Impact Factor ISSN: 1449-2288
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2016; 12(2):133-143. doi:10.7150/ijbs.14242 This issue Cite
Research Paper
“DNA Binding Region” of BRCA1 Affects Genetic Stability through modulating the Intra-S-Phase Checkpoint
Takaaki Masuda1, Xiaoling Xu2, Emilios K. Dimitriadis3, Tyler Lahusen1, Chu-Xia Deng1,2 ![]()
1. Genetics of Development and Disease Branch, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, USA.
2. Faculty of Health Sciences, University of Macau, Macau SAR, China.
3. Laboratory of Bioengineering and Physical Science, National Institute of Biomedical Imaging and Bioengineering, National Institutes of Health, USA.
Received 2015-10-27; Accepted 2015-12-9; Published 2016-1-1
Abstract

The breast cancer associated gene 1 (BRCA1) contains 3 domains: an N-terminal RING domain with ubiquitin E3 ligase activity, C-terminal BRCT protein interaction domain and a central region. RING and BRCT domains are well characterized, yet the function of the central region remains unclear. In this study, we identified an essential DNA binding region (DBR: 421-701 amino acids) within the central region of human BRCA1, and found that BRCA1 brings DNA together and preferably binds to splayed-arm DNA in a sequence-independent manner. To investigate the biological role of the DBR, we generated mouse ES cells, which lack the DBR (ΔDBR) by using the TALEN method. The ΔDBR cells exhibited decreased survival as compared to the wild type (WT) cells treated with a PARP inhibitor, however they have an intact ability to conduct DNA repair mediated by homologous recombination (HR). The ΔDBR cells continued to incorporate more EdU in the presence of hydroxyurea (HU), which causes replication stress and exhibited reduced viability than the WT cells. Moreover, phosphorylation of CHK1, which regulates the intra-S phase checkpoint, was moderately decreased in ΔDBR cells. These data suggest that DNA binding by BRCA1 affects the stability of DNA replication folks, resulting in weakened intra-S-phase checkpoint control in the ΔDBR cells. The ΔDBR cells also exhibited an increased number of abnormal chromosome structures as compared with WT cells, indicating that the ΔDBR cells have increased genetic instability. Thus, we demonstrated that the DBR of BRCA1 modulates genetic stability through the intra-S-phase checkpoint activated by replication stress.
Keywords: BRCA1, DNA binding, Intra-S-phase checkpoint, Genomic instability
Introduction
Breast cancer is the most common cancer and the second-leading cause of cancer mortality in women [1, 2]. BRCA1 was isolated as the gene responsible for increased susceptibility to familial breast and ovarian cancer [3]. Germline mutations of BRCA1 have been detected in the majority of familial breast and ovarian cancers and approximately one third of the cases of familial breast cancer [2, 4, 5]. Moreover, nearly 30-40% of sporadic malignancies are associated with impaired expression of BRCA1 [6].
BRCA1 is a large multifunctional protein implicated in DNA double-strand break repair, centrosome duplication, transcription regulation, DNA damage response, and cell cycle control, all of which are important for maintaining genomic stability [7-9].
In cancer patients, mutations in BRCA1 are most frequently observed in three domains: the N-terminal RING domain encoded by exons 2-7 with ubiquitin E3 ligase activity, coding regions of exons 11-13 (central region), and the C-terminal protein-interacting BRCT domain encoded by exons 16-24 [10]. RING and BRCT domains, which play a significant role in genomic integrity, are well characterized, and their protein structures have been determined [11-15]. However, these two domains cover only a small portion of the full length BRCA1 protein, whereas the central region encodes the majority of BRCA1 protein. However, the function of the central region of BRCA1 is still unclear [10]. It is one of the major challenges in the field of BRCA1 research to elucidate it [16].
Previously, we generated BRCA1 conditional knockout mice by deleting exon 11 (Δ11) [17, 18]. These mice form tumors as a result of genetic instability although both the RING finger and BRCT domains are conserved. BRCA1-Δ11 cells exhibited many phenotypes similar to those displayed by BRCA1 null cells, including defective homologous recombination, centrosome amplification, and abnormal cell cycle progression [17-19]. Thus, the functional domains of the central region of BRCA1 need to be further defined.
It was previously reported that BRCA1 binds DNA in the region encoded by exon 11 although the biological significance of this has not been well addressed [8, 20-22]. Therefore, we hypothesized that the DNA binding region (DBR) in the central region has a critical role for genomic integrity.
In this study, we identified an essential region responsible for BRCA1 binding to DNA. We then investigated the features of DNA binding by biochemical analysis, followed by an analysis of its biological significance using mouse ES cells, which are deficient in the DBR of BRCA1. We provide evidence that this DBR of BRCA1 modulates genetic stability through the intra-S-phase checkpoint that is activated by replication stress.
Materials and Methods
DNA constructs and protein preparation
A HA-tagged human BRCA1 expression vector pcDNA3 was obtained from Ralph Scully (Beth Israel Deaconess Medical Center, Boston, MA) [23]. The coding regions for BRCA1 protein were PCR amplified from this plasmid and subcloned downstream of a His8-MBP-PreScission cassette of the pLEX expression vector using AgeI and XhoI restriction sites [24]. The sequences of the resulting expression clones were verified. GST-BRCA1 fragments were obtained from Ralph Scully (Beth Israel Deaconess Medical Center, Boston, MA) [23]. Inverse PCR for Mutagenesis of BRCA1 constructs (mimics of phosphorylation (from serine (S) to aspartic acid (D)) and dephosphorylation (from serine (S) to alanine (A)) in S616 and S694 within DBR in BRCA1 using Phusion Site-Directed Mutagenesis Kit (Thermo Fisher Scientific) was performed according to the protocol. All primers for generating BRCA1 constructs were listed in Table S1.
With the exception of GST-BRCA1 fragments, all fragments were expressed in 293T cells (ATCC Manassas, VA). GST-BRCA1 fragments were expressed in E. coli BL21-CodonPlus (DE3)-RIL cells (Stratagene). 293T cells were grown in suspension culture at 37°C in 5% humidified CO2 atmosphere in Freestyle 293 medium (Invitrogen), supplemented with 1% fetal bovine serum (FBS) (Sigma, St. Louis, MO). E.coli for GST-BRCA1 fragments were grown at 37°C in LB medium supplemented with 15N ammonium chloride (1 g/L) to an absorbance of 0.8 (600nm) and induced with 0.5 mM IPTG for three hours at 30°C before harvesting. Cell pellets were thawed in 25 mM Tris-HCl pH 8.0, 500 mM NaCl, 2 mM MgCl2, 0.1mM ZnCl2, 0.4 mM EDTA, 5% (v/v) glycerol, 1mM DTT and proteinase inhibitors (1 µg/mL Pepstatin A, 10 µg/mL aprotinin, 10 µg/mL leupeptin, 100 µg/mL AEBSF) and lysed by sonication. The cellular extract was clarified by centrifugation at 12,000 rpm for 35 minutes at 4°C. In vitro dephosphorylation of BRCA1 protein was performed as previously [25]. Briefly, purified protein was incubated with λ protein phosphatase (New England Biolabs) for 30 min at 30°C.
