 | +1 | 5581-5633 | -4 | ||||
| tRNAGlu | J | 5643-5700 | -8 | 5624-5682 | -10 | ||||
| tRNAPhe | N | 5679-5732 | -22 | 5660-5714 | -23 | ||||
| ND5 | N | 5734-7377 | +1 | ATC/TAA | 5713-7356 | -2 | ATA/TAA | ||
| tRNAHis | N | 7372-7424 | -6 | 7354-7405 | -3 | ||||
| ND4 | N | 7425-8708 | 0 | ATA/TAA | 7426-8712 | +20 | TTG/TAA | ||
| ND4L | N | 8717-8976 | +8 | ATT/T | 8713-8977 | 0 | ATT/T | ||
| tRNAPro | N | 8963-9020 | -14 | 8965-9024 | -13 | ||||
| ND6 | J | 9033-9461 | +12 | ATA/TAA | 9034-9462 | +9 | ATA/TAA | ||
| tRNAIle | J | 9459-9518 | -3 | 9461-9513 | -2 | ||||
| Cytb | J | 9510-10643 | -9 | ATA/TAG | 9516-10643 | +2 | ATT/TAG | ||
| tRNASer(UCN) | J | 10639-10694 | -5 | 10652-10705 | +8 | ||||
| tRNAThr | J | 10695-10752 | 0 | 10706-10761 | 0 | ||||
| ND1 | N | 10742-11695 | -11 | ATT/TAG | 10743-11684 | -19 | ATA/TAA | ||
| tRNALeu(CUN) | N | 11690-11746 | -6 | 11693-11762 | +8 | ||||
| rrnL | N | 11747-12769 | 0 | 11763-12769 | 0 | ||||
| tRNAVal | N | 12770-12829 | 0 | 12770-12826 | 0 | ||||
| rrnS | N | 12830-13517 | 0 | 12827-13516 | 0 | ||||
| tRNAGln | N | 13518-13574 | 0 | 13517-13576 | 0 | ||||
| tRNATrp | J | - - | - - | 13588-13641 | +11 | ||||
| tRNAGly | J | - - | - - | 13632-13695 | -10 | ||||
| CR | J | 13575-14438 | 0 | 13696-14639 | 0 | ||||
| tRNATrp | J | 14439-14492 | 0 | - - | - - | ||||
| tRNAGly | J | 14484-14548 | -9 | - - | - - | ||||
* J and N refer to the major and minor strand, respectively. Position numbers refer to positions on the major strand.
The A+T contents of all the 13 mitochondrial PCGs, excluding stop codons, are 73.5% in T. maxillosa and 73.0% in T. nitens, respectively. The third codon position has a relatively higher A+T content (83.9% in T. maxillosa and 83.1% in T. nitens) than the first (68.7% in T. maxillosa and 68.5% in T. nitens) and second (67.9% in T. maxillosa and 67.3% in T. nitens) codon positions. In addition, both the second and third codon positions in T. nitens and all of the three codon positions in T. maxillosa have negative AT-skews. These AT-bias might affect codon usage in proteins, with ATT (encoding Ile) and TTT (encoding Phe) being the most frequently used codons in mitogenomes of T. maxillosa and T. nitens, respectively. The relative synonymous codon usage (RSCU) in 13 PCGs of the two spider mitogenomes is summarized in Fig. S1. The usage of both two-fold and four-fold degenerate codon is biased to use the codons which are abundant in A or T in the third position, and GC-rich codons are likely to be abandoned. These features are consistent with other arthropod mitogenomes [30].
