Impact Factor ISSN: 1449-2288
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2016; 12(2):172-183. doi:10.7150/ijbs.13420 This issue Cite
Research Paper
Caspase-3/7-mediated Cleavage of β2-spectrin is Required for Acetaminophen-induced Liver Damage
Hye Jung Baek1, Yong Min Lee1, Tae Hyun Kim1, Joo-Young Kim1, Eun Jung Park2, Kuniyoshi Iwabuchi3, Lopa Mishra4, Sang Soo Kim1 ![]()
1. Radiation Medicine Branch, and
2. Cancer Immunology Branch, National Cancer Center, Goyang, Gyeonggi, 410-769, Korea,
3. Department of Biochemistry I, School of Medicine, Kanazawa Medical University, Kahoku-gun, Ishikawa, 920-0293, Japan.
4. Department of Gastroenterology, Hepatology, and Nutrition, MD Anderson Cancer Center, Houston, Texas, 77030, USA
Received 2015-8-2; Accepted 2015-11-21; Published 2016-1-1
Abstract

The ubiquitously expressed β2-spectrin (β2SP, SPTBN1) is the most common non-erythrocytic member of the β-spectrin gene family. Loss of β2-spectrin leads to defects in liver development, and its haploinsufficiency spontaneously leads to chronic liver disease and the eventual development of hepatocellular cancer. However, the specific role of β2-spectrin in liver homeostasis remains to be elucidated. Here, we reported that β2-spectrin was cleaved by caspase-3/7 upon treatment with acetaminophen which is the main cause of acute liver injury. Blockage of β2-spectrin cleavage robustly attenuated β2-spectrin-specific functions, including regulation of the cell cycle, apoptosis, and transcription. Cleaved fragments of β2-spectrin were physiologically active, and the N- and C-terminal fragments retained discrete interaction partners and activity in transcriptional regulation and apoptosis, respectively. Cleavage of β2-spectrin facilitated the redistribution of the resulting fragments under conditions of liver damage induced by acetaminophen. In contrast, downregulation of β2-spectrin led to resistance to acetaminophen-induced cytotoxicity, and its insufficiency in the liver promoted suppression of acetaminophen-induced liver damage and enhancement of liver regeneration. Conclusions: β2-Spectrin, a TGF-β mediator and signaling molecule, is cleaved and activated by caspase-3/7, consequently enhancing apoptosis and transcriptional control to determine cell fate upon liver damage. These findings have extended our knowledge on the spectrum of β2-spectrin functions from a scaffolding protein to a target and transmitter of TGF-β in liver damage.
Keywords: β2-spectrin, acetaminophen, TGF-β, caspase-3/7, liver damage
Introduction
Spectrin, a tetrameric protein consisting of two antiparallel dimers of α-and β-subunits, acts as a molecular scaffold in the actin cytoskeletal network and plays a key role in membrane integrity, cell shape and polarity, and cell-cell interactions [1] [2]. β2-Spectrin (β2SP, SPTBN1) is ubiquitously expressed, and the most common non-erythrocytic member of the β-spectrin gene family. However, recent studies have revealed that β2-spectrin is more than just a simple component of the cytoskeleton. Mouse gene targeting experiments showed that β2-spectrin is involved in the regulation of transforming growth factor-β (TGF-β) signaling through interactions with Smad3/4 adaptor proteins [3] [4]. A β2-spectrin mutant embryo (β2sp-/-) exhibits distorted liver architecture with increased hepatocyte proliferation [3]. Moreover, mice with β2-spectrin haploinsufficiency (β2sp+/-) spontaneously display chronic liver disease and eventually develop hepatocellular cancer [5] [6]. Loss of β2-spectrin has been observed in many human cancer cell lines, and frequently, human HCC is associated with a significant reduction in β2-spectrin expression, suggesting that the protein acts as a tumor suppressor and inhibition of its function is a critical process by which normal cells escape regulation of proliferation in carcinogenesis [6] [7]. However, the exact mechanisms by which β2-spectrin regulates cellular proliferation and suppresses liver carcinogenesis remain to be clarified.