Protein purification
Full-length or fragments of MBP or GST-tagged BRCA1 were affinity purified using amylose resin (NEB) or glutathione-agarose beads (GE health care), respectively. The MBP tagged proteins were eluted by an amylose elution buffer containing (25 mM Tris-HCl pH 8.0, 500 mM NaCl, 2 mM MgCl2, 0.1mM ZnCl2, 0.1 mM EDTA, 1mM DTT, 5% (v/v) glycerol and 40mM maltose). The GST tagged proteins were eluted by an elution buffer containing (25 mM Tris-HCl pH 8.0, 150 mM NaCl, 2 mM MgCl2, 0.1mM ZnCl2, 0.1 mM EDTA, 1mM DTT, 5% (v/v) glycerol and 2mM glutathione). The MBP-tagged proteins were further purified by gel filtration (Superdex 200, GE Healthcare) in the buffer (25 mM Tris-HCl pH 8.0, 250 mM NaCl, 2 mM MgCl2, 0.1mM ZnCl2, 0.1 mM EDTA, 1mM DTT, 5% (v/v) glycerol). Some proteins were concentrated by ultrafiltration with Amicon Ultra (EMD Millipore). The concentrations were measured optically using each extinction coefficient at 280 nm. The purified proteins were stored at -80 °C.
SDS-PAGE
Cells were harvested from subconfluent plates and whole-cell lysates were prepared for immunoblot analysis. Cells were washed with cold PBS and lysed with lysis buffer containing: 0.1% NP-40, 25 mmol/L Tris-HCl, pH 7.5, 150 mmol/L NaCl, 5% glycerol, 1mM DTT, protein inhibitors (0.4 mmol/L EDTA, 1 µg/mL Pepstatin A, 10 µg/mL aprotinin, 10 µg/mL leupeptin, 100 µg/mL AEBSF) and phosphatase inhibitor PhoSTOP (Roche). The lysate was sonicated, and centrifuged at 14,000 rpm at 4°C for 10 minutes. The concentrations of the proteins were measured optically using each extinction coefficient at 280 nm. Protein lysates (1-35µg) were prepared in SDS-loading buffer containing the reducing agent β-mercaptoethanol (Sigma), heated at 99°C for 2 minutes, resolved by SDS-PAGE on 4% to 12% or 20% Tris-glycine or Bis-Tris gels (Invitrogen). For Coomassie staining, the gels were stained with PageBlue™ Protein Staining Solution (Thermo Fisher Scientific) according to the manufacturer's instructions. For western blotting, the proteins in the gels were transferred to polyvinylidene diflouride Immobilon-P membranes (EMD Millipore). The membranes were blocked with 5% nonfat milk in TBST (TBS, 0.1% Tween-20) for 40 minutes, incubated for 2 hours at room temperature or for overnight at 4°C with primary antibodies diluted in 1% milk in TBST, washed three times with TBST, and then incubated with either an anti-rabbit or anti-mouse fluorescent-conjugated secondary antibody (GE Healthcare) for 1 hour. The blots were washed three times with TBST, and the membranes were scanned with an Odyssey SA scanner (LICOR Biosciences, Lincoln, NE), and band intensities were quantified using the Odyssey software. Proteins were detected with the following antibodies: monoclonal anti-Flag, and monoclonal anti-β-Actin (Sigma) and polyclonal anti-pChk1 S345 and Chk1 (Cell Signaling Technology), monoclonal anti-HA (Sigma) and monoclonal anti-human BRCA1-AB2 (Millipore). Polyclonal anti-mouse BRCA1-AB1 antibody was obtained from Lewis A. Chodosh (University of Pennsylvania, Philadelphia, PA) [26].
Electrophoretic mobility shift assay
HPLC purified oligodeoxynucleotides used in EMSA were designed with Random DNA sequence generator (http://users-birc.au.du), and were purchased from eurofins MWG operon. The sequence of the oligodeoxynucleotides was shown in Table S2. The double stranded oligodeoxynucleotide probes were generated by annealing two complementary oligodeoxynucleotides. DNA oligonucleotides were labeled with γ32P-ATP (PerkinElmer) using T4 polynucleotide kinase, gel-purified and quantified. Purified protein was incubated with 32P-labeled DNA in a reaction buffer (25 mM Tris-HCl pH 8.0, 50 mM NaCl, 4 mM MgCl2, 0.1 mM EDTA, 4% (v/v) glycerol, 0.02% NP40 and 1mM DTT) and incubated for 15 min at room temperature. The complexes were resolved on a 6 or 8% (w/v) DNA-Retardation gel (Invitrogen) in 0.5X TBE at room temperature at 100V. The gels were vacuum dried and visualized by autoradiography.
Supershift assays were performed by preincubating purified full length BRCA1 protein with MBP antibody (NEB) before adding the labeled oligonucleotide.
To perform the competition assay to compare the binding affinity of various shaped DNA structures to BRCA1 protein, non-labeled oligonucleotides of the various shapes with various concentrations were added at the start of the incubation reaction with labeled double strand DNA and BRCA1 protein. We evaluated the binding affinity by measuring how much labeled double strand DNA was competed off by the non-labeled DNA. These images were analyzed to measure the radioactivity using the NIH ImageJ software (http://rsbweb.nih.gov/ij/index.html).
Atomic Force Microscopy
BRCA1 281-701aa or 421-988aa was used to see how BRCA1 binds to dsDNA by an atomic force microscopy (AFM). The proteins were obtained from MBP-BRCA1 281-701aa or 421-988aa by removing MBP with PreScission protease. Five microliters of diluted sample was deposited on mica pretreated with 1-(3-amino-propyl)-silatrane (APS) [27]. Imaging was performed with a commercial AFM (Multimode with NS-V controller, Bruker Nano Surfaces, Santa Barbara, CA) using ultrasharp probes (TESP-SS, Bruker Nano Surfaces) in the "tapping" mode. Typically, these probes have a 2-5 nm tip radius, a spring constant of 42 N/m and resonance frequency in the vicinity of 300 kHz. High resolution AFM images (1 nm/pixel) were collected and preprocessed with the AFM image processing software, Nanoscope v7.36. These images were then analyzed further to estimate the volumes and examine the geometry of the complexes using the NIH ImageJ software.