Transfer and ribosomal RNA genes
The complete set of 22 tRNA genes typically found in metazoan mitogenomes is present in the two spider mitogenomes: two tRNAs for each of serine and leucine, and one tRNA for each of the other 18 amino acids. All tRNAs are scattered throughout the circular mitogenome and varied in size from 51 bp (trnR) to 66 bp (trnA) in T. maxillosa and ranged from 51 bp (trnR) to 70 bp (trnI) in T. nitens. The concatenated sequence of all 22 tRNAs shows a high A+T biased, which accounts for 78.1% in T. maxillosa and 79.1% in T. nitens, and exhibits a negative AT-skew (-0.012) in T. maxillosa and a positive GC-skew (0.188) in T. nitens, respectively (Table S2). Among 22 tRNAs, 14 tRNAs are coded on the J-strand and the rest on the N-strand.
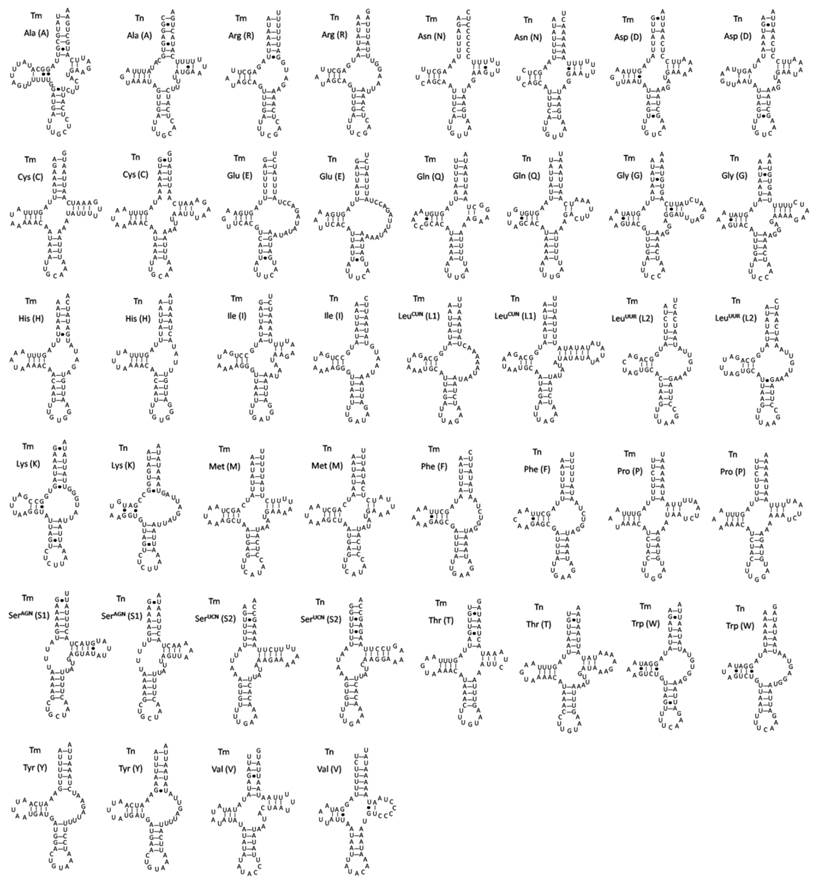
The predicted secondary structures of 22 tRNAs in two spider mitogenomes are shown in Fig. 2. Most of them cannot be folded into typical cloverleaf-shaped secondary structures. Nine of the tRNA sequences lack the potential to form TΨC arm stem pairings. Instead, they are inferred to have TV-replacement loops. These extremely truncated tRNAs might be resultant products of selection pressure for minimizing the size of Araneae mitogenomes. The trnSAGN and trnSUCN in both spider mitogenomes lack the dihydrouracil (DHU) arm, but are simplified down to a loop. Although the trnSAGN show typical clover-leaf structure in the mitogenomes of some exceptional species such as Adoxophyes honmai [38] and Pseudocellus pearsei [39], missing of the dihydrouracil (DHU) arm in the secondary structure of the trnSAGN are commonly observed in many metazoan mitogenomes including the sea spider Achelia bituberculata [40], the jumping spider H. oregonensis [41], and the camel spider Nothopuga sp. [42]. However, missing of the DHU-arm in trnSUCN is not a common feature observed in arthropod mitogenomes, though it has been found in some chelicerate mitogenomes [13]. Additionally, lots of unmatched base pairs and G-U wobble pairs scatter throughout all of tRNA secondary structures in both spider mitogenomes and most of the unmatched base pairs occur on the amino acid acceptor arm. Such aberrant secondary structures are also found in the spiders H. oregonensis [41], Heptathela hangzhouensis and O. huwena [12]. To be functional, these noncanonical tRNAs may require coevolved interacting factors or post-transcriptional RNA editing [41,43]. For examples, seryl-tRNA synthetase has evolved to be able to recognize the noncanonical tRNA-Ser in mammalian mitochondria [43] and a template-dependent RNA editing mechanism has been demonstrated in centipede Lithobius forficatus: the 3′ end sequences of the acceptor stem act as a template to synthesize the matching