Impact Factor ISSN: 1449-2288
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Whole genome sequence studies of...
Genome and transcriptome studies...
Whole microbiome studies of...
Summary and Outlook
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2016; 12(2):156-171. doi:10.7150/ijbs.13537 This issue Cite
Review
Recent Developments in Using Advanced Sequencing Technologies for the Genomic Studies of Lignin and Cellulose Degrading Microorganisms
Ayyappa kumar Sista Kameshwar, Wensheng Qin ![]()
Department of Biology, Lakehead University, 955 Oliver Road, Thunder Bay, Ontario, P7B 5E1, Canada.
Received 2015-8-12; Accepted 2015-11-3; Published 2016-1-1
Abstract

Lignin is a complex polyphenyl aromatic compound which exists in tight associations with cellulose and hemicellulose to form plant primary and secondary cell wall. Lignocellulose is an abundant renewable biomaterial present on the earth. It has gained much attention in the scientific community in recent years because of its potential applications in bio-based industries. Microbial degradation of lignocellulose polymers was well studied in wood decaying fungi. Based on the plant materials they degrade these fungi were classified as white rot, brown rot and soft rot. However, some groups of bacteria belonging to the actinomycetes, α-proteobacteria and β-proteobacteria were also found to be efficient in degrading lignocellulosic biomass but not well understood unlike the fungi. In this review we focus on recent advancements deployed for finding and understanding the lignocellulose degradation by microorganisms. Conventional molecular methods like sequencing 16s rRNA and Inter Transcribed Spacer (ITS) regions were used for identification and classification of microbes. Recent progression in genomics mainly next generation sequencing technologies made the whole genome sequencing of microbes possible in a great ease. The whole genome sequence studies reveals high quality information about genes and canonical pathways involved in the lignin and other cell wall components degradation.
Keywords: Lignocellulose, Fungi, Bacteria, 16s rRNA, Inter Transcribed Spacer, Biodegradation
Introduction
Drastic change in the climatic conditions and decrease in the fossil fuel reserves are the main motives for the extensive research being conducted in the field of bio refining industry. The abundance and availability of lignocellulosic biomass makes the cellulosic ethanol as a significant and immediate alternative for conventional fuels.
Lignocellulose - a potential source for future energy needs
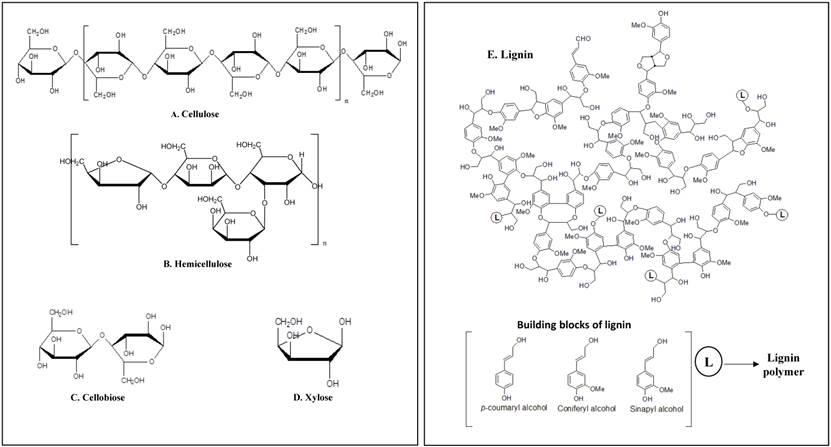
Plant cells are protected with three layers known as middle lamella, primary and secondary cell walls which are made up of carbohydrates (cellulose, hemicellulose and pectin) and lignin [1] [Figure 1] i) Cellulose is a plant polysaccharide and highly abundant organic polymer on the earth, composed of numerous glucose units linked in β (1→4) linkages, occurring in both crystalline and amorphous forms. Cellulose is present in plant cell walls, stems, straw, stalks and other woody portion of the plant, it alone constitutes to 40-50% of dry weight of plant material [2]. ii) Hemicellulose is a composite compound with a mixture of pentoses, hexoses and sugar acids [3-6]. Hemicellulose constitutes a significant component in cell wall connecting cellulose and lignin layers, thus totally imparting strength and rigidity to the whole lignocellulose network[6]. Hemicellulose is also studied for its derivative known as xylose, which is mainly used for the production of xylitol a valuable sucrose substituent. iii) Lignin is a complex organic biopolymer mainly present in plants in close association with carbohydrates like cellulose and hemicellulose, its main activity is to provide plants with structural stability, impermeability and resistance towards microbial attack [3]. It is made up of three phenylpropanoid units guaiacyl (G), p-hydroxyphenyl (H) and syringyl alcohols (S) also called as monolignols, inter linked by several carbon-carbon linkages [7, 8]. It is hard to effectively use cellulose without breaking and separating lignin efficiently. Extensive research has been carried out for separating lignin by using chemical, physical and biological methods [9, 10].
Schematic representation of plant cell wall components having high commercial importance a) Cellulose b) Hemicellulose c) Cellobiose d) xylose e) Lignin.
Microorganisms as possible degraders of lignocellulose
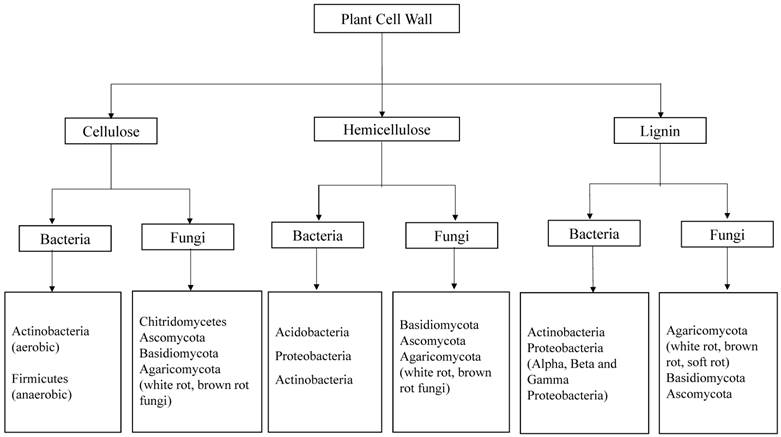
Fungi are greatly known for their ability to depolymerize lignocellulosic biomass compared to bacteria. Earlier studies convey that several fungal species like Trichoderma reesei, Phanerochaete chrysosporium, Fomitopsis palustris, Orpinomyces sp etc, were found to degrade cellulose by inhabiting the gastrointestinal tracts of several ruminating animals [11, 12]. Fungi produce variety of intracellular and extracellular enzymes such as cellulases and hemicellulases [11, 13, 14] lignin peroxidases, manganese peroxidases, versatile peroxidases and laccases which are collectively involved in the breakdown and utilization of lignocellulosic biomass [15]. The property, structure and function of fungal laccases have been studied and reported earlier [16, 17]. Compared to its counterparts bacterial enzymology for lignin degradation was not well understood. According to Ramachandran et.al (2000), Streptococcus viridosporus T7A, a lignin degrading bacterium produces several extracellular peroxidases which are similar to the peroxidases produced by fungi [18]. Recent advancements in the genome sequencing studies reveal that laccases are also widespread in several bacterial strains [19]. However the bacterial laccases were not well characterized and understood as fungal laccases [18-20]. Several molecular methods are applied in the modern microbiology to identify and classify the microorganisms such as bacteria and fungi. In order to find the molecular phylogeny of bacteria the 16s rRNA genes are used, because this part of DNA was undisturbed and conserved during the evolution. Similarly, Inter Transcribed Spacer (ITS) DNA is used for finding the molecular ecology of different fungal strains. Wide research has been carried out to find potential lignocellulosic biomass degrading microorganisms [Figure 2].
Represents the potential groups of bacteria and fungi, capable of degrading plant cell wall components (cellulose, hemicellulose and lignin). Compiled from [7, 10, 21-23]
Genome sequencing studies for the identification and characterization of microorganisms
Genome sequencing is a revolutionary method to analyze the DNA sequence of an organism efficiently. The era of sequencing can be divided into three successive generations such as:
First Generation Sequencing: Sanger sequencing and Maxam Gilbert sequencing comes under the first generation sequencing techniques. Sanger sequencing was invented by Frederick Sanger in the year 1977 based on the principle “Dideoxynucleotide” or “Chain termination” method. In this method the DNA strand is sequenced by using modified dNTP known as ddNTP (dideoxynucleotide triphosphates) which lacks 3'OH group. The 3'OH group is required for the formation of a phosphodiester bond between two nucleotides, however the absence of it leads to the cessation of the DNA strand elongation. The ddNTPs are either radioactively or fluorescently labeled for the detection by automated sequencer machines [24]. Sanger sequencing was the first successful method developed for DNA sequencing with innumerable applications in biology and medicine [24]. During 1976-1977, Maxam Gilbert introduced chemical method of DNA sequencing which involved the radioactive labeling of 5'OH group of DNA strand followed by base modification using chemicals and later the extracted DNA is subjected to electrophoresis and autoradiography for finding the sequence of the subject DNA. Maxam Gilbert method mainly have two disadvantages: a) It requires high amounts of quality DNA b) the process cannot be automated. Due to these drawbacks this method was supersede by Sanger's method of sequencing.
Next Generation Sequencing (NGS): It mostly refers to those high throughput technologies which facilitates parallel sequencing of DNA templates with millions of base pairs. NGS can be broadly divided as second generation sequencing and third generation sequencing methods based on their progressive evolution from the Sanger sequencing method. Three major companies such as Roche, Illumina and Life technologies can be considered as second generation sequencers. Second generation sequencing platforms are based on two principles: a) polymerase based clonal replication of single DNA molecules separated on a solid matrix either a bead (or) planar surface b) cyclic sequencing chemistries. Based on these principles, sequencing companies define their own protocol. Apart from the above mentioned similarities, all sequencing platforms follow same format of front-end library preparation methods such as adding universal adapter sequences to the DNA fragment on its terminal ends. The adapter sequences are complementary to polymerase chain reaction (PCR) primers (which are required for the amplification of the library) and also to the oligonucleotides adhered to a solid support (required for clonal DNA amplification). Sequencing platforms like Roche and ABi Solid use emulsion PCR and Illumina uses bridge PCR for its amplification process [25]. Next generation sequencing technology was extensively reviewed in the literature [23-28].