In vitro stability assay, and N-terminal sequencing
BRCA1 421-701aa was obtained from MBP-BRCA1 421-701aa by removing MBP with PreScission protease. The complex of BRCA1 421-701aa and splayed-arm DNA or the BRCA1 protein alone were incubated at room temperature. Then, the proteins were separated by SDS-PAGE, electroblotted to PVDF membranes, and the bands on the membrane were excised. N-terminal Edman sequencing was performed at the Mass Spectrometry and Proteomics Resource Laboratory at Harvard University (mcb.harvard.edu/microchem).
Generation of mouse ES cells containing BRCA1 wild type or mutants with C-terminal 3xFlag by TALEN-mediated Gene Targeting
Sequence alignment of BRCA1 from human (421-701 amino acids), cow, rat and mouse is shown in Figure S1A. This region is well conserved. Based on this alignment, the residue 422-693aa was deemed as a DBR in mouse BRCA1 (The sequences under gray background in Figure S1A).
Because current antibodies for mouse BRCA1 do not provide a clear Western blot using mouse ES cells, we have generated an ES cell line that carries a 3xFLAG tag in the C-terminal end of BRCA1 (WT) to facilitate the detection of BRCA1 (Xu and Deng unpublished data). This cell line was used to generate mouse ES cells that carry targeted deletion of the DBR (ΔDBR) or exon 11 (Δ11). Briefly, a FASTA file for the mouse BRCA1 DNA sequence was input into the ZiFit webtool (http://zifit.partners.org/ZiFiT) to retrieve a schematic for building TALEN constructs. First, we generated ES cells with BRCA1-ΔDBR containing a C-terminal 3xFlag. TALENs were constructed following the detailed instruction provided by the REAL Assembly Kit (Addgene) (Figure S1B). Next, a BRCA1 fragment, which lacks DBR with homology arms (Figure S1C) was cloned into pBluescript II SK+ (Stratagene) as a donor plasmid. A pair of TALEN constucts and the donor plasmid were electroporated into ES cells containing a C-terminal 3xFlag of BRCA1. Identity of correct targeted DNA clones was confirmed by genotyping PCR (Figure S1D) and sequence analysis (Eurofins MWG operon). Primers used for genotyping PCR were 816 mRNA-L (GCAGACCCAACCTCGAAAGA), 14 (CTGCGAGCAGTCTTCAGAAAG) [18], 18S-L (AGTCCCTGCCCTTTGTACACA) and 18s-R (CGATCCGAGGGCCTCACTA).
MTT Assay
1.0 x 104 cells were plated in 24-well plates. The cells treated with PARP inhibitor (AG-014361) or Hydroxyurea (HU) for 3 days were added with thiazolyl blue tetrazolium bromide (Sigma) working solution (0.5 mg/ml) for 1 h and with isopropanol for 15 min. The absorption was read with the Perkin-Elmer 1420 multi-label counter (Perkin-Elmer, Waltham, MA, USA) at 570 nm.
Generation of BRCA1 knockdown HeLa cells
Human BRCA1 shRNA constructs in the pLKO.1-based vector were obtained from Open Biosystems (Thermo Scientific). A control lentiviral shRNA vector, packaging vector pCMV-dR8.2 dvpr, and envelope vector VSV-G was obtained from Addgene. The BRCA1 shRNA construct (TRCN0000039833,CCCTAAGTTTACTTCTCTAAACTCGAGTTTAGAGAAGTAAACTTAGGGTTTTT), targeting the 3'UTR of BRCA1 was used to produce lentiviral particles for generation of stable BRCA1 knockdown cells. Lentivirus was produced in 293T cells and the media collected after 48 hours for transduction of HeLa cells (ATCC Manassas, VA). Cells were transduced with lentiviral supernatant and then selected with 2 µg/ml puromycin to generate cells with stable knockdown of BRCA1.
Homologous recombination assay
A green fluorescent protein (GFP) reporter gene within which has embedded an I-SceI site were analyzed for HR repair for DNA double strand break as described previously [28]. Briefly, ES cells were transfected with the GFP reporter gene containing an I-SceI site (DR-GFP) or wild type GFP and an I-SceI expressing plasmid using XtremeGene9 (Roche), and were harvested 2 days later. HeLa cells stably knocked down with BRCA1 by shRNA (Figure S2A) was transfected with HA-BRCA1-WT or HA-BRCA1-ΔDBR construct in addition to DR-GFP and I-Sce1 vector. GFP positive cells were analyzed with a FACScan flow cytometer (BD).
EdU Incorporation Assay
For EdU incorporation, we used Click-iT® EdU Alexa Fluor® 647 Flow Cytometry Assay Kit (invitrogen) according to the manufacturer's instructions. EdU positive cells were analyzed with a FACScan flow cytometer (BD). EdU incorporation was normalized with cells without EdU labelling.
Chromosome Spreads from ES cells
Chromosome spreads from ES cells were performed as described previously [29, 30]. Briefly, embryos were incubated with 100 ng/ml colcemid for 1.5 hr. The hypotonic treatment was carried out for 20 min at room temperature in 0.56% KCl. The ES cells were transferred to methanol:acetic acid (3:1) for fixation. The ES cells were then disaggregated under a dissection microscope in 60% acetic acid. The disaggregated ES cells were spun down, suspended in methanol:acetic acid, and dropped onto slides. All chromosome spreads were stained with Giemsa, and chromosome number and morphology was assessed using a Leica microscope with a 1003 objective and Olympus camera with MagnaFire software (Optronics).
Statistical Analysis. Student's t-test was used to compare differences between samples analyzed with the JMP 11.0 software (SAS Institute). All data were expressed as means + s.d.. Statistical differences were deemed significant at the level of P < 0.05.