strand [14]. Since organisms that possessed the similar non-canonial tRNA structures might be share the evolutionary history and have a much closer lineage [44], the information of tRNA secondary structures could be used for deep-level phylogenetic inference. Masta and Boore (2008) have reported that T-arm loss in trnR, trnK and trnM occurred only once and could be a synapomorphy for Opisthothelae spiders [44]. In case of the two Tetragnatha mitogenomes, however, the typical cloverleaf structure could be predicted for trnM gene, although the stem is short with only two complementary base pairs in T. nitens (Fig. 2).
Inferred secondary structure of 22 tRNAs of T. maxillosa (Tm) and T. nitens (Tn) mitogenomes. The tRNAs are labeled with the abbreviations of their corresponding amino acids. Dashed (-) lines indicate Watson-Crick base pairing and centered dots (●) indicate G-U base pairing.
The ends of rRNAs are difficult to be precisely determined by DNA sequencing alone, so they are assumed to extend to the boundaries of flanking genes. As in mitogenomes of H. oregonensis [41] and H. hangzhouensis [12], the two rRNAs are located between trnLCUN and trnV, and between trnV and trnQ, respectively. The length of rrnL is 1023 bp with an A+T content of 79% in T. maxillosa and 1007 bp with an A+T content of 78.3% in T. nitens, respectively. The rrnS is 688 bp with an A+T content of 81.2% in T. maxillosa and 690 bp with the A+T content of 82.5% in T. nitens, respectively. In addition, both rRNAs exhibit a negative AT-skew and a positive GC-skew in T. maxillosa. In T. nitens, however, only the rrnL shows a negative AT-skew and a positive GC -skew, whereas rrnS exhibits a positive AT-skew and a negative GC-skew.
The A+T-rich region
The A+T-rich region in mitogenome is also called control region (CR), and essential for the initiation of replication in vertebrates [45,46]. The A+T-rich region in T. maxillosa is located between trnQ and trnW, spans 864 bp with 73.3% A+T content and shows a negative AT-skew (-0.055). However, this region is flanked by trnG and trnM in T. nitens, spans 690 bp and contains 71.5% A+T content with both positive AT-skew and GC-skew. The sizes of A+T-rich region in T. maxillosa and T. nitens are consistent with those of the A+T-rich region of other spiders, which range from 387 bp in Phyxioschema suthepium to 1078 bp in Pholcus phalangioide, but relatively shorter than those of most insect mitogenomes [30]. Moreover, two spider species share 55.0% sequence identity in this region and both have a conserved T-stretch (28 bp) in the initial quarter of the A+T-rich region on the N-strand, which is thought to be responsible for the control of replication and transcription of arthropod mitogenomes [47].