Acetaminophen (APAP), chemically known as N-acetyl-p-aminophenol, is a widely utilized over-the-counter analgesic and antipyretic drug. APAP is safe at the recommended doses. However, overdose of acetaminophen can cause potentially fatal liver damage, which is considered one of the most common reasons of acute hepatic injury [8, 9]. Damage to the liver or hepatotoxicity results not from acetaminophen itself, but one of its metabolites, N-acetyl-p-benzoquinoneimine, which depletes the liver's natural antioxidant glutathione and directly damages hepatic cells, leading to liver failure [10]. Apoptosis is proposed to play a critical role in acetaminophen-induced injury, which enables us to hypothesize its inhibition in preventing the development of acute liver failure [11]. The mechanisms underlying apoptosis induction by acetaminophen that lead to predisposition to either acute liver failure or cirrhosis are yet to be identified.
The current study was performed to determine the potential involvement of β2-spectrin in acetaminophen-induced liver damage in human HCC cell lines and mutant mice, with the aim of exploring the molecular consequences of the acetaminophen-induced liver damage response and establishing the causal link between β2-spectrin and liver injury. Our experiments demonstrated that during acetaminophen-induced liver damage, β2-spectrin is cleaved by caspase-3/7. The cleaved fragments of β2-spectrin performed separate functions through regulation of apoptosis and transcription at different locations, implying that its cleavage facilitates liver response to acetaminophen-induced damage. Furthermore, downregulation of β2-spectrin led to a marked decrease in cytotoxicity and increased regeneration of liver cells damaged by acetaminophen. These findings collectively suggest that β2-spectrin is not only a scaffolding protein that interacts with Smad proteins but also a target molecule of TGF-β for transmission and amplification of signaling to generate the physiological response to liver damage.
Materials and Methods
Mice and drug administration
Generation of β2sp+/- knockout mice has been described previously [12]. Four month-old male C57BL/6 (Charles River Laboratories) or β2sp-targeted mice were intraperitoneally injected with Solutol HS-15-dissolved acetaminophen (200 mg/kg) after overnight starvation, and their livers collected at indicated periods after acetaminophen administration. All animal procedures were approved by the Institutional Animal Care and Use Committee of the National Cancer Center, in accordance with the NIH Guide for the Care and Use of Animals.
Cell culture and analysis
HepG2, U-2 OS, HeLa, COS7 and 293T cells were obtained from American Type Culture Collection, and Huh7 and SNU-761 cells from the Korean Cell Line Bank in Seoul National University (SNU). Several forms of human β2-spectrin were cloned into pcDNA3.1/V5-His plasmid (Invitrogen) or pCAGG-FLAG using PCR. Cells were plated and transfected the next day with plasmids containing β2-spectrin or empty vector using Lipofectamine 2000 (Invitrogen). Transfected cells were further incubated for 48 h and treated with TGF-β (100 pM) for one day. In flow cytometry analysis, cells were harvested using trypsin, fixed in 70% ethanol, resuspended in propidium iodide solution, and cellular fluorescence measured using a FACSCalibur flow cytometer (BD Biosciences). DNA content and cell cycle distribution were analyzed using CellQuest (BD Biosciences). To eliminate β2-spectrin expression, HepG2 cells were transduced with lentivirus containing pGIPZ shRNAmir for β2-spectrin or empty vector as control (Open Biosystems). Transduction was confirmed by monitoring GFP expression in puromycin selection. For estimating acetaminophen-induced cytotoxicity, the MTT-based cytotoxicity kit (Sigma) was used to measure cell survival according to the manufacturers' instructions. For assay of TGF-β-dependent transcription, various β2-spectrin-containing or empty vectors were co-transfected into Huh7 cells with p3TP-lux (Addgene) and reference pRL (Promega) vectors for 2 days, and transcriptional activities measured using the dual-luciferase reporter assay kit (Promega), in keeping with the manufacturers' instructions.