Results
BRCA1 directly binds DNA in a sequence-independent manner in vitro
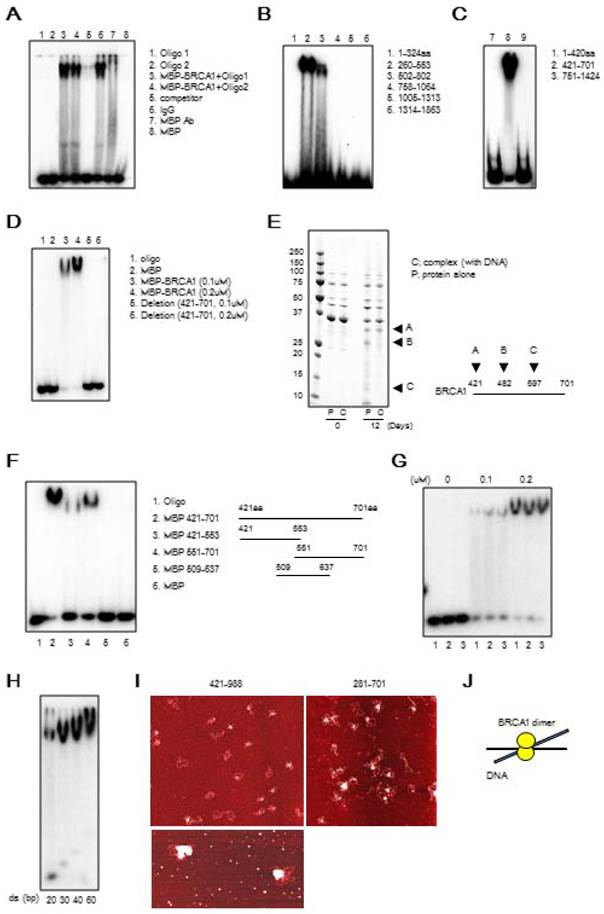
It has previously been reported that BRCA1 binds directly to DNA [8, 20-22]. To narrow down the DNA binding region in the BRCA1, we generated varying lengths of BRCA1 fragment (Figure S3) and performed binding assay. We confirmed the direct binding of BRCA1 to dsDNA by EMSA using MBP-tagged full length BRCA1 protein (Figure 1A). Next, to determine the precise DBR, we performed EMSA using 6 GST-tagged BRCA1 fragments and found that the fragments 260-553aa and 502-802aa interacted with DNA (Figure 1B). Further analysis using 3 MBP-tagged BRCA1 fragments indicated that BRCA1 fragment 421-701aa interacted with DNA (Figure 1C) while a full length form of BRCA1 lacking residues 421-701aa failed to do so (Figure 1D), revealing that this region was critical for DNA binding (Figure 1D). A stability assay using BRCA1 alone and in complex with dsDNA was performed, which narrowed the binding region, and 3 fragments came out (A, B, C) (Figure 1E). The N-terminal sequence showed that A, B, and C starts at 421aa, 482aa and 597aa, respectively. Performing EMSA using small BRCA1 fragments showed that the N- and C-terminus of 421-701aa binds to dsDNA (Figure 1F). Next, we performed EMSA using 3 oligos with random sequences of various lengths of dsDNA. All 3 oligos bound to dsDNA and the binding affinity increased in proportion to concentration and length, respectively (Figures 1G, H). Finally, atomic force microscopy (AFM) was performed to see how BRCA1 binds to dsDNA. Images produced by AFM showed that BRCA1 fragment 421-988aa (63.3 kDa) forms a dimer (~130kDa), and the dimer is located at a DNA crossing point. Therefore, a BRCA1 dimer brings dsDNA together (Figures 1I, J). The images using BRCA1 281-701aa showed the same results.
BRCA1 directly binds to DNA in vitro. (A) Binding of full-length MBP-BRCA1 to dsDNA by EMSA. The identity of the samples is shown on the figure. The concentration of labeled dsDNA and protein are 40nM and 100nM, respectively. (B) Identification of the DBR of BRCA1 using GST-BRCA1 fragments. The identities of the samples are shown on the figure. The concentration of labeled dsDNA and protein are 40nM and 100nM, respectively. (C) Identification of the DBR of BRCA1 using MBP-BRCA1 fragments. The identities of the samples are shown on the figure. The concentration of labeled dsDNA and protein are 40nM and 100nM, respectively. (D) Validation of the DBR of BRCA1 using full-length BRCA1 deficient in the DBR (421-701aa) by EMSA. The identities of the samples are shown on the figure. The concentration of labeled dsDNA is 40nM. (E) In vitro stability of complex of the DBR of BRCA1 421-701aa and splayed-arm DNA or the DBR alone, and N-terminus sequencing of 3 BRCA1 fragments cleaved from the DBR alone (A, B, C). The DNA-BRCA1 complex and BRCA1 alone were incubated at room temperature for 12 days. The proteins were separated by SDS-PAGE, electroblotted to PDVF membranes, and the bands on the membrane were excised for N-terminus sequencing to identify the N-terminal sequence of the cleaved fragments. (F) N-and C-terminus of DBR (421-701aa) binds to dsDNA in EMSA. The identities of the samples are shown on the figure. The concentration of labeled dsDNA and proteins are 40nM and 100nM, respectively. (G) BRCA1 binds dsDNA in a sequence-independent manner in EMSA. The sequences of 3 dsDNA with random sequences are listed in Table S1. The concentration of labeled dsDNA and proteins are 40nM and 0, 100 or 200nM, respectively. (H) The binding affinity of BRCA1 421-701aa to various lengths of dsDNA in EMSA. The concentration of labeled dsDNA and proteins are 40nM and 100nM, respectively. (I) The image of binding of BRCA1 421-988aa or 281-701aa to plasmid DNA by atomic force microscopy. The white dots are the protein and the red structures are plasmid DNA. (J) A model of BRCA1 binding to dsDNA.
BRCA1 preferably binds to DNA under DNA damage condition with splayed-arm shaped DNA
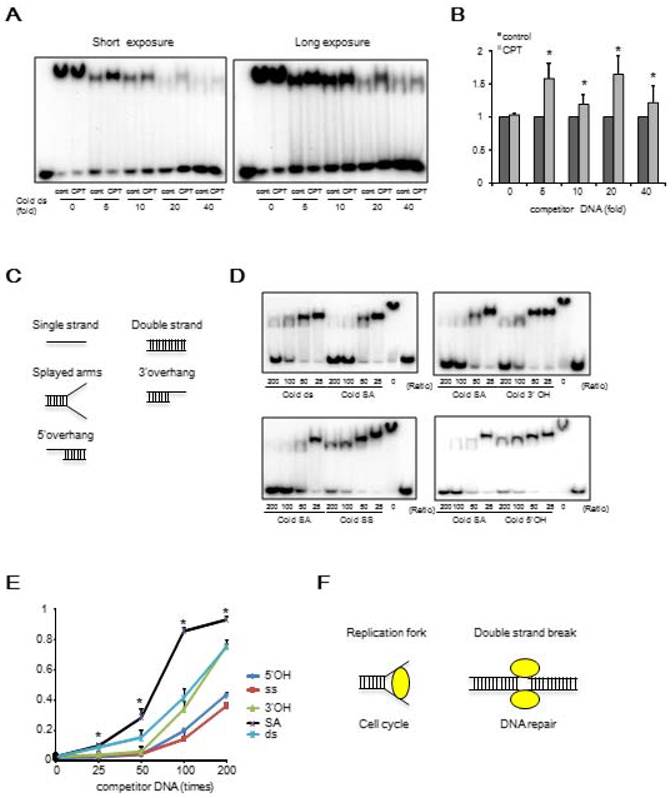
We determined whether the binding affinity of BRCA1 to DNA was affected by DNA damage. MBP-421-701aa (DBR) was purified from 293T cells treated with or without the DNA damage agent, camptothecin (CPT11), which is an inhibitor of topoisomerase 1. Our data indicated that upon DNA damage, the affinity of BRCA1 binding to DNA in vitro was increased using BRCA1 protein purified from cells treated with CPT11 (Figures 2A, B). It is known that protein phosphorylation may affect their ability to interact with DNA. To investigate this, we treated the DBR with λ phosphatase, and our data indicated that dephosphorylation of the DBR increased its binding to DNA (Figure S4A). We further showed that dephosphorylation mimics of S616 and S694 within BRCA1, which have been reported to be phosphorylation sites within the DBR [31, 32] (S616A and S694A), increased the binding while the phosphorylation mimics (S616D and S694D) decreased it (Figure S4B, C). Thus, it is conceivable that DNA damage caused by CPT11 may cause decreased phosphorylation of DBR, leading to the increased DNA binding as previously reported [33]. Next, we examined which structure of DNA preferably binds BRCA1 using 5 possible structures: single strand (SS), 5' overhang (5'OH), 3' overhang (3'OH), splayed arms (SA) and double strand (DS) (Figure 2C). Through a competition assay in EMSA for binding affinities using labeled dsDNA and unlabeled DNA with various structures, we found that the splayed-arm shaped DNA had the most affinity among all the structures tested (Figure 2D, E). Considering that the SA structure resembles DNA replication fork and a part of the DNA double strand break (DSB), and that the affinity of BRCA1 to DNA increases upon DNA damage, we hypothesized that BRCA1 would bind to the DNA replication fork or DSBs under DNA damage condition (Figure 2F).