As the largest noncoding part of the metazoan mitogenome, the A+T-rich region shows a higher size variation than the other regions of the mitogenome due to both length variation with tandem repeat units (TRs) and differences in their copy numbers. There are four long TRs present in both spider mitogenomes. The length of the first three TRs are 172 bp in T. maxillosa and 180 bp in T. nitens, respectively, and the fourth TR is slightly shorter, which is 152 bp (1 bp overlapped with adjacent trnW) in T. maxillosa and 174 bp (6 bp overlapped with downstream trnM) in T. nitens, respectively. The TR motifs of the two spiders share 63.0% sequence identity and are rich in GC in the beginning of this region. The TRs could be duplicated through slipped-strand mispairing and differences in their numbers were considered as the source of size variation in the entire mitogenome [30]. Stem-loop structures were also detected in the TR region of the two spider species. Two large hairpin structures could be potentially formed in the TR of T. maxillosa, and similar complicated hairpin structures were also shown in the TR of T. nitens (Fig. S2).
Phylogenetic analysis
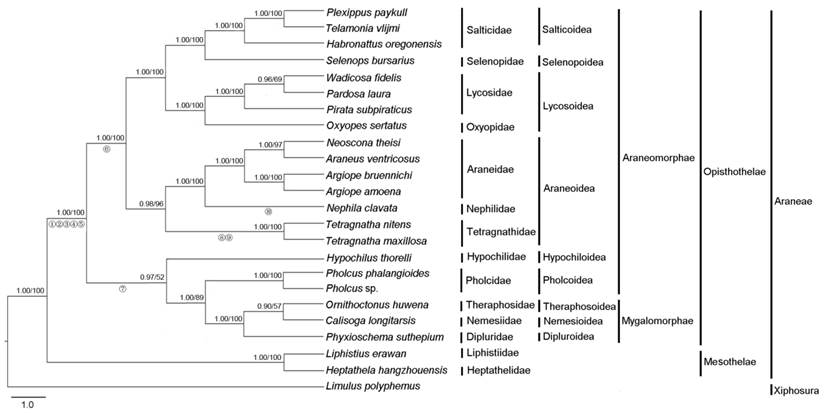
Phylogenetic analysis was performed on three concatenated nucleotide datasets from 23 Araneae species and an outgroup species. The final alignment resulted in 11,241 sites for dataset PCG123, 7,494 sites for dataset PCG12 and 9,529 sites for dataset PCG12R, respectively. The topologies of phylogenetic trees inferred from two methods (ML and BI) and three datasets were almost identical (Fig. 4, Fig. S3). The results clearly indicated that T. maxillosa and T. nitens share a close ancestry with N. clavata. Tetragnathidae as sister group of other araneoids (Nephilidae and Araneidae) is quite well supported both by BI and ML analysis, consistent with earlier studies that used morphological and molecular data [7]. Our findings also provide strong support for the monophyly of Araneoidea [8], which is placed as a sister relationship to Lycosoidea + Selenopoidea + Salticoidea (RTA-clade, named for the retrolateral tibal apophysis on the male copulatory organs). Moreover, the results also strongly supported the monophyly of two suborders (Opisthothelae and Mesothelae) in Araneae with high supporting values. However, the subdivision of Opisthothelae into two infraorders (Araneomorphae and Mygalomorphae) was not supported in the phylogenetic trees, although the monophyly of the two infraorders has been broadly accepted by most of the arachnologists [48]. As illustrated in Fig. 4, the super-families of Hypochilidae and Pholcoidea belonging to Araneomorphae and the super-families of Theraphosoidea, Nemesioidea and Dipluroidea belonging to Mygalomorphae formed a phylogenetically closely related clade that was well supported by BI tree (0.97), in spite of the lower bootstrap supporting value (52) in ML tree. Increased taxon sampling can significantly reduce phylogenetic error. Hence, additional mitogenomes data from the infraorders Araneomorphae and Mygalomorphae are required to better establish the phylogeny of Opisthothelae.