Western blot, immunocytochemistry, and histology
Primary antibodies against Smad2, Smad3, phospho-Rb (Ser807/811), phospho-Smad2 (Ser465/467), phospho-Smad3 (Ser423/425), cleaved caspase-3, caspase-7, cleaved caspase-7, phospho-histone H3 (Ser10), PARP, and cleaved PARP were obtained from Cell Signaling Technologies. Antibodies against β-actin, cyclin D1, CKD4, HA-epitope, Rb, β2-spectrin, TGF-β receptor II, phospho-TGF-β receptor II (Tyr424), α-tubulin, and V5-epitope were obtained from Santa Cruz, anti-FLAG from Sigma, and antibody to Ki67 from Novus. These antibodies were used for Western blot, immunocytochemistry and immunohistochemistry experiments. In histological analysis, liver specimens were fixed in 10% formalin, blocked in paraffin, sectioned, stained with hematoxylin-eosin (H&E), and examined using light microscopy. Immunohistochemical analysis was performed using the ZYMED Histomouse Kit (Zymed), as described previously [13]. Western blot and immunohistochemical findings were quantified using AlphaEaseFC software (ver. 4.0, Cell Bioscience) and Leica Application Suite image analysis (ver. 4.2, Leica), respectively.
Purification of β2-spectrin recombinant protein and in vitro cleavage assay
293T cells were transfected with pCAGG vector containing FLAG-tagged β2-spectrin using Lipofectamine 2000 (Invitrogen). Transfected 293T cells were further cultured for 48 h, washed three times with ice-cold PBS, harvested using a cell scraper, and centrifuged at 5,000 x g for 5 min for collection. Cells were homogenized by sonication in buffer A consisting of 50 mM Tris-HCl (pH 7.5), 5 mM MgCl2, 1 mM dithiothreitol, 10% (v/v) glycerol, and 1x protease inhibitor cocktail (Roche). After centrifugation at 15,000 x g for 30 min, supernatant fractions were collected and protein concentrations determined using the method of Bradford with BSA as the standard. The resulting lysates were loaded onto an anti-FLAG M2 affinity gel (0.7 X 2.5, Sigma) equilibrated with buffer A. After washing the column with the same buffer, proteins were eluted with 1 ml FLAG-tag peptide (5 mg/ml, Sigma). Eluants were analyzed using SDS-PAGE, followed by Coomassie blue R-250 staining. Fractions containing β2-spectrin were pooled and dialyzed against 20 mM Tris-HCl (pH 7.5). The purity of β2-spectrin was monitored via SDS-PAGE and Western blot analysis.
β2-Spectrin cleavage assays were performed by incubation of 2 μg purified β2-spectrin and 1 unit of the indicated protease in reaction buffer containing 20 mM Tris-HCl (pH 7.5), 100 mM NaCl, 5 mM MgCl2, 5 mM dithiothreitol, 5 mM EDTA, and 10% (v/v) glycerol, while the calpain reactions used 2 mM CaCl2 instead of EDTA. After incubation for 2 h at 37°C, the reaction was terminated by adding Laemmli sampling buffer, electrophoresed using 4-20% (w/v) SDS-PAGE (Novex), and stained with Coomassie blue R-250. All purified recombinant proteases were purchased from Calbiochem.
Statistical analysis
All data are expressed as means ± SE or SD. Student's t-test (http://www.physics.csbsju.edu/stats/t-test.html) was used to compare differences.