Third Generation Sequencing: Though NGS techniques are effortless and cost effective for sequencing a whole genome, it also has some drawbacks such as i) Short read lengths ii) Inaccurate sequencing methods for homopolymer stretches. Third Generation sequencing platforms like PacBio, Helicos Biosciences are developing new methods to resolve these drawbacks [25-31] (Figure 3).
Illustrates the methods used for the characterization of lignocellulose degrading bacteria and fungi using traditional and next generation sequencing methods.
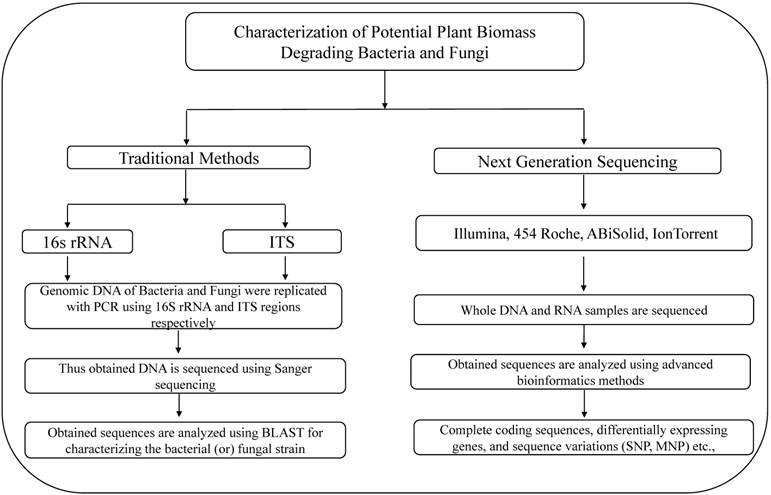
Although research was conducted since few decades, very little information is revealed about the microbial degradation of lignocellulosic biomass. However, recent sequencing studies based on potential lignocellulose degrading microorganisms revealed high quality information about the genes and molecular pathways underlying lignocellulose degradation. In this review we broadly discussed about the outcomes of these sequencing projects.
CAZy and FOLy Databases for Genome wide Enzyme Analysis
a) Carbohydrate Active Enzyme Database (CAZy) for sequence based classification of structurally related families of catalytic and functional domains (carbohydrate binding modules) of enzymes which are required for the creation, modification and breakdown of glycosidic bonds [32]. CAZy database forms a single platform for the display and analysis of functional and genomic aspects of carbohydrate active enzymes. Monosaccharides, the basic unit of the carbohydrates tend to join with other monosaccharides giving rise to a chain of saccharides called oligosaccharides through glycosidic linkages. These oligosaccharides also join with other non-carbohydrate molecules resulting in complex glycoconjugates. Different classes of enzymes are required for the biosynthesis, modification and catabolism of carbohydrates. CAZy database can be accessed by searching the genomic contents (or) sequence based families, the genomes of different organisms are updated based on the releases of GenBank. CAZy database [32] is currently classified CAZy enzymes into six families they are:
I. Glycoside Hydrolases (GH): (Also called as glycosidase) this group of enzymes catalyzes the hydrolytic cleavage of the glycosidic bond. Currently the glycoside hydrolase family is classified in to 134 sub families. Total number of classified enzymes are 256280 and non-classified enzymes are 3767.
II. Glycosyl Transferases (GT): Glycosyl transferases are required for the biosynthesis of the di, oligo and poly saccharides. These enzymes are required for the transfer of sugar molecules between two conjugates by forming a glycosidic bond. Currently glycosyl hydrolase family is classified in to 97 sub families. Total number of classified enzymes are 208338 and non-classified enzymes are 3734.
III. Polysaccharide Lyases (PL): Enzymes belonging to this group are involved in breaking down glycosidic linkages in uronic-acid containing polysaccharides by a special mechanism called β-elimination mechanism resulting in hexenuronic acid and a new reducing sugar end. Currently there are 23 subfamilies in polysaccharide lyases family, with total number of 6137 classified enzymes and 422 non-classified enzymes.
IV. Auxiliary Activity Enzymes (AA): These class of enzymes cover the redox enzymes which work in combinations with the CAZymes. Auxiliary activity enzymes presently classified in to three subfamilies of polysaccharide monoxygenases and eight subfamilies containing lignolytic enzymes. Currently there are 10455 enzymes classified in to 13 sub families and 304 non-classified enzymes.
V. Carbohydrate Esterases (CE): Carbohydrate esterases catalyzes the de-O and de-N-acylation of saccharides. Carbohydrate esterases mainly considers two classes of substrates a) sugar plays the role of acid (pectin methyl esters) b) sugar plays the role of alcohol (acetylated xylan). Present CAZy database has divided the carbohydrate esterases in to 16 sub families with total number of 28450 classified enzymes and 834 non classified enzymes.
VI. Carbohydrate Binding Modules (CBM): Group of amino acids present within carbohydrate active enzymes with a distinct folding patterns to accommodate a carbohydrate molecule, thus possessing a carbohydrate-binding activity. Initially these modules are classified as cellulose-binding modules (CBDs), however due to the discovery of new modules which bind to different carbohydrates other than cellulose was the main reason to be rename this class as carbohydrate binding modules. Currently there are 59498 enzymes classified into 71 subfamilies and 283 non classified enzymes.
b) Fungal Oxidative Lignin Enzymes (FOLy) is a database developed for the enumeration and classification of enzymes involved in the breakdown lignin. FOLy has divided lignin degrading enzymes in to two main classes as Lignin Oxidases (LOs) and Lignin Degrading Auxiliary enzymes (LDAs) [33].
Lignin Oxidases (LO): Lignin oxidases are further divided into three main classes as LO1 (represent laccases), LO2 (Lignin peroxidases, Manganese peroxidases, Chloro peroxidases) LO3 (Cellobiose dehydrogenase).
Lignin Degrading Auxiliary Enzymes (LDA): The enzymes classified under lignin degrading auxiliary enzymes are mostly hydrogen peroxide producers. LDAs are further classified into six classes LDA1 (Aryl alcohol oxidase) LDA2 (Vanillyl alcohol oxidase) LDA3 (Glyoxal Oxidase) LDA4 (Pyranose Oxidase) LDA5 (Galactose Oxidase) LDA6 (Glucose Oxidase) and LDA7 (Benzoquinone reductase).
Whole genome sequence studies of lignocellulose degrading bacteria
Molecular methods like sequencing 16s rRNA have helped scientific community in identification and classification of the bacteria efficiently. 16s ribosomal RNA (16s rRNA) is a component of the 30s small subunit of prokaryotic ribosomes. 16s rRNA is highly considered for the identification and classification of bacteria due to its subdued rates of evolution [34]. Research study conducted by Pold et al for finding lignin degrading bacteria involved the usage of Next Generation Sequencing techniques like Illumina MiSeq for sequencing 16s rRNA to rebuild the molecular phylogenies of bacteria [35]. Whole genome sequencing studies of the several potential lignocellulose degrading bacteria had been carried out during recent years (Table 1). These studies reveal tremendous amount of high quality information about the lignin degrading mechanisms adopted by several microorganisms. In most of the studies combinatorial sequencing methods were simultaneously applied to sequence and analyze the genomes of the microorganisms. The following are the outcomes from the whole genome sequence of the bacterial strains:
Whole genome sequencing studies of different bacterial strains
| Phylum | Bacterial Strain | Sequencing Platform | Layout | Submission and Reference |
|---|---|---|---|---|
| Actinobacteria | Arthrobacter sp. Rue61a | 454 Pyrosequencing | SE | CP003203; CP003205 [37] |
| Amycolatopsis sp. Strain ATCC 39116 | Illumina, 454 Pyrosequencing | PE | AFWY00000000[41] | |
| α-proteobacteria | Novosphingobium sp. Strain MBES04 | 454 Pyrosequencing, Ion Torrent PGM | MPE | BBNP00000000 [42] |
| Rhizobium sp. strain YS-1r | Illumina MiSeq | PE | JPYQ00000000 [43] | |
| β-proteobacteria | Burkholderia sp. Strain LIG30 | Illumina MiSeq | PE | JGVW00000000 [44] |
| Cupriavidus basilensis B-8 | Illumina HiSeq | PE | AKXR00000000 [45] | |
| γ-proteobacteria | Halomonas sp. strain KO116 | Illumina MiSeq PacBio RSII | PE | CP011052 [46] |
| Pseudomonas sp. Strain YS-1p | Illumina MiSeq | PE | PYP00000000 [43] | |
| Klebsiella sp. strain BRL6-2 | Illumina, PacBio Sequencing | MPE | ARVT00000000 [47] | |
| Raoultella ornithinolytica Strain S12 | Illumina MiSeq, 454 Pyrosequencing | PE | CP010557 [48] |
Sequencing studies of actinobacterial strains
Arthrobacter strain Rue61a: Arthrobacter species is widely present in the soil environments. Based on the studies conducted by Niewerth et.al (2012) Arthrobacter strain Rue61a isolated from sewage sludge was found to utilize several lignin derivatives like quinaldine as sole carbon source for its energy. Whole genome sequencing was performed to analyze the molecular basis of the versatility of Arthrobacter strain Rue16a [35]. The genome of Arthrobacter sp. Rue61a consists of a single circular chromosome of 4,736,495 bp with usual G+C content of 62.32%, 231,551-bp of circular plasmid known as pARUE232, and a linear plasmid of 112,992-bp known as pARUE113. The pARUE232 plasmid contribute to Arthrobacter sp. Rue61a resistance towards arsenate and Pb2+, whereas the linear plasmid facilitates for the conversion of quinaldine to anthranilate. According to Niewerth et.al (2012; 2013) Arthrobacter strain Rue61a expresses genes encoding quinaldine catabolism which clustered on a linear plasmid known as pAL1 with two upper operon pathways called meqABC and meqDEF. These cluster of genes codes for the conversion of quinaldine to anthranilate [36, 37]. The study also confirms that Arthrobacters outstanding degradation of anthranilate exclusively continues through CoA-thioester pathway. Apart from quinaldine utilization, strain Rue61a has a set of aromatic degradation pathways, enabling the utilization of 4-hydroxy-carboxylic acids, which are characteristic products of lignin depolymerization, via ortho cleavage of protocatechuate [36, 37].