Phylogenetic tree from Araneae species based on nucleotide sequence of 13 mitochondrial protein-coding genes using Bayesian inference (BI) and maximum likelihood (ML). Limulus polyphemus was used as an outgroup. Numbers above the nodes refer to Bayesian posterior probabilities in percentages (left) and ML bootstrap values (right). Hypothesized gene translocations for spiders are denoted on the phylogenetic tree by numbered circles as shown in Fig. 3.
Rearrangement of mitochondrial gene order in Araneae
When compared with the putative ancestral arthropod mitogenome as presented by L. polyphemus [23], eight tRNAs are found to translocate their positions in both Tetragnatha spider mitogenomes (Fig. 3). The trnI changes its position to a new location between ND6 and Cytb. This tRNA rearrangement is also found in most of Araneomorphae spiders, such as H. oregonensis [41], T. vlijmi [49] and N. clavata [27]. Additionally, trnLCUN, trnN and trnSAGN rearrange their positions in both Tetragnatha spider mitogenomes, and these three tRNAs translocations are seem to be a conserved rearrangement event in all Opisthothelae species. Tandem duplication random deletion events and transposition of single tRNAs are considered to be the predominant mechanism of mitogenome rearrangement [50]. When the total 21 spider mitogenomes that have been sequenced to date were analyzed, the arthropod ground pattern is found to be retained in two Mesothelae spiders (Liphistius erawan and H. hangzhouensis). Six spider species (Calisoga longitarsis, Hypochilus thorelli, O. huwena, P. phalangioides, Pholcus sp. and P. suthepium) share the same six translocated tRNAs, and a subsequent trnI translocation in nine spider species (Argiope bruennichi, H. oregonensis, T. vlijmi, A. amoena, Selenops bursarius, Plexippus paykulli, Oxyopes sertatus, Pardosa laura and Pirata subpiraticus) and trnM is rearranged in N. clavata. The new finding in present study is the novel gene order rearrangement of trnG and trnW, which has not been observed in all above Araneae mitogenomes yet. The trnW and trnG are translocated to the new position between the A+T-rich region and trnM in T. maxillosa, and between trnQ and A+T-rich region in T. nitens, respectively. Whether this rearrangement event is limited to only the spiders belonging to Tetragnathidae? To answer this, further mitogenomes sequencing of closely related taxa would be required. Moreover, our data suggest that the phylogenetic relationships among the Araneae can be also distinguished by gene order synapomorphies. We present a parsimonious scenario of gene order changes and map these rearrangement events on the phylogenetic trees inferred from the nucleotide dataset of PCG123. As shown in Fig. 4, the taxa from Mesothelae in basal clade have the same gene order as L. polyphemus, suggesting that gene order rearrangements might occur after the divergence of the Araneae suborders. Translocation of four tRNAs (trnN, trnQ, trnS1 and trnL2) and inversion of two tRNAs (between trnY and trnC) could be a synapomorphy for Opisthothelae spiders. Likewise, reverse translocation of trnI in species of RTA-clade + Araneoidea and translocation of trnI in species belong to Hypochiloidea, Dipluridae, Theraphosidae, Nemesiidae and Plolicidae likely experienced parallel evolution after Opisthothelae divergence. Additionally, translocation of trnM is unique to a particular spider taxon (N. clavata) and rearrangement of trnW and trnG seems to be a synapomorphy for the genus Tetragnatha. Overall, our results imply that the complicated gene rearrangement in mitochondria could be considered as one of key characters in inferring higher-level phylogenetic relationship of Araneae.
Schematic representation of mitochondrial gene arrangements in Araneae species compared to the putative ancestral arthropod gene order. Underlined protein coding genes and tRNAs are encoded in the minor strand. Genes that changed the positions were marked by numbers below, and the numbers refer to putative rearrangement events as follows: ①Translocation of trnL2; ②Translocation of trnN; ③Translocation of trnS1; ④Translocation of trnQ; ⑤Inversion between trnY and trnC; ⑥Reverse translocation of trnI; ⑦Translocation of trnI; ⑧Translocation of trnW; ⑨Translocation of trnG; ⑩Translocation of trnM.