Results
Cleavage of β2-spectrin is induced by acetaminophen
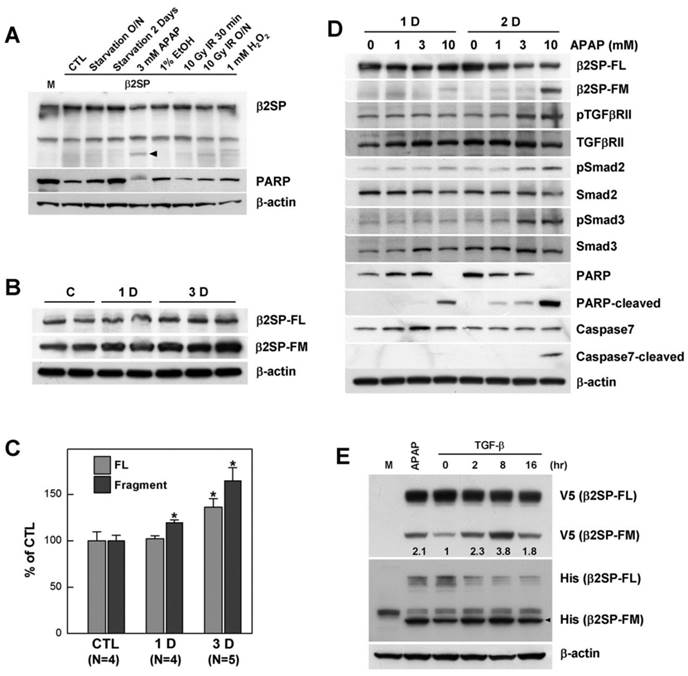
We previously reported that loss or downregulation of β2-spectrin expression leads to liver abnormalities, including cirrhosis and hepatocellular cancer [5, 14]. These results suggest that β2-spectrin is required for liver homeostasis, and its loss influences proper function of liver. However, the mechanisms by which β2-spectrin contributes to liver maintenance are currently unclear. To elucidate the role of β2-spectrin in liver damage, we examined the patterns of protein expression following treatment with several reagents and conditions that induce liver injury in β2-spectrin-transfected Huh7 cells. As shown in Fig. 1A, β2-spectrin protein levels in Huh7 cells were not significantly altered in the majority of liver damage conditions. However, upon treatment with acetaminophen that frequently caused acute liver injury in high dose, we observed the most significant reduction of β2-spectrin protein and a newly appeared faster migrating band on Western blot using an antibody against β2-spectrin. We further examined whether the β2-spectrin level is altered upon intraperitoneal injection of acetaminophen (200 mg/kg) in normal livers of C57BL/6 mice. Similar to data obtained with Huh7 cells, acetaminophen-induced liver damage led to rapid and significant accumulation of the faster migrating form of β2-spectrin (β2-spectrin-FM) (Fig. 1B, C). To ascertain whether the appearance of the fast migrating band is related to acetaminophen-induced liver damage, we examined the patterns of β2-spectrin, TGF-β regulation and apoptosis-related proteins, following treatment with various concentrations of acetaminophen in Huh7 cells at 24 and 48 h (Fig. 1D). Treatment with 3 mM acetaminophen for 2 days induced marked phosphorylation of TGF-β receptor II, Smad2. At a higher dose of acetaminophen (10 mM), we observed significant appearance of the faster migrating form, simultaneously accompanied by a sharp increase in apoptotic signals, such as cleaved PARP and caspase-7. Next, to examine whether TGF-β is responsible for the appearance of β2-spectrin-FM, C-terminal V5/His-tagged β2-spectrin-transfected Huh7 cells were incubated in the absence or presence of TGF-β (100 pM) for 16 h and analyzed via Western blot using antibodies against both V5 and His epitopes. As shown in Fig. 1E, the level of the fast migrating form of β2-spectrin was dramatically increased and reached a peak at 8 h after TGF-β treatment. In addition, the newly appeared β2-spectrin band was reactive to both C-terminal-tagged antibodies, indicating generation from fragmentation of full-length β2-spectrin by TGF-β. Next, we also tested whether inhibition of TGF-β signal suppresses the acetaminophen-induced cleavage of β2-spectrin. However, inhibition of TGF-β signaling by LY2019761, an inhibitor of TGF-β receptor I/II, did not showed the remarkable reduction of β2-sepctrin-cleavage (Supplementary Fig. 1A). These results suggest that β2-spectrin cleavage caused by acetaminophen is mediated by TGF-β in part.