Amycolatopsis sp. ATCC 39116: Amycolatopsis species is a soil dwelling microorganism previously known as Streptomyces setonii. According to Antai and Crawford, this species was able to depolymerize lignin and its resulting downstream products like benzoate, catechol, gentisate, guaiacol, p-coumarate, protocatechuate, ferulate, producing several commercially valuable products like vanillin [38-40]. The genome sequencing of Amycolatopsis was performed by Davis et.al to understand the molecular mechanisms employed by Amycolatopsis for the breakdown of lignocellulose.
The complete genome size is 8,442,518 bp with a G+C content of 71.9%, total of 8,264 candidate protein-encoding genes were identified. Genome sequencing study of Amycolatopsis sp. ATCC 39116 was consistent with earlier reports. The presence of genes coding for lignin-depolymerizing enzymes such as heme peroxidases, laccases, catalases, and oxidases confirms its ability to use lignin. According to Fleige et.al (2013), Amycolatopsis ATCC 39116 strain was successful in conversion of ferrulic acid to vanillin. From genomic sequences, it was noticed that ATCC 39116 strain expressed vdh (Vanillin dehydrogenase) gene which is required for the conversion vanillin to vanillic acid [49]. Similarly, there are several genes coding for pathways required for the catabolism of catechol, benzoate, protocatechuate, phenylacetate, and other methylated aromatic compounds. Sequencing studies also convey that Amycolatopsis sp. ATCC 39116 lacks genes coding for cellulases but has many other genes coding for carbohydrate degrading enzymes [41].
Sequence studies of Alphaproteobacterial strains
Novosphingobium sp. Strain MBES04: According to Ohta et.al (2015) Novosphingobium sp. Strain MBES04 was a marine strain isolated from sunken wood. This strain was found to depolymerize a broad range of lignin related compounds [42, 50-52]. Genome sequencing studies were conducted by Ohta Y et.al in order to understand the underlying mechanisms involved behind its ability to utilize plant derived compounds as complete energy source [42, 53]. Draft genome of Novosphingobium sp. Strain MBES04 consists of 5,361,448 nucleotides with a G+C content of 65.4%. Based on the genomic data, 4,728 gene coding sequences (CDSs) were obtained. The draft genome sequence of strain MBES04 showed the existence of benzoate degrading gene cluster and also genes coding for benzoate transporter proteins, 2-chlorobenzoate 1, 2-dioxygenase, and for the catechol branch of the ketoadipate pathway. Genome sequence of MBES04 also revealed information about the genes involved in the aromatic compound degradation such as p-hydroxybenzoate 3-monooxygenase and vanillate monooxygenase. MBES04 strain also showed three copies of the protocatechuate 4, 5-dioxygenase α and β subunits required for meta cleavage pathway of protocatechuate, which is an important compound in lignin related aromatic compound degrading pathways [42].
Rhizobium sp. strain YS-1r: According to Prabhakaran et.al Rhizobium sp. Strain YS-1r was isolated from a decaying wood sample from a thermal pond situated in Yellowstone National Park, Wyoming. The primary goal of Prabhakaran et.al study was to isolate and characterize bacteria which are able to utilize lignin and its derivatives as complete carbon source for their growth and metabolism. Rhizobium sp. Strain YS-1r genome sequencing was performed to understand the molecular basis of lignin degradation. Sequence analysis revealed that YS-1r expresses several genes coding for the enzymes such as laccase, DyP-peroxidase, β-etherase, vanillate O-demethylase, feruloyl esterase, carboxyl esterase, cytochrome P450, and chloroperoxidase which are involved in the degradation of lignin and its derived aromatic compounds. YS-1r strain also codes for enzymes like phenol 2 monooxygenase, 4-hydroxybenzoate-3-monooxygenase, catechol-2,3-dioxygenase, protocatechuate 3, 4-dioxygenase, and gentisate 1,2-dioxygenase which are involved in aromatic ring-oxidation and ring-cleavage reactions [43].
Sequence studies of Betaproteobacterial strains
Burkholderia sp. Strain LIG30: Burkholderia species is a widely present bacterial species present in the environment playing different roles in the ecological niche [54]. Burkholderia species secrete several extracellular enzymes which are capable of degrading plant cell wall components like cellulose, hemicellulose, lignin and xylose [54-56]. According to Woo et.al (2014) strain LIG30 was isolated from the rainforest soil using the alkali lignin as sole carbon source [44]. Thus genome sequencing was performed to get the insights into novel lignin degrading enzymes. The draft genome of strain LIG30 was found to be 5.5 Mb, with a 66.4% G+C content and 4,996 candidate protein-encoding gene models. The putative functional annotations were obtained from Cluster of orthologous groups (COGs) analysis based on which functional groups were assigned to 73% of the genes; 305 of the allocated genes were connected with secondary metabolite biosynthesis, transport, and catabolism. The enzyme analysis was performed using PRIAM (method for automated enzyme analysis using genome sequences) EC numbers resulted in 2 predicted genes which encode multicopper oxidases and 22 putative genes coding for catalases and peroxidases which might contribute to the lignin degrading ability of Burkholderia sp. strain LIG30 [44].
Cupriavidus basilensis B-8: Studies have proved that Cupriavidus basilensis has the ability to metabolize several toxic intermediates of lignocellulosic biomass including furfural [57]. According to Qian et al, Cupriavidus basilensis species was able to mineralize organic phosphate [58]. According to Shi et al, Cupriavidus basilensis B-8 isolated from erosive bamboo slips has exhibited significant ability to degrade kraft lignin, it was able to degrade 44.4% of kraft lignin with an initial concentration of 2g per liter by highly producing manganese peroxidase, lignin peroxidase and laccase. In order to complete the metabolic reconstruction of lignin degrading mechanisms by C. basilensis B-8, genome sequencing was performed by Shi et.al. A total of 495 Mb of sequence data with 8448 coding sequences were analyzed. Several predicted genes which are said to be involved in the lignin degradation pathways were identified using the genome analysis of C. basilensis B-8. Based on the genome and proteome studies of C. basilensis B-8, three major lignin degrading pathways known as Β-ketoadipate, phenol degradation and gentisate pathways were reconstructed [45].
Sequence studies of γ-proteobacterial strains
Raoultella ornithinolytica Strain S12: Raoultella ornithinolytica S12 is a facultative anaerobic organism having both a respiratory and fermentative type of metabolism mostly producing acid, gas (or) 2, 3-butanediol as major end products of glucose metabolism [59]. According to Bao et.al, R.ornithinolytica S12 was screened by growing on alkali lignin as sole carbon source under aerobic conditions. R. ornithinolytica S12 genome has one chromosome, which is 5,522,044 bp in size with a G+C content of 57.47%. The whole genome has 4,875 predicted coding sequences (CDS), 86 pseudogenes, and 18 frameshifted genes making it to a total of 5075 genes. Protein BLAST (BLASTp) analysis revealed that about 78.4% of the genes were assigned to specific COGs, and out of which 52.6% were involved in 196 predicted metabolic pathways. Genome sequencing analysis of S12 strain exposed many genes involved in several pathways, like aromatic compounds degradation, starch, sucrose and methane metabolic pathways, providing the source for lignin degradation mechanisms [48].
Klebsiella sp. strain BRL 6-2: According to Woo et.al (2014) Klebsiella species strain BRL 6-2 was isolated from the soil collected from a watershed. It was initially isolated based on its ability to grow on lignin as sole carbon source under anaerobic conditions [47, 56]. Genome sequencing was performed by Woo et.al (2014) to understand the underlying lignin utilization mechanisms of BRL62. The sequence consists of one circular chromosome of 5,801,355 bp with no distinct plasmids. The G+C content of BRL6-2 was found to be 55.24%. Gene prediction analysis was performed using Integrated Microbial Genomes (IMG-ER) platform, majority of the genes coding for proteins (86.3%) and their putative functions were identified. Based on the analysis, 5,495 predicted genes were detected out of which 5,296 were protein coding genes, 199 were RNAs and 64 were pseudogenes. The lignin degrading ability of BRL6-2 strain can be attributed to its fully functional protocatechuate pathway and high expression of six putative genes coding for peroxidase, two putative genes coding for lactate dehydrogenase and two putative genes coding for catalase enzymes. Sequence analysis also revealed that BRL6-2 strain has several genes coding for cytochrome oxidase, which suggests the occurrence of both dissimilatory and assimilatory lignin degrading pathways [47].
Halomonas sp. strain KO116: O'Dell et.al have isolated Halomonas sp. strain KO116, a halotolerant bacteria from the surface sea water, which is able to grow on insoluble organosolvent lignin and further characterized using alkali lignin minimal salt medium [60]. Genome sequencing of KO116 was performed to understand its molecular mechanisms behind lignin degradation. KO116 genome consists of circular sequence corresponding to 4.6-Mb of circular genome, 2 mega plasmids with a length of 313 kb and 205 kb with a G+C content of 54.3%. Gene prediction analysis was performed using COG, which resulted in total of 4298 protein coding genes of which 926 genes were involved in information storage and processing. 1,430 and 3244 genes were found to be involved in cellular and metabolic processes respectively, and gene functions of 1,228 genes were not well characterized. Genes like catalases, peroxidases, and enzymes involved in aromatic compound degradation and ketoadipate pathways and several other relevant genes required for lignin degradation were highly observed in KO116 genome. Thus the genome analysis explains the ability of KO116 to inhabit insoluble organosolvent lignin and also its tolerance towards ionic solvents. [46].
Pseudomonas sp. strain YS-1p: Pseudomonas species is a well-known for its high versatility of metabolic activities in different ecosystems including soil [61]. Several Pseudomonas species were highly studied for their ability to metabolize several polymers like cellulose, lignin etc. According to Akinosho et al (2015), Pseudomonas sp. Strain 10-1B isolated from polluted soil was able to degrade several polycyclic aromatic hydrocarbons (PAH) by releasing several enzymes involved in the cleavage of aromatic rings [62]. According to Prabhakaran et al, Pseudomonas sp. strain YS-1p isolated from a decaying wood samples were able to utilize lignin and its derivatives as primary source of carbon [43]. Genome sequencing was performed to understand the molecular basis of lignin degradation by these organisms. Genes coding for enzymes such as laccase, DyP-peroxidase, β-etherase, vanillate O-demethylase, feruloyl esterase, carboxyl esterase, cytochrome P450, and chloroperoxidase were highly observed in YS-1P genome. Enzymes involved in aromatic ring oxidation and cleavage such as phenol 2-monooxygenase, 4-hydroxybenzoate 3-monooxygenase, catechol 2, 3-dioxygenase, protocatechuate 3, 4-dioxygenase, and gentisate 1,2-dioxygenase were highly detected in YS-1P genome sequence [43].