Conclusion
The complete mitogenomes of two long jawed spiders T.maxillosa and T. nitens were characterized and compared with those of other Araneae species. The complete mitogenomes of both Tetragnatha spiders reveal a typical circular molecule with a similar gene content and orientation as found in other metazoans. The comparative analysis suggests that the gene orders of the Tetragnatha mitogenomes are moderately rearranged and represent new patterns. With exceptions of the similar translocation events shared by other sequenced spider mitogenomes, two tRNAs (trnG and trnW) in the two Tetragnatha mitogenomes were found in different positions. Most of the tRNAs lose TΨC arm stems and have TV-replacement loops instead. Likely to the trnS (AGN), the trnS(UCN) lacks the DHU arm in both spider mitogenomes. Phylogenetic analysis indicates that mitogenome sequences are useful in resolving higher-level relationship of Araneae. Data on genome arrangement provide further information to help resolve relationships among the spiders. The present results of T. maxillosa and T. nitens mitogenomes will elevate our knowledge on mitogenomes of Araneae. However, future research including additional taxon sampling is needed to determine rearrangement mechanisms and evolutionary processes.
Supplementary Material
Supplementary Tables and Figures.
Acknowledgements
We thank Chao-Feng Lu, Wei Cao and Dr. Guang-Fu Liu (Zhejiang Provincial Key Laboratory of Biometrology and Inspection and Quarantine, College of Life Sciences, China Jiliang University) for their help with the collection of the spider specimen. This work was financially supported by the National High Technology Research and Development Program of China (863 Program) (Grant No. 2012AA021601) and the Scientific Research Fund of Zhejiang Provincial Education Department (Grant No.: Y201431574).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Boore JL. Animal mitochondrial genomes. Nucleic Acids Res. 1999;27:1767-1780
2. Simon C, Buckley TR, Frati F, Stewart JB, Beckenbach AT. Incorporating molecular evolution into phylogenetic analysis, and a new compilation of conserved polymerase chain reaction primers for animal mitochondrial DNA. Annu Rev Ecol Evol S. 2006;37:545-579
3. Boore JL, Lavrov DV, Brown WM. Gene translocation links insects and crustaceans. Nature. 1998;392:667-668
4. Dowton M, Castro LR, Austin AD. Mitochondrial gene rearrangements as phylogenetic characters in the invertebrates: The examination of genome 'morphology'. Invertebr Syst. 2002;16:345-356
5. Coddington JA, Levi HW. Systematics and evolution of spiders. Ann Rev Ecol Syst. 1991;22:569-592
6. Platnick NI. The World Spider Catalog, Version 13.5; American Museum of Natural History, 2014. http://research.amnh.org/iz/spiders/catalog/
7. Hormiga G, Griswold CE. Systematics, phylogeny, and evolution of orb-weaving spiders. Annu Rev Entom. 2014;59:487-512
8. Fernández R, Hormiga G, Giribet G. Phylogenomic analysis of spiders reveals nonmonophyly of orb weavers. Curr Biol. 2014;24:1772-1777
9. Fang K, Yang CC, Lue BW, Chen SH, Lue KY. Phylogenetic corroboration of superfamily Lycosoidae spiders (Araneae) as inferred from partial mitochondrial 12S and 16S ribosomal DNA sequences. Zool Stud. 2000;39:107-113
10. Arnedo MA, Coddington J, Agnarsson I, Gillespie RG. From a comb to a tree: Phylogenetic relationships of the combfooted spiders (Araneae, Theridiidae) inferred from nuclear and mitochondrial genes. Mol Phylogenet Evol. 2004;31:225-245