BRCA1 preferably binds to DNA after CPT11 treatment and Splayed-arm shaped DNA. (A) The comparison in DNA binding affinity of BRCA1 with or without CPT11 treatment in EMSA. Various amounts of unlabeled dsDNA (40bp) were added to see the difference of binding affinity with or without CPT11 treatment. C, control; T, CPT11 treatment. The concentration of labeled dsDNA and proteins are 40nM and 100nM, respectively. (B) Graphical representation and statistical analysis of data from Figure 2A (*P < 0.05, N = 3). (C) Various structures of DNA for competition assay in EMSA. Each DNA is 40bp in length. (D) Competition assay in EMSA to compare the binding affinity using labeled dsDNA and unlabeled DNA with various structures. (E) Graphical representation and statistical analysis of data from Figure 2D (*P < 0.05, N = 3). (F) Candidates for biological function of BRCA1.
The BRCA1 DNA binding region affects the stability of the DNA replication fork
These in vitro data suggest that the binding of BRCA1 to DNA may affect DNA repair or protection of the DNA replication fork. To identify the biological function of the DBR, we generated mouse ES cells with the following BRCA1 constructs containing a C-terminal 3xFlag: BRCA1-WT (two independent clones: #75, and #140) and BRCA1-ΔDBR (#34, and #121) using TALEN technology (Figure S1B, C). We also generated BRCA1-Δ11 as a control because BRCA1-Δ11 cells exhibited many phenotypes similar to those displayed by BRCA1 null cells, including defective homologous recombination (HR) and abnormal cell cycle progression [17-19]. Genotyping by RT-PCR was shown in Figure S1D. BRCA1 protein was analyzed by Western blot (Figure S1E). The region 422-693aa binds to DNA (Figure S1F).
We then determined whether there was any difference in survival between the WT and mutant ES cells after treatment with a PARP1 inhibitor. We observed that the ΔDBR cells exhibited decreased survival as compared to WT cells treated with a PARP1 inhibitor; however, they were more resistant than Δ11 cells (Figure 3A). This observation suggests that HR repair may be partially lost in the ΔDBR cells [34]. To further investigate this, we conducted a HR assay using a sensitive GFP reporter system (Figure 3B) [28]. We failed to detect an obvious difference between WT cells and ΔDBR cells, although marked differences between these cells and Δ11 cells were detected (Figure 3C). There was not statistical difference with a HR assay using HeLa cells between WT cells and ΔDBR cells either (Figure S2B). Altogether, these data demonstrate that the ΔDBR cells are capable of HR and the partial sensitivity to a PARP1 inhibitor may be due to other defects.
Mouse ES cells with BRCA1 deficient in DBR are sensitive to a PARP inhibitor and HU, but maintain similar ability for HR repair as WT cells. (A) Survival MTT assay using BRCA1-WT cells and BRCA1-ΔDBR cells treated with a PARP inhibitor for 3 days. *P < 0.05, N = 3 (ΔDBR vs WT) (B) Graphical representation of HR assay. (C) HR ability of BRCA1-WT, BRCA1-ΔDBR and BRCA1-Δ11. (MT vs Δ11, *P < 0.01, N = 3); (WT vs Δ11, **P < 0.01, N = 3) (D) Survival MTT assay of BRCA1-WT, BRCA1-ΔDBR and BRCA1-Δ11 after HU for 3 days. *P < 0.05, N = 3 (ΔDBR vs WT)
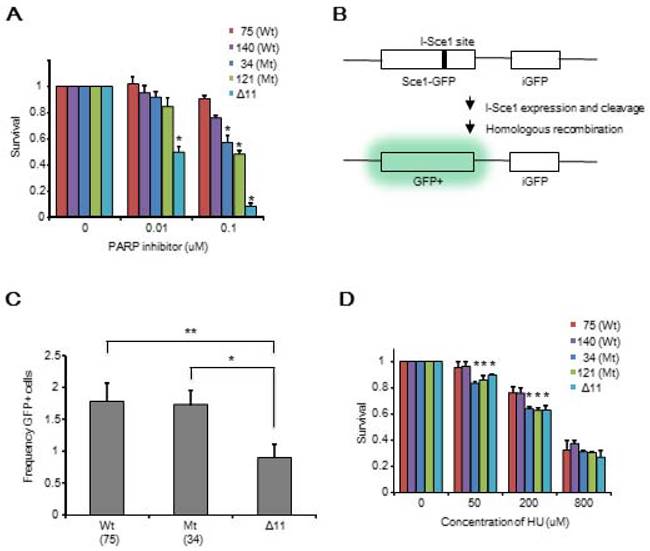
Then, we aimed to identify the other defects by studying the behavior of ΔDBR cells with hydroxyurea (HU) treatment, which causes replication stress. We found that the mutant cells were more sensitive than WT cells to HU treatment (Figure 3D). Indeed both ΔDBR cells and Δ11 cells were indistinguishable in their response to HU treatment. This data suggests that DNA binding by BRCA1 affects the stability of DNA replication forks followed by sensitivity to HU treatment. Since stalled replication forks lead to DNA DSBs, we hypothesized that partial sensitivity of the ΔDBR cells to the PARP1 inhibition may be due to their inability in handling replication stress [35].