Genome and transcriptome studies of lignocellulose degrading fungi
Most considered molecular method for characterizing fungi was in vitro replication (PCR) of fungal DNA with ITS primers followed by DNA sequencing. Inter Transcribed Spacer (ITS) regions are the non-coding DNA sequences which are located between small subunit and large subunits of ribosomal RNA genes. ITS regions are widely used for identification and classification of fungi. ITS regions are considered as the universal barcodes for fungi. Several studies were conducted on the PCR amplification of the fungal DNA using the ITS primers followed by Sanger's sequencing and analyzing thus obtained sequence using the NCBI BLAST for finding the similarity matches with other species [63, 64]. Similar work was carried by Ortiz et al for the characterization of wood decaying Basidiomycetes from Chiloe National Park. Out of 14 pure cultures isolated from wood decaying microbes 12 strains belonged to Basidiomycota phylum. Fungal DNA was isolated and sequenced using the ITS DNA sequence as primers, replicated and sequenced using the Sanger's method. The cultures had a BLAST match of 97 % and more of the sequences in the GenBank database were identified as T. versicolor, C. subvermispora, P. radiata, P. affinis, P. cinerea P. setulosa P. chrysocreas, M. fuscoatra, H. radula, B. adusta, , S. gausapatum and P. sordida, [65]. Compared to Sanger sequencing whole genome sequence studies using next generation sequencers revealed high amount of quality information in the recent years, thus slowly we notice increase in the whole genome sequence studies using NGS methods. Recent fungal genome sequencing studies were shown in Table 2.
List of some of the sequencing studies performed on different classes of fungi strains
| Phylum | Fungal Strain | Sequencing Platform | Layout | Submission and Reference |
|---|---|---|---|---|
| Ascomycota | Aspergilus Niger | ABi SOLiD | SE | GSE33852 [66] |
| Trichoderma reesei | ABi SOLiD | SE | GSE44648 [67] | |
| Basidiomycota | Ganoderma lucidum | Illumina HiSeq 2000 | PE | SRA036392 [68] |
| Rhizoctonia solani AG1-IB 7/3/14 | 454 Pyro Sequencing | --- | [69] | |
| Phanerochaete chrysosporium | 454 Pyro Sequencing | --- | [70] | |
| Agaricomycota | Fistulina hepatica | Illumina HiSeq PacBio SMRT | PE | JYFI00000000 [71] |
| Cylindrobasidium torrendii | Illumina HiSeq PacBio SMRT | PE | JYFH00000000 [71] |
Genome and transcriptome studies of Ascomycota fungi
Whole transcriptome analysis of Aspergillus Niger: Aspergilus niger is a filamentous black spored fungus well known for its commercial applications [72]. Because of its high commercial importance, whole genome studies were conducted on the enzymes producing A. niger strain [73] and citric acid producing A. niger strain [74] to find the molecular aspects of A. niger metabolism. According to Delmas et al (2012), whole transcriptome analysis of Aspergillus niger was performed to understand the response of the strain N402 upon treating with wheat straw as sole carbon source incubated for 24 and 48 hours [67]. RNA sequencing was performed to understand the gene expression of N402 strain at different time periods. The reads obtained from Aspergilus niger N402 strain RNA sequencing were mapped to ATCC1015 and later to CBS513.88 genome sequences based on its phylogeny, to obtain maximum information from N402 transcriptome. Results obtained from the whole transcriptome of N402 strain were divided in to two classes CAZy genes and Non-CAZy genes. Results show that 65 percent of the transcriptome belonged to five classes of CAZy genes (GH7, GH11, GH61, GH62, CE) coding for enzymes such as cellobiohydrolases, acetylxylanesterases arabinofuranosidases, polysaccharide monoxygenases and xylanases. Transcriptome of N402 strain has also revealed novel lipases, esterases and surface interacting proteins which might be involved in the plant biomass degradation [66]. Further studies were conducted to study the responses of N402 strain to both low carbon source and lignocellulosic substrates by conducting transcription deletion mutant studies using creA and xlnR. The sequencing study conducted by Delmas et.al has revealed an interesting fact that, during carbon starvation strain N402 has expressed a set of degradative enzymes. These released enzymes further confirm the presence and start the process of degradation locally, the releasing carbohydrates from plant cell walls initiate large scale degradation [67].
Transcriptome analysis of Trichoderma reesei and comparison of Aspergillus niger: Several species belonging to Trichoderma and Aspergilus genera are known to secrete high range of enzymes for the degradation of biomass [67]. Industrially Trichoderma reesei and Aspergilus niger are two prominent fungi, because of their ability to grow on wide variety of habitats and secrete arsenal of commercially relevant enzymes with varied activities [66, 75, 76]. Trichoderma reesei genome is about 33.9 Mb with the total sequence covering 9129 predicted genes CAZy analysis of the genome resulted in a total of 228 polysaccharides degrading enzymes which represents around 61 enzyme families which is almost similar to that of total number of enzymes in A. niger [77].The whole transcriptome study of T. reesei by Ries et al (2013) was conducted to study the molecular mechanisms employed by T. reesei to degrade lignocellulose using different substrates (wheat straw, Glucose), and also to compare its efficiency with Aspergilus niger. Trichoderma reesei has expressed a wide variety of CAZy genes, analysis of the T. reesei QM6a genome identified 22 Carbohydrate Esterases (CE) encoding genes representing 8 families, 5 Polysaccharide Lyases (PL) encoding genes representing 3 families, 195 Glycoside Hydrolases (GH) encoding genes, representing 49 families and 6 Auxiliary Activities (AA) encoding genes, representing one family. Transcriptome comparisons of T. reesei and A. niger has revealed that both the fungi expressed a mixture of expected GHs, CEs, AAs and other species dependent families. Gene expression studies of wheat straw degradation showed that both the fungi employed a similar set of essential enzymes from GH and AA families and dissimilar set of enzymes belonging to CE family [68]. In T. reesei transcriptome, the gene expression patterns of 32 genes coding for non-CAZy enzymes are similar to that of CAZy genes. Transcriptome study of A. niger the genes coding for non-CAZy enzymes were divided as lipases, surface interacting proteins, esterases and carbon-nitrogen metabolism were also highly expressed in T. reesei transcriptome [67]. This study suggests that both T. reesei and A. niger follow a similar degradation patterns for the lignocellulosic substrate [68].
Transcriptome studies of Basidiomycota
Transcriptome analysis of Rhizoctonia solani AG1-IB 7/3/14: Rhizoctonia solani is a soil borne basidiomycete, it is a plant pathogenic fungus affecting several agriculturally important crops [78]. The draft genome of Rhizoctonia solani AG1-IB 7/3/14 was performed by Wibberg et al (2013), which provided several molecular insights into the structure and function of its genome [79]. Whole transcriptome analysis of Rhizoctonia solani AG1-IB 7/3/14 was performed by Wibberg et al (2014) using a combination of Next Generation Sequencing (454 FLX Roche sequencer) and bioinformatics method for sequencing of the coding units to find the Expressed Sequence Tags (EST) from two different growth conditions, which uncovered the gene expression patterns of this fungus [69]. The transcriptome of Rhizoctonia shows high expression of several genes involved in the degradation of lignin such as laccases and glyoxal oxidases, it also encodes for several other enzymes like glucose oxidases and alcohol oxidases. It was seen that it also expresses several CAZy genes (Glycoside Hydrolases, Carbohydrate Esterases and Polysaccharide Lyases). Beside these it also expressed genes coding for mitogen activated protein kinase cascades, 4-aminobutyric acid (GABA) metabolism, melanin synthesis, plant defence antagonism, phytotoxin, and mycotoxin synthesis were detected [69, 79].
Transcriptome Analysis of Ganoderma lucidum: Ganoderma lucidum is a white rot fungus belonging to Basidiomycetes phylum, which is well known for its medicinal properties in Asian countries [80]. Since many years G. lucidum is considered as a potential resource for the lignocellulose degrading enzymes [81, 82]. Transcriptome analysis of G. lucidum was performed by Yu GuoJun et al (2012) using the Illumina HiSeq 2000 sequencing platform, to study the expression profile of metabolic pathways and lignocellulose degrading pathways during different developmental stages of Ganoderma lucidum. Total of 6,416,670 and 6,439,690 high-quality reads were obtained from the mycelium and fruiting bodies, later these reads were assembled which resulted in 27,408 and 18,892 unigenes respectively. The unigenes were further annotated using Gene Ontology, Kyoto Encyclopedia of Genes Eukaryotic Orthologous Group terms and Genomes. In order to find the expression of lignin degrading enzymes (lignin oxidases and lignin degrading auxiliary enzymes) in Ganoderma lucidum transcriptome BLASTx was performed against fungal oxidative lignin enzymes (FOLymes) database. From the analysis it was seen that G. lucidum expressed 13 potential genes coding for lignin oxidases (LO) and 9 genes coding for lignin degrading auxiliary enzymes (LDA). Similarly, it also expressed genes coding for the carbohydrate metabolism analyzed using BLASTx against CAZy database, which showed that 120 unigenes from G. lucidum transcriptome had high identity to CAZymes. In which 78 homologs belonged to glycoside hydrolases, 40 candidates to glycosyl transferases and 2 candidates to carbohydrate esterases [68].