11. Wheeler WC, Hayashi CY. The phylogeny of extant chelicerate orders. Cladistics. 1998;14:173-192
12. Qiu Y, Song D, Zhou K, Sun H. The mitochondrial sequences of Heptathela hangzhouensis and Ornithoctonus huwena reveal unique gene arrangements and atypical tRNAs. J Mol Evol. 2005;60:57-71
13. Wang ZL, Li C, Fang WY, Yu XP. The complete mitochondrial genome of the wolf spider Wadicosa fidelis (Araneae: Lycosidae). Mitochondr DNA. 2014 [Epub ahead of print]
14. Lavrov DV, Brown WM, Boore JL. A novel type of RNA editing occurs in the mitochondrial tRNAs of the centipede Lithobius forficatus. Proc Natl Acad Sci USA. 2000;97:13738-13742
15. Staden R, Beal KF, Bonfield JK. The Staden package, 1998. Methods Mol Biol. 2000;132:115-130
16. Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G. et al. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 2013;69:313-319
17. Cameron SL. How to sequence and annotate insect mitochondrial genomes for systematic and comparative genomics research. Syst Entomol. 2014;39:400-411
18. Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955-964
19. Laslett D, Canback B. ARWEN: a program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics. 2008;24:172-175
20. Kumar S, Nei M, Dudley J, Tamura K. MEGA: a biologist-centric software for evolutionary analysis of DNA and protein sequences. Brief Bioinform. 2008;9:299-306
21. Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573-580
22. Perna NT, Kocher TD. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J Mol Evol. 1995;41:353-358
23. Lavrov DV, Boore JL. Brown WM. The complete mitochondrial DNA sequence of the horseshoe crab Limulus polyphemus. Mol Biol Evol. 2000;17:813-824
24. Posada D, Crandall KA. Modeltest: Testing the model of DNA substitution. Bioinformatics. 1998;14:817-818
25. Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696-704
26. Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572-1574
27. Pan WJ, Fang HY, Zhang P, Pan HC. The complete mitochondrial genome of Nephila clavata (Araneae: Nephilidae) Chinese population. Mitochondr DNA. 2014 [Epub ahead of print]
28. Zhang P, Fang HY, Pan WJ, Pan HC. The complete mitochondrial genome of the wasp spider Argiope bruennichi (Araneae: Araneidae). Mitochondr DNA. 2014 [Epub ahead of print]
29. Hassanin A, Léger N, Deutsch J. Evidence for multiple reversals of asymmetric mutational constraints during the evolution of the mitochondrial genome of metazoa, and consequences for phylogenetic inferences. Syst Biol. 2005;54:277-298
30. Ma C, Liu C, Yang P, Kang L. The complete mitochondrial genomes of two band-winged grasshoppers, Gastrimargus marmoratus and Oedaleus asiaticus. BMC Genomics. 2009;10:156
31. Masta SE, Longhorn SJ, Boore JL. Arachnid relationships based on mitochondrial genomes: asymmetric nucleotide and amino acid bias affects phylogenetic analyses. Mol Phylogenet Evol. 2009;50:117-128
32. Ovchinnikov S, Masta SE. Pseudoscorpion mitochondria show rearranged genes and genome-wide reductions of RNA gene sizes and inferred structures, yet typical nucleotide composition bias. BMC Evol Biol. 2012;12:31
33. Jühling F, Pütz J, Bernt M, Donath A, Middendorf M, Florentz C, Stadler PF. Improved systematic tRNA gene annotation allows new insights into the evolution of mitochondrial tRNA structures and into the mechanisms of mitochondrial genome rearrangements. Nucleic Acids Res. 2012;40:2833-2845
34. Ojala D, Montoya J, Attardi G. tRNA punctuation model of RNA processing in human mitochondria. Nature. 1981;290:470-474