BRCA1 regulates the intra-S-phase checkpoint by modulating CHK1 phosphorylation
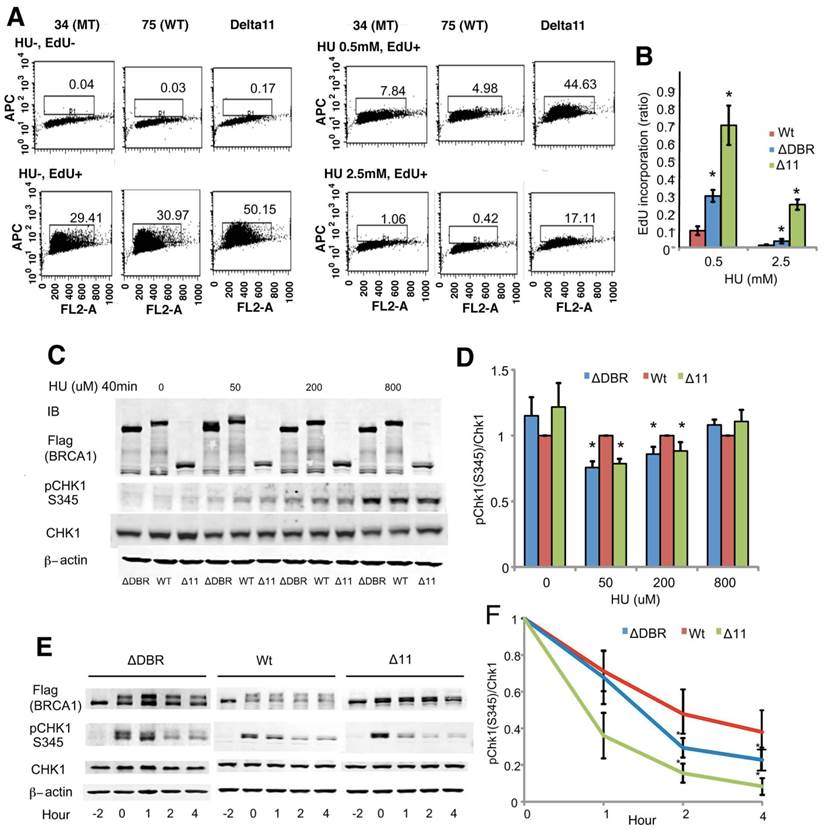
Therefore, we focused on the intra-S-phase checkpoint, which inhibits DNA replication in case of stalled replication forks to allow for repair of DNA damage. We assessed DNA synthesis after cells were treated with HU for 2 hours followed by 30 min labeling with the nucleotide analog EdU. The ΔDBR cells incorporated significantly more EdU than the WT cells (Figure 4A, B) at both low 0.5mM and high 2.5mM concentrations, although the levels of EdU incorporation was lower than Brca11-Δ11 cells, which suffer defects in multiple cell cycle checkpoints [17-19]. Our data also indicated that the ratio of CHK1 phosphorylation, which plays an essential role in regulating the intra-S phase checkpoint, vs total CHK1 protein was decreased in the mutant cells compared with WT cells after a 40min treatment of 50 or 200µM HU (Figure 4C, D) and upon a 2h treatment with 5mM HU followed by varying hours after washing out the HU (Figure 4E, F). Of note, the reduction of pCHK1 is moderate, however comparable with that displayed by BRCA1-Δ11 cells, which is known to be defective in intra-S phase checkpoint control [35]. These data suggest that intra-S-phase checkpoint control was impaired in the ΔDBR cells.
Mouse ES cells homozygous for BRCA1-ΔDBR are impaired in intra-S-phase cell cycle checkpoint. (A) EdU incorporation for 30 min after 2 hours treatment with/without HU. (B) Graphical representation and statistical analysis of data from Figure 4A. Each treatment was normalized to 0 hours of HU treatment. *P < 0.01, N = 3 (ΔDBR vs WT). (C) Phosphorylation status (pChk1 S345) of CHK1 in mouse ES cells after treatment with various concentrations of HU treatment for 40min. ΔDBR:#34, WT:#75 (D) Graphical representation of CHK1 phosphorylation (pChk1 S345) status from Figure 4C. The status was normalized to CHK1. *P < 0.05, N = 3 (ΔDBR or Δ11 vs WT) (E) Time course of CHK1 phosphorylation status (pChk1 S345) in mouse ES cells treated with 5mM HU for 2 hours and then analyzed for up to 4 hours after HU wash out. ΔDBR: #34, WT: #75 (F) Graphical representation of CHK1 phosphorylation status (pChk1 S345) normalized to CHK1 from Figure 4F. *P < 0.05, N = 3 (ΔDBR vs WT)
BRCA1 cells that are deficient in the DNA binding region exhibit moderate chromosomal abnormalities
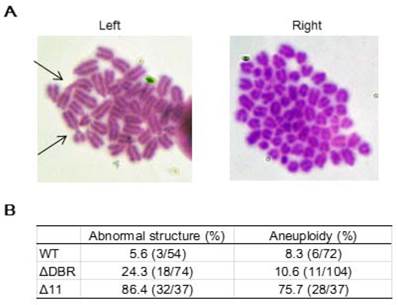
We performed karyotyping analysis to check for genetic instability using WT and ΔDBR ES cells. The data indicated that the ΔDBR cells had a markedly increased number of abnormal chromosome structures as compared with WT cells (Figure 5A Left). The major type of abnormal structures detected in ΔDBR ES cells is Robinson fusion, in which 2 chromosomes are fused at the short arms (Figure 5B Left, arrows). 10.6% ΔDBR ES cells also displayed chromosome aneuploidy (Figure 5B Right), which is not significantly different from that found in WT cells (Figure 5A Right). Altogether, these data indicated that BRCA1-ΔDBR cells exhibit moderate genetic instabilities.
Chromosomal aberrations in ES cells. (A) Summary of chromosome abnormalities detected in BRCA1-WT (#75), and BRCA1-ΔDBR (#34) cells. (B) Chromosome spread by Giemsa staining. Left: Arrows, chromosome with abnormal structure (Robinson fusion) in ΔDBR cells. Right: Aneuploidy in ΔDBR cells (#34).
Discussion
Replication stress, which impairs DNA synthesis at replication forks or interferes with their progression, is a complex phenomenon that has serious implications for genome stability, cell survival, and cancer [36, 37]. Recent evidence suggests that replication stress can be a major source of spontaneous genomic instability driving malignant transformation of pre-cancerous cells [38]. Understanding the process of the intra-S-phase checkpoint, which regulates DNA replication may be key to diagnosing and treating human cancers caused by defective responses to replication stress.