Phanerochaete chrysosporium model organism for wood degradation: Phanerochaete chrysosporium is a well-studied white rot fungi since five decades. Phanerochaete chrysosporium produces most stable lignin and manganese peroxides which are deployed during the lignin degradation, several studies were conducted for understanding the molecular and functional stability of these enzymes [83, 84]. Phanerochaete chrysosporium was first Basidiomycetes fungus to get sequenced using shot gun sequencing method by Martinez et al (2004). The 30-Mbp genome of Phanerochaete chrysosporium revealed several molecular facts on lignocellulose degradation. The majority of the protein coding genes corresponded to the cytochrome P450 superfamily, which explains its ability on complex aromatic compounds degradation [85]. It was reported that it secretes several extracellular enzymes such as manganese dependent peroxidases (MnP) and lignin peroxidases (LiP), which are hydrogen peroxide dependent enzymes. These genome studies also reported that P. chrysosporium does not express any conventional laccases, however the genes coding for multicopper oxidases were observed [85]. Whole transcriptome study conducted by Sato et al (2009) revealed important facts on its wood decaying mechanisms, P. chrysosporium expressed important cellulose degrading enzymes (endocellulase, exocellulase CBHI, exocellulase CBHII, and β-glucosidase) hemicellulose degrading enzymes (endoxylanases, acetyl xylan esterases and mannosidases) degrading enzymes [70]. Lignin degrading enzymes such as lignin peroxidase, manganese dependent peroxidase, and hydrogen peroxide generating enzyme such as alcohol oxidase are highly expressed. This study also emphasized that hydrogen peroxide generation and utilization play a central part in wood degradation by P. chrysosporium [70, 86]
Genome and Transcriptome studies Agaricomycota
Wood decaying mechanisms of Agaricales: Wood decaying microorganisms from Agaricomycota phylum can be divided into White rot and brown rot fungi. It was studied that white rot fungus degrades several wood polymers including lignin and crystalline cellulose [87, 88]. In contrast with white rot fungi, brown rot fungi can efficiently degrade polysaccharides but partially degrades the lignin [89-91]. Recent sequencing studies have questioned the division of this fungus based on their wood decaying mechanisms [85, 92-94]. The sequencing studies have shown that, fungal genome has suffered with numerous gene loss which has resulted in the transition of white rot to a brown rot lifestyle. The draft genome sequence studies of Cylindrobasidium torrendii and Fistulina hepatica [95] was performed using Illumina HiSeq platform and PacBio SMRT technologies by Floudas et.al (2015) were both the species belongs to Agaricales but the former species causes white rot while the later species causes brown rot on hard wood. This study was conducted to compare the wood degradation mechanisms by analyzing the genomes of both fungi, and also to explore the evolution of plant cell wall degradation in Agaricales. The draft genomes of F. hepatica and C. torrendii show that wood decay gene networks of these fungi diverge from characteristic brown-rot and white-rot species respectively. F. hepatica has progressively suffered with an extensive loss of genes coding for the enzymes involved in the degradation of lignocellulose. Genes required for the degradation crystalline cellulose were found to be expressed in F. hepatica genome. The genome of C. torrendii is enriched in CAZY genes similar to white-rot species, but lacks most of the genes associated with lignin degradation similar to brown rot species. The results also suggested that both the fungi are progressively losing their distinctive wood decaying properties as both species do not show complete brown-rot and white-rot characteristics, since they are comparatively weak wood decayers and found to decay wood only locally [71].
Whole microbiome studies of lignin degrading higher organisms
Whole microbiome sequencing studies were performed to understand the gut microbiome of several organisms like termites, moths, wood boring beetles, yak etc. Earlier research studies involved in the sequencing of 16s rRNA and ITS regions for characterizing bacterial and fungal colonies present in gut of several wood boring insects. According to Dantur et al (2015), sequencing was conducted to isolate efficient lignocellulose degrading bacteria from the intestine of sugarcane fed larvae “Diatraea saccharalis”. In this study, a total of 118 bacterial strains were isolated from the intestine of D. Saccharalis among them 38 bacteria were found to be potential lignocellulose degraders. The phylogenetic study was conducted using 16s rRNA sequences showed that the cellulolytic strains were belonged to the phyla Gammaproteobacteria, Actinobacteria and Firmicutes. Further investigation revealed that these bacteria belonged majorly to 5 genera Klebsiella, Stenotrophomonas, Microbacterium, Bacillus and Enterococcus, which mainly possessed cellulolytic activity [96]. However whole microbiome studies using Next Generation Sequencing methods reveal high level of quality information about the microorganisms habituating higher organisms gut or intestines. Compared to the conventional sequencing methods NGS techniques will help the research communities for understanding and revealing several molecular mechanisms employed by gut microbes in degrading wood polymers. The details on microbiome studies conducted using next generation sequencing are shown in Table 3.
Illustration of Whole microbiome sequencing studies performed
| Organism | Microbial Strain | Sequencing Platform | Layout | Submission and Reference |
|---|---|---|---|---|
| Nasutitermes Ephratae | Treponema Fibrobacteres | Sanger Sequencing, | PE | ABDH00000000; ABDH01000000 |
| Nasutitermes Corniger | 454 Roche Pyrosequencing | EF453758, EF455009, EU024891, EU024927 cellulase EF428062-EF428109 [98] | ||
| Bos grunniens | Bacteroidetes (Cytophaga hutchinsonii) (Flavobacterium johnsoniae) | 454 Roche Pyrosequencing | SE | [100] |
| Firmicutes (Fibrobacter succinogenes) | Illumina Solexa GAII | PE | ||
| Macrotermes annandalei | Listeria Bacteroides | 454 Roche Pyrosequencing | SE | HM483387, HM483386 [101] |
| Coptotermes gestroi | Spirochaetales, Lactobacillales, Bacteroidales, Clostridiales, Enterobacteriales, Pseudomonades, Synergistales, Desulfovibrionales, Xanthomonadales, Burkholderiales Bacillales, and Actinomycetales | Illumina HiSeq2000 | PE | PRJNA217852 [102] |
| Reticulitermes flavipes | Protozoan symbiont | 454 Roche Pyrosequencing | SE | KA650619e, KA659892 [103] |
| Diatraea saccharalis | Klebsiella, Stenotrophomonas, Microbacterium, Bacillus, Enterococcus | ABI 3130 Capillary DNA analyzer | ----- | KM096608,KM096599,KM096598,KM09660,KM096602,KM096605,KM096606,KM096603,KM096601 [96] |
Metagenomic studies of wood degrading termites: All species of termites are mainly associated with various microbial species forming a unusual style of nutritional mutualism [97]. During the past century several studies were conducted to understand these mutualistic relationships. However, very little information was revealed about the fundamental molecular mechanisms applied for lignocellulose degradation and its conversion [98]. Termites can be divided into two major communities as lower and higher wood degrading termites based on their gut microbial diversity. Higher termites generally do not have flagellate protozoans in their hindguts [99]. However, these flagellate protozoans are considered to be the source of cellulases and hemicellulases in lower termites [99]. Recent metagenomic studies revealed tremendous amount of information about the microbial diversity of both lower and higher termites.
Meta-transcriptome analysis of Reticulitermes flavipes : Reticulitermes flavipes is a lower termite known to digest lignocellulose in symbiotic relationship with lower prokaryotes [103, 104]. A meta transcriptome analysis of Reticulitermes flavipes was performed by Tartar A et al (2009), the sequence analysis revealed two types of insights into its lignocellulose metabolism a) genes coding for the enzymes involved in the degradation of cellulose, carbohydrates and pectin which are coded by both host and symbiont genomes. Similarly genes coding for enzymes degrading hemicellulose and lignin were found to be from symbiont genome host genome respectively [103]. Similar study was conducted by Amit et al (2013) to understand the effects of diets containing varying degrees of lignin complexity on host-symbiont digestome composition, to explore deep sample host and symbiont lignocellulase diversity, and to identify promising lignocellulase candidates for functional characterization [105]. Sequencing was performed on Reticulitermes flavipes and its protozoan symbiont using 454 Pyrosequencing method. As a part of this study the termites were selectively fed with diets containing different degrees of lignin complexity. Later followed by sequencing, which resulted around 9500 lignin associated host and symbiont transcripts that include over 300 candidate genes coding for the ligninases, detoxification, antioxidant, hemicellulases and cellulase transcripts. These results convey that termites employ complex enzymatic machinery to digest dietary lignocellulose and also the xenobiotic challenges that termites undergo during lignin degradation process. The sequence analysis conducted by Sethi Amith et al (2013) reveals that the transcripts coding for recombinant AKR, CAT and LacA proteins are induced by lignin feeding. These proteins enhances lignocellulose saccharification by enhancing host and symbiont cellulases (or) xylanases. However, these proteins are apparently not involved in cellulose and hemicellulose metabolism. [105]
Metagenomic analysis of Nasutitermes ephratae and N. corniger: Metagenomic analysis of the Nasutitermes species hindgut was performed by Warnecke et al, to study the potential lignocellulose degrading bacterial community [98] and to study the set of enzymes are required for its degradation [105, 106]. Metagenome of Nasutitermes P3 luminal community revealed no evidence for lignin degradation. However, a conventional analysis detecting all the domains by aligning the reads globally has identified many genes and modules of this analysis were homologous to more than 700 catalytic domains of glycoside hydrolase (GH) which corresponds to 45 different CAZy families. Thus, including a rich diversity of putative cellulases and hemicellulases [98]. Sequencing and the parallel PCR cycles with (16s rRNA) show that Nasutitermes species highly expressed bacterial genes coding for the enzymes involved in degradation of cellulose and hemicellulose. Bacterial species belonging to the phylum Spirochete and Fibrobacteres are involved in the gut lignocellulose degradation. Results show that termite hindgut reservoirs several highly motile bacteria around 1500 genes related to chemotaxis and chemosensation were identified from the sequence analysis, as motile bacteria show chemotactic behavior. Thus, metagenomic analysis of Nasutitermes ephratae and N. corniger revealed new intuitions into other symbiotic functions involved in the lignocellulose degradation such as H2 metabolism, CO2 reductive acetogenesis and nitrogen fixation [98].
Microbiome of fungus growing termites: Some termites belonging to the sub phylum Macrotermitinae, commonly cultivate fungi in their gut. Termites belonging to this group are considered as major decomposers in tropical and subtropical regions [107]. These termites specifically habitat fungi belonging to the genus Termitomyces which are further classified under basidomycetes phylum. These termites usually develop a sophisticated three staged symbiotic relationship with fungi in two ways as a part of their intestinal microbiota and also as ectosymbiotic fungi [108-110]. Liu et al (2011), has conducted a metagenomic study of Macrotermes annandalei to reveal the importance of fungi growing in the gut or intestine of M. annandalei for lignocellulose degradation and carbon mineralization. Sequencing was performed using 454 pyrosequencing method these results show that intestinal microbes residing in termite gut preferentially express β-glucosidases for the efficient breakdown of cellobiose to glucose monomers. Fosmid (constructed from total microbial DNA isolated from fungi) were screened for the cellulose and hemicellulose degrading enzymes such as (Xylanase, β-glucosidase, endoglucanase and cellobiohydrolase) which resulted in 13 clone sequences (1 xylanase and 12 β-glucosidase positive clones). The xylanase clone sequence resulted in 1,818 bp ORF (Open Reading Frames) which expressed genes for coding several CAZymes such as endo -1, 4-β xylanase (Xyl6E7) consisting glycoside hydrolase (GH11) catalytic domains and a carbohydrate-binding module 4_9 domain. These studies have also confirmed that endo -1,4-β xylanase was remarkably active which might be imparted to its high substrate specificity and pH tolerance, thus proving its potential applications in food, fermentation and pharmaceutical industries [101, 108].