35. Mans BJ, de Klerk D, Pienaar R, de Castro MH, Latif AA. The mitochondrial genomes of Nuttalliella namaqua (Ixodoidea: Nuttalliellidae) and Argas africolumbae (Ixodoidae: Argasidae): estimation of divergence dates for the major tick lineages and reconstruction of ancestral blood-feeding characters. PLoS One. 2012;7:e49461
36. Fenn JD, Cameron SL, Whiting MF. The complete mitochondrial genome sequence of the Mormon cricket (Anabrus simplex: Tettigoniidae: Orthoptera) and an analysis of control region variability. Insect Mol Biol. 2007;16:239-252
37. Rand DM. Endotherms, ectotherms, and mitochondrial genome-size variation. J Mol Evol. 1993;37:281-295
38. Lee ES, Shin KS, Kim MS, Park H, Cho S, Kim CB. The mitochondrial genome of the smaller tea tortrix Adoxophyes honmai (Lepidoptera: Tortricidae). Gene. 2006;373:52-57
39. Fahrein K, Talarico G, Braband A, Podsiadlowski L. The complete mitochondrial genome of Pseudocellus pearsei (Chelicerata: Ricinulei) and a comparison of mitochondrial gene rearrangements in Arachnida. BMC Genomics. 2007;8:386
40. Park SJ, Lee YS, Hwang UW. The complete mitochondrial genome of the sea spider Achelia bituberculata (Pycnogonida, Ammotheidae): arthropod ground pattern of gene arrangement. BMC Genomics. 2007;8:343
41. Masta SE, Boore JL. The complete mitochondrial genome sequence of the spider Habronattus oregonensis reveals rearranged and extremely truncated tRNAs. Mol Biol Evol. 2004;21:893-902
42. Masta SE, Klann AE, Podsiadlowski L. A comparison of the mitochondrial genomes from two families of Solifugae (Arthropoda: Chelicerata): Eremobatidae and Ammotrechidae. Gene. 2008;417:35-42
43. Chimnaronk S, Jeppesen MG, Suzuki T, Nyborg J, Watanabe K. Dual-mode recognition of noncanonical tRNAsSer by seryl-tRNA synthetase in mammalian mitochondria. EMBO J. 2005;24:3369-3379
44. Masta SE, Boore JL. Parallel evolution of truncated transfer RNA genes in arachnid mitochondrial genomes. Mol Biol Evol. 2008;25:949-959
45. Fernandez-Silva P, Enriquez JA, Montoya J. Replication and transcription of mammalian mitochondrial DNA. Exp Physiol. 2003;88:41-56
46. Zhang DX, Szymura JM, Hewitt GM. Evolution and structural conservation of the control region of insect mitochondrial DNA. J Mol Evol. 1995;40:382-391
47. Saito S, Tamura K, Aotsuka T. Replication origin of mitochondrial DNA in insects. Genetics. 2005;171:1695-1705
48. Coddington JA, Giribet G, Harvey MS, Prendini L, Walter DE. Arachnida. In: (ed.) Cracraft J, Donoghue MJ. Assembling the tree of life. New York: Oxford University Press. 2004:296-318
49. Kim JY, Yoon KB, Park YC. The complete mitochondrial genome of the jumping spider Telamonia vlijmi (Araneae: Salticidae). Mitochondr DNA. 2014 [Epub ahead of print]
50. Moritz C, Dowling TE, Brown WM. Evolution of animal mitochondrial DNA-relevance for population biology and systematics. Annu Rev Ecol Syst. 1987;18:269-292
Author contact
![]() Corresponding author: X.-P. Yu, Zhejiang Provincial Key Laboratory of Biometrology and Inspection and Quarantine, College of Life Sciences, China Jiliang University, Hangzhou, Zhejiang, People's Republic of China. Phone and fax: 86-571-86836006. E-mail: yxpedu.cn.
Corresponding author: X.-P. Yu, Zhejiang Provincial Key Laboratory of Biometrology and Inspection and Quarantine, College of Life Sciences, China Jiliang University, Hangzhou, Zhejiang, People's Republic of China. Phone and fax: 86-571-86836006. E-mail: yxpedu.cn.