In this study, we aimed to clarify the function of the DBR of BRCA1. First, we identified an essential region (421-701aa) for BRCA1 binding to DNA using various short fragments of BRCA1 by EMSA in vitro. Although previous studies showed a DBR in vitro [8, 20-22], we have further clarified the binding region to a narrower region, in which we validated by deleting this region in the context of full length BRCA1.
Next, we found that BRCA1 brings DNA together and preferably binds to splayed-arm DNA in a sequence-independent manner. Some studies also demonstrated that BRCA1 preferably binds to branched DNA [8, 20, 21]. Therefore, we hypothesized that BRCA1 may affect the DNA replication fork which is in a splayed-arm structure.
Our study is the first study to address the biological significance of the DBR of BRCA1 in vivo, through analyzing BRCA1-ΔDBR ES cells, and compared them with BRCA1-WT or BRCA1-Δ11 ES cells which are defective in multiple biological processes [17-19]. Surprisingly, ΔDBR did not affect the activity of HR repair, but there was an effect on the intra-S-phase checkpoint as a result of replication stress by treatment with HU via CHK1 phosphorylation, followed by the formation of abnormal chromosomal structures.
Generation of aberrant replication fork structures containing single-strand DNA activates the replication stress response, which is primarily mediated by ATR (ATM- and Rad3-related) [39]. Along with its downstream effectors such as the TOPBP1/CLASPIN/CHK1 axis, ATR stabilizes and initiates the restart of stalled replication forks to avoid generation of DNA damage and genome instability by suppressing late replication origin activation to allow the DNA damage repair machinery to repair DNA (called intra-S-phase checkpoint) [40]. The ATR downstream gene CLASPIN interacts with ATR, TOPBP1, CHK1, and BRCA1, and regulates CHK1 phosphorylation, which induces Cdc25 degradation, followed by inhibition of cyclin-dependent kinases and cell cycle delay [41]. Our data suggests that BRCA1 protects the replication fork from replication stress by regulating the intra-S-phase checkpoint through the DBR.
The mechanism of how DNA binding of BRCA1 affects the phosphorylation of CHK1 remains unclear although it was previously shown that BRCA1 is required for CHK1 phosphorylation and activation following ionizing radiation [42]. The binding of BRCA1 to the DNA replication fork may stabilize the complex of TOPBP1, CLASPIN and CHK1, and increase the phosphorylation of CHK1. Further work is needed to clarify this.
This DBR in human BRCA1 includes “hot spots” of missense mutations in breast and ovarian cancer patients [8, 43]. These mutations may affect the function of DBR, and regulation of intra-S-phase checkpoint. The future efforts will be focused on the identification of the biological significance of the mutations by creating knock-in mutations introduced by the CRISPR/Cas9 system.
It has been known that BRCA1-Δ11 cells are defective in both HR and the DNA replication checkpoint [9, 18]. Here we have demonstrated a specific role of the BRCA1-DBR/CHK1 signaling in modulating DNA replication through modulating the intra-S-phase checkpoint resulting from replication stress, while its role in HR repair is dispensable. Our study supports a role of BRCA1-DBR in BRCA1's tumor suppressor properties, as loss of this BRCA1 function leads to impairment of the intra-S-phase checkpoint and genetic instability. Thus, our finding refines a functional domain of BRCA1, BRCA1-DBR, deficiency of which results in the failure of DNA replication upon replicative stress, leading to accumulative DNA mutations and cancer development.
Supplementary Material
Supplementary Table and Figures.
Acknowledgements
We thank members of the Deng laboratory for critical discussions. This work was supported, in part, by the Intramural Research Program of DIDDK, NIH, Bethesda, MD, USA, and the Faculty of Health Sciences, University of Macau, Macau SAR, China.
Funding
This work was supported, in part, by the Intramural Research Program of the National Institutes of Health/National Institute of Diabetes and Digestive and Kidney Diseases, and by start-up funds to CXD of the Faculty of Health Sciences, University of Macau SAR, China.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Alberg AJ, Helzlsouer KJ. Epidemiology, prevention, and early detection of breast cancer. Curr Opin Oncol. 1997;9(6):505-11
2. Hill AD, Doyle JM, McDermott EW, O'Higgins NJ. Hereditary breast cancer. Br J Surg. 1997;84(10):1334-9
3. Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S. et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266(5182):66-71
4. Casey G. The BRCA1 and BRCA2 breast cancer genes. Curr Opin Oncol. 1997;9(1):88-93
5. Alanee SR, Glogowski EA, Schrader KA, Eastham JA, Offit K. Clinical features and management of BRCA1 and BRCA2-associated prostate cancer. Front Biosci (Elite Ed). 2014;6:15-30
6. Paul A, Paul S. The breast cancer susceptibility genes (BRCA) in breast and ovarian cancers. Front Biosci (Landmark Ed). 2014;19:605-18
7. Brazda V, Jagelska EB, Liao JC, Arrowsmith CH. The central region of BRCA1 binds preferentially to supercoiled DNA. J Biomol Struct Dyn. 2009;27(1):97-104
8. Mark WY, Liao JC, Lu Y, Ayed A, Laister R, Szymczyna B. et al. Characterization of segments from the central region of BRCA1: an intrinsically disordered scaffold for multiple protein-protein and protein-DNA interactions? J Mol Biol. 2005;345(2):275-87
9. Deng CX. BRCA1: cell cycle checkpoint, genetic instability, DNA damage response and cancer evolution. Nucleic Acids Res. 2006;34(5):1416-26
10. Clark SL, Rodriguez AM, Snyder RR, Hankins GD, Boehning D. Structure-Function Of The Tumor Suppressor BRCA1. Comput Struct Biotechnol J. 2012:1 (1)
11. Brzovic PS, Rajagopal P, Hoyt DW, King MC, Klevit RE. Structure of a BRCA1-BARD1 heterodimeric RING-RING complex. Nat Struct Biol. 2001;8(10):833-7