Metagenome of free living bacteria in the gut of the lower termite Coptotermes gestroi: Lower termites belonging to the genus Cryptotermes, Heterotermes, and Coptotermes were considered to be invasive wood eating urban pests with a wide habitat range all over the world [111]. Li et.al, have performed the metagenomic analysis of Coptotermes gestroi (a lower termite) gut microbial diversity using Illumina HiSeq 2000 technology [102]. C. gestroi metagenome generated about 5.4 GB of short reads, comprising about 125,431 Open reading frames (ORFs) covering 78,271,365 bp, out of which 80% of the reads were derived from bacteria. Total of 1460 bacterial species were identified in the metagenome of C. gestroi, most of these bacterial sequences obtained from the metagenome belonged to 12 major orders they are Spirochaetales, Synergistales, Pseudomonades, Bacteroidales, Clostridiales, Enterobacteriales, Xanthomonadales, Burkholderiales, Bacillales, Desulfovibrionales, Actinomycetales and Lactobacillales, [102]. The gene prediction analysis of 12,000 ORFs sequences from the metagenome showed that majority of the genes are involved in carbohydrate metabolism, 587 genes encoded for hydrolytic enzymes which are directly involved in the degradation of cellulose, hemicellulose, and pectin. Out of which 316 ORFs expressed the genes coding for cellulose degradation, such as β glucosidases, 6-phospho β glucosidases, glucan endo-1,3 β D-glucosidases, glucan 1,3 β glucosidases, licheninases, endoglucanases, cellulase 1,4-b-cellobiosidases, , and cellobiose phosphorylases. 259 ORFs expressed the genes encoding for hemicellulose degradation such as endo-1,4 β xylanases, endo-1,4 β mannanases, xylan 1,4 β xylanosidases, endo-1,5 α L arabinosidases, and α glucuronidases, a-N-arabinofuranosidases, α galactosidases and 12 ORFs encoded for the enzymes like pectin esterases and pectate lyases which are required for the breakdown of pectin. According Do et al, this study was the first successful application using Illumina-based de novo sequencing for analyzing the lignocellulose degrading mechanisms employed by bacterial communities in the gut of C. gestroi [102].
Metagenomic analysis of fibrolytic microbiome in yak rumen: Several studies were conducted to understand the complex lignocellulase systems employed by the microorganisms present in the rumen. Different metagenomic approaches were applied to understand the microbial diversity of rumen of higher organisms, at the same time these studies were found to be promising in revealing several mechanisms [112-114]. Dai et.al, have performed the metagenomic study of yak rumen to understand the microbial diversity, by sequencing both the metagenomic DNA and fibrolytic BAC clones. From the sequence it was noticed that several fibrolytic genes and their corresponding fibrolytic gene clusters were highly expressed which represents the lignocellulose degradation mechanisms employed in the yak rumen [100]. Quality reads obtained from the BAC clones in this study has provided good insights about the gene and operon assemblies, this study has also helped in analyzing the organizational patterns of fibrolytic gene, and fibrolytic gene clusters in the genomes. This information will further help the scientific community in understanding the coordination, expression and regulation of fibrolytic genes and particularly the gene clusters. Sequencing results of BAC libraries also showed that, genes coding for GH proteins were obtained from the sequences of bacterial strains belonging to the phylum bacteroidetes and firmicutes. Thus bacterial strains belonging to the phylum bacteroidetes and firmicutes play a key role in yak rumen for the degradation of plant cell walls, similar rumen studies conducted on herbivorous animals also proved the presence of the bacterial strains belonging to these phylum in cell wall degradation [100].
Summary and Outlook
In this review we extensively discussed about the recent advancements and applications made in deciphering complex molecular mechanisms involved in the breakdown of lignocellulose polymers using next generation sequencing methods. Traditional methods like sequencing genomic DNA using 16s rRNA and ITS regions is widely used in the scientific community for finding and characterizing the bacteria and fungi respectively. However, these methods convey very little information about the genomic insights of these microorganisms. Different NGS protocols like DNA, RNA, ChIP, Methyl and several other sequencing methods reveal the underlying molecular mechanisms employed by the microorganisms for the lignocellulose degradation. At the same time, NGS methods offer great advantages when compared to the traditional sequencing methods. NGS methods are efficient by cost, labor and time. In this review, we have broadly described the outcomes of recent genome, transcriptome and metagenome sequence analysis projects performed on potential lignin degrading microorganisms. Bacterial genome sequencing studies have revealed that bacterial strains belonging to Actinomycetes, α-proteobacteria, β-proteobacteria and γ-proteobacteria phylum are efficient lignocellulose degraders, several genes encoding for enzymes involved in cellulose, hemicellulose and lignin degradation are highly observed. Fungal genome and transcriptome studies have revealed about the complex pathways, differential gene expression and regulation patterns involved during lignocellulosic biomass degradation. Whole gut microbiome (Metagenome) sequencing studies of wood boring insects conducted in the recent years have revealed several aspects such as wide microbial diversity, mutualistic behaviors, host and symbiotic relationships. Using metagenomic sequence analysis efficient lignin degrading microorganisms were easily identified and differentiated from the wide microbial communities present in the gut (or) rumen of higher organisms. The genes and gene clusters involved in degradation of lignocellulose were identified and characterized, thus revealing about the expression, regulation and synchronization of several genes involved in this process. Extensive usage of different NGS methods, and Mass spectrometry methods combined with (HPLC and GC) will help further in understanding the genomic proteomic and metabolomics insights of lignin degrading microorganisms.
Acknowledgements
This work was supported by NSERC-RDF Fund to Wensheng Qin and Ontario Trillium Scholarship (OTS) to Ayyappa Kumar Sista Kameshwar.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Streffer F. Lignocellulose to Biogas and other Products. JSM Biotechnology & Biomedical Engineering. 2014;2:1023
2. Crawford RL. Lignin biodegradation and transformation. Wiley New York. 1981
3. Fengel D, Wegener G. Wood: chemistry, ultrastructure, reactions. Walter de Gruyter. 1983
4. Van Wyk JP. Biotechnology and the utilization of biowaste as a resource for bioproduct development. TRENDS in Biotechnology. 2001;19:172-7
5. Knauf M, Moniruzzaman M. Lignocellulosic biomass processing: a perspective. International Sugar Journal. 2004;106:147-50
6. Laureano-Perez L, Teymouri F, Alizadeh H, Dale BE. Understanding factors that limit enzymatic hydrolysis of biomass. Applied Biochemistry and Biotechnology. 2005;124:1081-99
7. Bugg TD, Ahmad M, Hardiman EM, Singh R. The emerging role for bacteria in lignin degradation and bio-product formation. Current opinion in biotechnology. 2011;22:394-400
8. Chen Y, Chai L, Zhu Y, Yang Z, Zheng Y, Zhang H. Biodegradation of kraft lignin by a bacterial strain Comamonas sp. B-9 isolated from eroded bamboo slips. Journal of applied microbiology. 2012;112:900-6
9. Argyropoulos DS, Menachem SB. Lignin. Biotechnology in the Pulp and Paper Industry. Springer. 1997:127-58
10. Kumar R, Singh S, Singh OV. Bioconversion of lignocellulosic biomass: biochemical and molecular perspectives. Journal of industrial microbiology & biotechnology. 2008;35:377-91
11. Rubin EM. Genomics of cellulosic biofuels. Nature. 2008;454:841-5
12. Ljungdahl LG. The Cellulase/Hemicellulase System of the Anaerobic FungusOrpinomycesPC-2 and Aspects of Its Applied Use. Annals of the New York Academy of Sciences. 2008;1125:308-21
13. Yoon J-J, Cha C-J, Kim Y-S, Son D-W, Kim Y-K. The brown-rot basidiomycete Fomitopsis palustris has the endo-glucanases capable of degrading microcrystalline cellulose. Journal of microbiology and biotechnology. 2007;17:800-5
14. Bayer EA, Chanzy H, Lamed R, Shoham Y. Cellulose, cellulases and cellulosomes. Current opinion in structural biology. 1998;8:548-57
15. Weng J-K, Li X, Bonawitz ND, Chapple C. Emerging strategies of lignin engineering and degradation for cellulosic biofuel production. Current opinion in biotechnology. 2008;19:166-72
16. Guillén F, Martínez MJ, Gutiérrez A, Del Rio J. Biodegradation of lignocellu-losics: microbial, chemical, and enzymatic aspects of the fungal attack of lignin. Int Microbiol. 2005;8:187204Minami
17. Thurston CF. The structure and function of fungal laccases. Microbiology. 1994;140:19-26
18. Ramachandran S, Magnuson TS, Crawford DL. Isolation and analysis of three peroxide sensor regulatory gene homologs ahpC, ahpX and oxyR in Streptomyces viridosporus T7A-a lignocellulose degrading actinomycete. DNA Sequence. 2000;11:51-60