12. Henderson JP, Byun J, Williams MV, Mueller DM, McCormick ML, Heinecke JW. Production of brominating intermediates by myeloperoxidase. A transhalogenation pathway for generating mutagenic nucleobases during inflammation. J Biol Chem. 2001;276(11):7867-75
13. Williams RS, Lee MS, Hau DD, Glover JN. Structural basis of phosphopeptide recognition by the BRCT domain of BRCA1. Nat Struct Mol Biol. 2004;11(6):519-25
14. Campbell SJ, Edwards RA, Glover JN. Comparison of the structures and peptide binding specificities of the BRCT domains of MDC1 and BRCA1. Structure. 2010;18(2):167-76
15. Coquelle N, Green R, Glover JN. Impact of BRCA1 BRCT domain missense substitutions on phosphopeptide recognition. Biochemistry. 2011;50(21):4579-89
16. Huen MS, Sy SM, Chen J. BRCA1 and its toolbox for the maintenance of genome integrity. Nat Rev Mol Cell Biol. 2010;11(2):138-48
17. Xu X, Wagner KU, Larson D, Weaver Z, Li C, Ried T. et al. Conditional mutation of Brca1 in mammary epithelial cells results in blunted ductal morphogenesis and tumour formation. Nat Genet. 1999;22(1):37-43
18. Xu X, Qiao W, Linke SP, Cao L, Li WM, Furth PA. et al. Genetic interactions between tumor suppressors Brca1 and p53 in apoptosis, cell cycle and tumorigenesis. Nat Genet. 2001;28(3):266-71
19. Xu X, Weaver Z, Linke SP, Li C, Gotay J, Wang XW. et al. Centrosome amplification and a defective G2-M cell cycle checkpoint induce genetic instability in BRCA1 exon 11 isoform-deficient cells. Mol Cell. 1999;3(3):389-95
20. Paull TT, Cortez D, Bowers B, Elledge SJ, Gellert M. Direct DNA binding by Brca1. Proc Natl Acad Sci U S A. 2001;98(11):6086-91
21. Naseem R, Sturdy A, Finch D, Jowitt T, Webb M. Mapping and conformational characterization of the DNA-binding region of the breast cancer susceptibility protein BRCA1. Biochem J. 2006;395(3):529-35
22. Naseem R, Webb M. Analysis of the DNA binding activity of BRCA1 and its modulation by the tumour suppressor p53. PLoS One. 2008;3(6):e2336
23. Scully R, Chen J, Plug A, Xiao Y, Weaver D, Feunteun J. et al. Association of BRCA1 with Rad51 in mitotic and meiotic cells. Cell. 1997;88(2):265-75
24. Aricescu AR, Lu W, Jones EY. A time- and cost-efficient system for high-level protein production in mammalian cells. Acta Crystallogr D Biol Crystallogr. 2006;62(Pt 10):1243-50
25. Sakasai R, Sakai A, Iimori M, Kiyonari S, Matsuoka K, Kakeji Y. et al. CtIP- and ATR-dependent FANCJ phosphorylation in response to DNA strand breaks mediated by DNA replication. Genes Cells. 2012;17(12):962-70
26. Huber LJ, Yang TW, Sarkisian CJ, Master SR, Deng CX, Chodosh LA. Impaired DNA damage response in cells expressing an exon 11-deleted murine Brca1 variant that localizes to nuclear foci. Mol Cell Biol. 2001;21(12):4005-15
27. Shlyakhtenko LS, Gall AA, Filonov A, Cerovac Z, Lushnikov A, Lyubchenko YL. Silatrane-based surface chemistry for immobilization of DNA, protein-DNA complexes and other biological materials. Ultramicroscopy. 2003;97(1-4):279-87
28. Moynahan ME, Chiu JW, Koller BH, Jasin M. Brca1 controls homology-directed DNA repair. Mol Cell. 1999;4(4):511-8
29. Shen SX, Weaver Z, Xu X, Li C, Weinstein M, Chen L. et al. A targeted disruption of the murine Brca1 gene causes gamma-irradiation hypersensitivity and genetic instability. Oncogene. 1998;17(24):3115-24
30. Deng CX, Xu X. Generation and analysis of Brca1 conditional knockout mice. Methods Mol Biol. 2004;280:185-200
31. Moritz A, Li Y, Guo A, Villen J, Wang Y, MacNeill J. et al. Akt-RSK-S6 kinase signaling networks activated by oncogenic receptor tyrosine kinases. Science signaling. 2010;3(136):ra64
32. Nelson AC, Lyons TR, Young CD, Hansen KC, Anderson SM, Holt JT. AKT regulates BRCA1 stability in response to hormone signaling. Mol Cell Endocrinol. 2010;319(1-2):129-42
33. Kehn K, Berro R, Alhaj A, Bottazzi ME, Yeh WI, Klase Z. et al. Functional consequences of cyclin D1/BRCA1 interaction in breast cancer cells. Oncogene. 2007;26(35):5060-9
34. De Soto JA, Deng CX. PARP-1 inhibitors: are they the long-sought genetically specific drugs for BRCA1/2-associated breast cancers? Int J Med Sci. 2006;3(4):117-23
35. Willis NA, Chandramouly G, Huang B, Kwok A, Follonier C, Deng C. et al. BRCA1 controls homologous recombination at Tus/Ter-stalled mammalian replication forks. Nature. 2014;510(7506):556-9
36. Rosen EM. BRCA1 in the DNA damage response and at telomeres. Front Genet. 2013;4:85
37. Wang RH, Lahusen TJ, Chen Q, Xu X, Jenkins LM, Leo E, Fu H, Aladjem M, Pommier Y, Appella E, Deng CX. SIRT1 deacetylates TopBP1 and modulates intra-S-phase checkpoint and DNA replication origin firing. Int J Biol Sci. 2014;10(10):1193-202
38. Bartek J, Lukas J. DNA damage checkpoints: from initiation to recovery or adaptation. Curr Opin Cell Biol. 2007;19(2):238-45
39. Zeman MK, Cimprich KA. Causes and consequences of replication stress. Nat Cell Biol. 2014;16(1):2-9
40. Errico A, Costanzo V. Mechanisms of replication fork protection: a safeguard for genome stability. Crit Rev Biochem Mol Biol. 2012;47(3):222-35
41. Liu S, Bekker-Jensen S, Mailand N, Lukas C, Bartek J, Lukas J. Claspin operates downstream of TopBP1 to direct ATR signaling towards Chk1 activation. Mol Cell Biol. 2006;26(16):6056-64
42. Yarden RI, Pardo-Reoyo S, Sgagias M, Cowan KH, Brody LC. BRCA1 regulates the G2/M checkpoint by activating Chk1 kinase upon DNA damage. Nat Genet. 2002;30(3):285-9
43. Rebbeck TR, Mitra N, Wan F, Sinilnikova OM, Healey S, McGuffog L. et al. Association of type and location of BRCA1 and BRCA2 mutations with risk of breast and ovarian cancer. JAMA. 2015;313(13):1347-61
Author contact
![]() Corresponding author: Chu-Xia Deng, Faculty of Health Sciences, University of Macau, Macau SAR, China. cxdengmo
Corresponding author: Chu-Xia Deng, Faculty of Health Sciences, University of Macau, Macau SAR, China. cxdengmo