19. Ausec L, Zakrzewski M, Goesmann A, Schlüter A, Mandic-Mulec I. Bioinformatic analysis reveals high diversity of bacterial genes for laccase-like enzymes. PLoS One. 2011;6:e25724
20. Kellner H, Luis P, Zimdars B, Kiesel B, Buscot F. Diversity of bacterial laccase-like multicopper oxidase genes in forest and grassland Cambisol soil samples. Soil Biology and Biochemistry. 2008;40:638-48
21. Dashtban M, Schraft H, Qin W. Fungal bioconversion of lignocellulosic residues; opportunities & perspectives. International Journal of Biological Sciences. 2009;5:578
22. Sánchez C. Lignocellulosic residues: biodegradation and bioconversion by fungi. Biotechnology advances. 2009;27:185-94
23. Maki M, Leung KT, Qin W. The prospects of cellulase-producing bacteria for the bioconversion of lignocellulosic biomass. International Journal of Biological Sciences. 2009;5:500
24. Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proceedings of the National Academy of Sciences. 1977;74:5463-7
25. Ansorge WJ. Next-generation DNA sequencing techniques. New biotechnology. 2009;25:195-203
26. MacLean D, Jones JD, Studholme DJ. Application of'next-generation'sequencing technologies to microbial genetics. Nature Reviews Microbiology. 2009;7:287-96
27. Mardis ER. The impact of next-generation sequencing technology on genetics. Trends in genetics. 2008;24:133-41
28. Metzker ML. Sequencing technologies—the next generation. Nature reviews genetics. 2010;11:31-46
29. Morozova O, Marra MA. Applications of next-generation sequencing technologies in functional genomics. Genomics. 2008;92:255-64
30. Shendure J, Ji H. Next-generation DNA sequencing. Nature biotechnology. 2008;26:1135-45
31. Liu L, Li Y, Li S, Hu N, He Y, Pong R. et al. Comparison of next-generation sequencing systems. BioMed Research International. 2012. 2012
32. Lombard V, Ramulu HG, Drula E, Coutinho PM, Henrissat B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic acids research. 2014;42:D490-D5
33. Levasseur A, Piumi F, Coutinho PM, Rancurel C, Asther M, Delattre M. et al. FOLy: an integrated database for the classification and functional annotation of fungal oxidoreductases potentially involved in the degradation of lignin and related aromatic compounds. Fungal genetics and biology. 2008;45:638-45
34. Woese CR, Fox GE. Phylogenetic structure of the prokaryotic domain: the primary kingdoms. Proceedings of the National Academy of Sciences. 1977;74:5088-90
35. Pold G, Melillo JM, DeAngelis KM. Two decades of warming increases diversity of a potentially lignolytic bacterial community. Frontiers in Microbiology. 2015;6:480
36. Niewerth H, Parschat K, Rauschenberg M, Ravoo BJ, Fetzner S. The PaaX-Type Repressor MeqR2 of Arthrobacter sp. Strain Rue61a, Involved in the Regulation of Quinaldine Catabolism, Binds to Its Own Promoter and to Catabolic Promoters and Specifically Responds to Anthraniloyl Coenzyme A. Journal of bacteriology. 2013;195:1068-80
37. Niewerth H, Schuldes J, Parschat K, Kiefer P, Vorholt JA, Daniel R. et al. Complete genome sequence and metabolic potential of the quinaldine-degrading bacterium Arthrobacter sp. Rue61a. BMC genomics. 2012;13:534
38. Antai SP, Crawford DL. Degradation of softwood, hardwood, and grass lignocelluloses by two Streptomyces strains. Applied and environmental microbiology. 1981;42:378-80
39. Ma Xk, Daugulis AJ. Transformation of ferulic acid to vanillin using a fed-batch solid-liquid two-phase partitioning bioreactor. Biotechnology progress. 2014;30:207-14
40. Fleige C, Steinbüchel A. Construction of expression vectors for metabolic engineering of the vanillin-producing actinomycete Amycolatopsis sp. ATCC 39116. Applied microbiology and biotechnology. 2014;98:6387-95
41. Davis JR, Goodwin LA, Woyke T, Teshima H, Bruce D, Detter C. et al. Genome sequence of Amycolatopsis sp. strain ATCC 39116, a plant biomass-degrading actinomycete. Journal of bacteriology. 2012;194:2396-7
42. Ohta Y, Nishi S, Kobayashi K, Tsubouchi T, Iida K, Tanizaki A. et al. Draft genome sequence of Novosphingobium sp. strain MBES04, isolated from sunken wood from Suruga Bay, Japan. Genome announcements. 2015;3:e01373-14
43. Prabhakaran M, Couger MB, Jackson CA, Weirick T, Fathepure BZ. Genome sequences of the lignin-degrading Pseudomonas sp. strain YS-1p and Rhizobium sp. strain YS-1r isolated from decaying wood. Genome announcements. 2015;3:e00019-15
44. Woo HL, Utturkar S, Klingeman D, Simmons BA, DeAngelis KM, Brown SD. et al. Draft genome sequence of the lignin-degrading Burkholderia sp. strain LIG30, isolated from wet tropical forest soil. Genome announcements. 2014;2:e00637-14
45. Shi Y, Chai L, Tang C, Yang Z, Zhang H, Chen R. et al. Characterization and genomic analysis of kraft lignin biodegradation by the beta-proteobacterium Cupriavidus basilensis B-8. Biotechnol Biofuels. 2013:6
46. O'Dell KB, Woo HL, Utturkar S, Klingeman D, Brown SD, Hazen TC. Genome sequence of Halomonas sp. strain KO116, an ionic liquid-tolerant marine bacterium isolated from a lignin-enriched seawater microcosm. Genome announcements. 2015;3:e00402-15
47. Woo HL, Ballor NR, Hazen TC, Fortney JL, Simmons B, Davenport KW. et al. Complete genome sequence of the lignin-degrading bacterium Klebsiella sp. strain BRL6-2. Stand Genomic Sci. 2014;9:19
48. Bao W, Zhou Y, Jiang J, Xu Z, Hou L, Leung FC-C. Complete genome sequence of Raoultella ornithinolytica strain S12, a lignin-degrading bacterium isolated from forest soil. Genome announcements. 2015;3:e00104-15
49. Fleige C, Hansen G, Kroll J, Steinbüchel A. Investigation of the Amycolatopsis sp. strain ATCC 39116 vanillin dehydrogenase and its impact on the biotechnical production of vanillin. Applied and environmental microbiology. 2013;79:81-90
50. Aylward FO, McDonald BR, Adams SM, Valenzuela A, Schmidt RA, Goodwin LA. et al. Comparison of 26 sphingomonad genomes reveals diverse environmental adaptations and biodegradative capabilities. Applied and environmental microbiology. 2013;79:3724-33
51. Looper JK, Cotto A, Kim B-Y, Lee M-K, Liles MR, Chadhain SMN. et al. Microbial community analysis of Deepwater Horizon oil-spill impacted sites along the Gulf coast using functional and phylogenetic markers. Environmental Science: Processes & Impacts. 2013;15:2068-79
52. Sohn JH, Kwon KK, Kang J-H, Jung H-B, Kim S-J. Novosphingobium pentaromativorans sp. nov, a high-molecular-mass polycyclic aromatic hydrocarbon-degrading bacterium isolated from estuarine sediment. International Journal of Systematic and Evolutionary Microbiology. 2004;54:1483-7
53. Ohta Y, Nishi S, Haga T, Tsubouchi T, Hasegawa R, Konishi M. et al. Screening and phylogenetic analysis of deep-sea bacteria capable of metabolizing lignin-derived aromatic compounds. Open Journal of Marine Science. 2012. 2012
54. Coenye T, Vandamme P. Diversity and significance of Burkholderia species occupying diverse ecological niches. Environmental Microbiology. 2003;5:719-29
55. Vial L, Groleau M-C, Dekimpe V, Déziel É. Burkholderia diversity and versatility: an inventory of the extracellular products. Journal of microbiology and biotechnology. 2007;17:1407-29
56. Woo HL, Hazen TC, Simmons BA, DeAngelis KM. Enzyme activities of aerobic lignocellulolytic bacteria isolated from wet tropical forest soils. Systematic and applied microbiology. 2014;37:60-7
57. Wierckx N, Koopman F, Bandounas L, De Winde JH, Ruijssenaars HJ. Isolation and characterization of Cupriavidus basilensis HMF14 for biological removal of inhibitors from lignocellulosic hydrolysate. Microbial biotechnology. 2010;3:336-43
58. Qian Y, Shi J, Chen Y, Lou L, Cui X, Cao R. et al. Characterization of phosphate solubilizing bacteria in sediments from a shallow eutrophic lake and a wetland: isolation, molecular identification and phosphorus release ability determination. Molecules. 2010;15:8518-33
59. Drancourt M, Bollet C, Carta A, Rousselier P. Phylogenetic analyses of Klebsiella species delineate Klebsiella and Raoultella gen. nov, with description of Raoultella ornithinolytica comb. nov, Raoultella terrigena comb. nov. and Raoultella planticola comb. nov. International Journal of Systematic and Evolutionary Microbiology. 2001;51:925-32
60. Dobson S, Franzmann P. Unification of the genera Deleya (Baumann et al. 1983), Halomonas (Vreeland et al. 1980), and Halovibrio (Fendrich 1988) and the species Paracoccus halodenitrificans (Robinson and Gibbons 1952) into a single genus, Halomonas, and placement of the genus Zymobacter in the family Halomonadaceae. International Journal of Systematic Bacteriology. 1996;46:550-8
61. Peix A, Ramírez-Bahena M-H, Velázquez E. Historical evolution and current status of the taxonomy of genus Pseudomonas. Infection, Genetics and Evolution. 2009;9:1132-47
62. Bello-Akinosho M, Adeleke R, Swanevelder D, Thantsha M. Draft genome sequence of Pseudomonas sp. strain 10-1B, a polycyclic aromatic hydrocarbon degrader in contaminated soil. Genome announcements. 2015;3:e00325-15
63. Bruns TD, White TJ, Taylor JW. Fungal molecular systematics. Annual Review of Ecology and Systematics. 1991:525-64
64. Gardes M, Bruns TD. ITS primers with enhanced specificity for basidiomycetes-application to the identification of mycorrhizae and rusts. Molecular ecology. 1993;2:113-8
65. Ortiz R, Navarrete J, Oviedo C, Párraga M, Carrasco I, de la Vega E. et al. White rot Basidiomycetes isolated from Chiloé National Park in Los Lagos region, Chile. Antonie van Leeuwenhoek. 2013;104:1193-203
66. Delmas S, Pullan ST, Gaddipati S, Kokolski M, Malla S, Blythe MJ. et al. Uncovering the genome-wide transcriptional responses of the filamentous fungus Aspergillus niger to lignocellulose using RNA sequencing. PLoS Genet. 2012;8:e1002875
67. Ries L, Pullan ST, Delmas S, Malla S, Blythe MJ, Archer DB. Genome-wide transcriptional response of Trichoderma reesei to lignocellulose using RNA sequencing and comparison with Aspergillus niger. BMC genomics. 2013;14:541
68. Yu G-J, Wang M, Huang J, Yin Y-L, Chen Y-J, Jiang S. et al. Deep insight into the Ganoderma lucidum by comprehensive analysis of its transcriptome. PloS one. 2012;7:e44031
69. Wibberg D, Jelonek L, Rupp O, Kröber M, Goesmann A, Grosch R. et al. Transcriptome analysis of the phytopathogenic fungus Rhizoctonia solani AG1-IB 7/3/14 applying high-throughput sequencing of expressed sequence tags (ESTs). Fungal biology. 2014;118:800-13
70. Sato S, Feltus FA, Iyer P, Tien M. The first genome-level transcriptome of the wood-degrading fungus Phanerochaete chrysosporium grown on red oak. Current genetics. 2009;55:273-86
71. Floudas D, Held BW, Riley R, Nagy LG, Koehler G, Ransdell AS. et al. Evolution of novel wood decay mechanisms in Agaricales revealed by the genome sequences of Fistulina hepatica and Cylindrobasidium torrendii. Fungal Genetics and Biology. 2015;76:78-92
72. Archer DB. Filamentous fungi as microbial cell factories for food use. Current opinion in biotechnology. 2000;11:478-83
73. Pel HJ, de Winde JH, Archer DB, Dyer PS, Hofmann G, Schaap PJ. et al. Genome sequencing and analysis of the versatile cell factory Aspergillus niger CBS 513.88. Nature biotechnology. 2007;25:221-31
74. Andersen MR, Salazar MP, Schaap PJ, van de Vondervoort PJ, Culley D, Thykaer J. et al. Comparative genomics of citric-acid-producing Aspergillus niger ATCC 1015 versus enzyme-producing CBS 513.88. Genome Research. 2011;21:885-97
75. Banerjee G, Scott-Craig JS, Walton JD. Improving enzymes for biomass conversion: a basic research perspective. Bioenergy research. 2010;3:82-92
76. Schuster A, Schmoll M. Biology and biotechnology of Trichoderma. Applied microbiology and biotechnology. 2010;87:787-99
77. Häkkinen M, Arvas M, Oja M, Aro N, Penttilä M, Saloheimo M. et al. Re-annotation of the CAZy genes of Trichoderma reesei and transcription in the presence of lignocellulosic substrates. Microb Cell Fact. 2012;11:134
78. García VG, Onco MP, Susan VR. Review. Biology and systematics of the form genus Rhizoctonia. Spanish Journal of Agricultural Research. 2006;4:55-79
79. Wibberg D, Jelonek L, Rupp O, Hennig M, Eikmeyer F, Goesmann A. et al. Establishment and interpretation of the genome sequence of the phytopathogenic fungus Rhizoctonia solani AG1-IB isolate 7/3/14. Journal of biotechnology. 2013;167:142-55
80. Paterson RRM. Ganoderma-a therapeutic fungal biofactory. Phytochemistry. 2006;67:1985-2001
81. Ko E-M, Leem Y-E, Choi H. Purification and characterization of laccase isozymes from the white-rot basidiomycete Ganoderma lucidum. Applied microbiology and biotechnology. 2001;57:98-102
82. Wang H, Ng T. A laccase from the medicinal mushroom Ganoderma lucidum. Applied microbiology and biotechnology. 2006;72:508-13
83. Ulmer DC, Leisola MS, Schmidt BH, Fiechter A. Rapid degradation of isolated lignins by Phanerochaete chrysosporium. Applied and environmental microbiology. 1983;45:1795-801
84. Kirk TK, Schultz E, Connors W, Lorenz L, Zeikus J. Influence of culture parameters on lignin metabolism byPhanerochaete chrysosporium. Archives of Microbiology. 1978;117:277-85
85. Martinez D, Larrondo LF, Putnam N, Gelpke MDS, Huang K, Chapman J. et al. Genome sequence of the lignocellulose degrading fungus Phanerochaete chrysosporium strain RP78. Nature biotechnology. 2004;22:695-700
86. Kersten P, Cullen D. Extracellular oxidative systems of the lignin-degrading Basidiomycete Phanerochaete chrysosporium. Fungal Genetics and Biology. 2007;44:77-87
87. Baldrian P, Valášková V. Degradation of cellulose by basidiomycetous fungi. FEMS microbiology reviews. 2008;32:501-21
88. Blanchette RA. Degradation of the lignocellulose complex in wood. Canadian Journal of Botany. 1995;73:999-1010
89. Niemenmaa O, Uusi-Rauva A, Hatakka A. Demethoxylation of [O14CH3]-labelled lignin model compounds by the brown-rot fungi Gloeophyllum trabeum and Poria (Postia) placenta. Biodegradation. 2008;19:555-65
90. Yelle DJ, Ralph J, Lu F, Hammel KE. Evidence for cleavage of lignin by a brown rot basidiomycete. Environmental microbiology. 2008;10:1844-9
91. Martinez D, Challacombe J, Morgenstern I, Hibbett D, Schmoll M, Kubicek CP. et al. Genome, transcriptome, and secretome analysis of wood decay fungus Postia placenta supports unique mechanisms of lignocellulose conversion. Proceedings of the National Academy of Sciences. 2009;106:1954-9
92. Eastwood DC, Floudas D, Binder M, Majcherczyk A, Schneider P, Aerts A. et al. The plant cell wall-decomposing machinery underlies the functional diversity of forest fungi. Science. 2011;333:762-5
93. Floudas D, Binder M, Riley R, Barry K, Blanchette RA, Henrissat B. et al. The Paleozoic origin of enzymatic lignin decomposition reconstructed from 31 fungal genomes. Science. 2012;336:1715-9
94. Ginns J, Lefebvre M.N.L. Lignicolous corticioid fungi (Basidiomycota) of North America. Systematics, distribution, and ecology. Mycologia Memoir 19 The American Phytopathological Society Press, St Paul, Minnesota. 1993:19
95. Schwarze F, Baum S, Fink S. Dual modes of degradation by Fistulina hepatica in xylem cell walls of Quercus robur. Mycological Research. 2000;104:846-52
96. Dantur KI, Enrique R, Welin B, Castagnaro AP. Isolation of cellulolytic bacteria from the intestine of Diatraea saccharalis larvae and evaluation of their capacity to degrade sugarcane biomass. AMB Express. 2015;5:15
97. Brune A. Symbiotic associations between termites and prokaryotes. The prokaryotes. Springer. 2006:439-74
98. Warnecke F, Luginbühl P, Ivanova N, Ghassemian M, Richardson TH, Stege JT. et al. Metagenomic and functional analysis of hindgut microbiota of a wood-feeding higher termite. Nature. 2007;450:560-5
99. Abe T, Bignell DE, Higashi M. Termites: evolution, sociality, symbioses, ecology. Springer Science & Business Media. 2000
100. Dai X, Zhu Y, Luo Y, Song L, Liu D, Liu L. et al. Metagenomic insights into the fibrolytic microbiome in yak rumen. PLoS One. 2012;7:e40430
101. Liu N, Yan X, Zhang M, Xie L, Wang Q, Huang Y. et al. Microbiome of fungus-growing termites: a new reservoir for lignocellulase genes. Applied and environmental microbiology. 2011;77:48-56
102. Li H-F, Fujisaki I, Su N-Y. Predicting habitat suitability of Coptotermes gestroi (Isoptera: Rhinotermitidae) with species distribution models. Journal of economic entomology. 2013;106:311-21
103. Tartar A, Wheeler MM, Zhou X, Coy MR, Boucias DG, Scharf ME. Parallel metatranscriptome analyses of host and symbiont gene expression in the gut of the termite Reticulitermes flavipes. Biotechnol Biofuels. 2009;2:25
104. Raychoudhury R, Sen R, Cai Y, Sun Y, Lietze VU, Boucias D. et al. Comparative metatranscriptomic signatures of wood and paper feeding in the gut of the termite Reticulitermes flavipes (Isoptera: Rhinotermitidae). Insect molecular biology. 2013;22:155-71
105. Sethi A, Slack JM, Kovaleva ES, Buchman GW, Scharf ME. Lignin-associated metagene expression in a lignocellulose-digesting termite. Insect biochemistry and molecular biology. 2013;43:91-101
106. Davies GJ. Structural genomics of carbohydrate-active enzymes: implications for the synthesis of plant polysaccharides. Biochemical Society Transactions. 2002;30:A24-A
107. Breznak JA, Brune A. Role of microorganisms in the digestion of lignocellulose by termites. Annual review of entomology. 1994;39:453-87
108. Liu N, Zhang L, Zhou H, Zhang M, Yan X, Wang Q. et al. Metagenomic insights into metabolic capacities of the gut microbiota in a fungus-cultivating termite (Odontotermes yunnanensis). PLoS One. 2013;8:e69184
109. Wood T, Thomas R. The mutualistic association between Macrotermitinae and Termitomyces. Insect-fungus interactions. 1989:69-92
110. Hinze B, Crailsheim K, Leuthold R. Polyethism in food processing and social organisation in the nest of Macrotermes bellicosus (Isoptera, Termitidae). Insectes sociaux. 2002;49:31-7
111. Evans TA, Forschler BT, Grace JK. Biology of invasive termites: a worldwide review. Annual review of entomology. 2013;58:455
112. Do TH, Nguyen TT, Nguyen TN, Le QG, Nguyen C, Kimura K. et al. Mining biomass-degrading genes through Illumina-based de novo sequencing and metagenomic analysis of free-living bacteria in the gut of the lower termite Coptotermes gestroi harvested in Vietnam. Journal of bioscience and bioengineering. 2014;118:665-71
113. Hess M, Sczyrba A, Egan R, Kim T-W, Chokhawala H, Schroth G. et al. Metagenomic discovery of biomass-degrading genes and genomes from cow rumen. Science. 2011;331:463-7
114. Brulc JM, Antonopoulos DA, Miller MEB, Wilson MK, Yannarell AC, Dinsdale EA. et al. Gene-centric metagenomics of the fiber-adherent bovine rumen microbiome reveals forage specific glycoside hydrolases. Proceedings of the National Academy of Sciences. 2009;106:1948-53
Author contact
![]() Corresponding author: Email: wqinca, Tel: 807-343-8467
Corresponding author: Email: wqinca, Tel: 807-343-8467