Cleavage of β2-spectrin is induced by acetaminophen in a TGF-β-dependent manner. (A) Various liver-damaging conditions and reagents were tested in exogenous β2-spectrin (β2SP)-expressing Huh7 cells. The arrow indicates an acetaminophen-induced fast migrating immunoreactive signal against the β2-spectrin antibody. (B, C) C57BL/6 mice were treated with acetaminophen (200 mg/kg, ip), and their livers collected at 1 and 3 days post-injection. The patterns of β2-spectrin expression were analyzed using Western blot, and the amounts measured as described in Materials and Methods. Relative amounts of full-length (β2SP-FL) and fast migrating β2-spectrin (β2SP-FM) are presented on the histogram. (D) The patterns of TGF-β signaling and apoptosis proteins were analyzed in Huh7 cells treated with various concentrations of acetaminophen at the indicated days. (E) C-terminal V5/His-tagged β2-spectrin- transfected Huh7 cells were incubated in the absence or presence of TGF-β (100 pM) in for the indicated times, and analyzed via Western blot using antibodies against the V5 and His epitopes. β2-Spectrin-transfected Huh7 cells treated with 10 mM acetaminophen for 16 h were applied as the positive control for cleavage of β2-spectrin. The numbers indicate relative amounts of β2SP-FM compared untreated. β-actin was used as the loading control.
Acetaminophen-induced β2-spectrin cleavage is mediated by caspase-3/7
To investigate the cleavage pattern of β2-spectrin, we cloned full-length human β2-spectrin cDNA into vectors tagged with N-terminal FLAG or C-terminal V5. These plasmids were transfected into COS-7 cells, and Western blot analysis performed with antibodies against FLAG and V5 epitopes. Overexpression of exogenous β2-spectrin in COS-7 cells resulted in spontaneous cleavage by endopeptidase, yielding N-terminal and C-terminal fragments with molecular masses of 160 and 80 kDa, respectively, that were distinguishable with gel electrophoresis (Fig. 2A). Previous reports have suggested that β2-spectrin is a substrate of two endopeptidases, caspase-3 and calpain, and its proteolytic products are important biomarkers in injury [15] [16, 17]. Accordingly, to establish whether acetaminophen-induced cleavage of β2-spectrin is mediated by caspase-3 or calpain, we purified FLAG-tagged β2-spectrin using affinity chromatography, performed the in vitro cleavage assay with recombinant human calpain-1, calpain-2 and caspase-3, and compared the fragments separated with gel electrophoresis. The C-terminal fragment of β2-spectrin cleaved by caspase-3 exhibited similar migration to the acetaminophen-induced cleavage fragment of β2-spectrin, while patterns from calpain-mediated cleavage were different in terms of number and migration of fragments (Fig. 2B). The sum of the molecular weights of the two fragments was equal to the size of full-length protein without the presence of additional fragments, suggesting that β2-spectrin is cleaved by caspase-3 at a single position. An in vitro assay with different calpain amounts indicated different patterns of intermediate fragments from calpain cleavage to acetaminophen-cleaved β2-spectrin (data not shown).
Acetaminophen-induced β2-spectrin cleavage is mediated by caspase-3/7. (A) COS7 cells were transfected with N-terminal FLAG- (F) or C-terminal V5 (V)-tagged β2-spectrin constructs. Two days later, cells were harvested and subjected to Western blot by reprobing using antibodies against FLAG and V5 epitopes in identical membranes. (B) Purified human recombinant β2-spectrin (2 μg) was incubated with 1 unit of purified recombinant caspase-3, calpain-1 or calpain-2 at 37°C for 2 h. The reaction was terminated by adding Laemmli sampling buffer for SDS-PAGE, followed by Coomassie staining. The arrows indicate the fragment of β2-spectrin migrating similarly with acetaminophen treatment. (C) Acetaminophen (3 mM)-treated Huh7 cells were incubated in the absence or presence of increasing amounts of Ac-DEVD-CHO, a caspase-3 inhibitor, or Z-VAD-FMK, a pan-caspase inhibitor. The resulting cell lysates were subjected to SDS-PAGE, followed by Western blot using the β2-spectrin antibody with β-actin as the loading control. (D) Purified human recombinant β2-spectrin was incubated with various caspases, and further analyzed with SDS-PAGE and Coomassie staining.
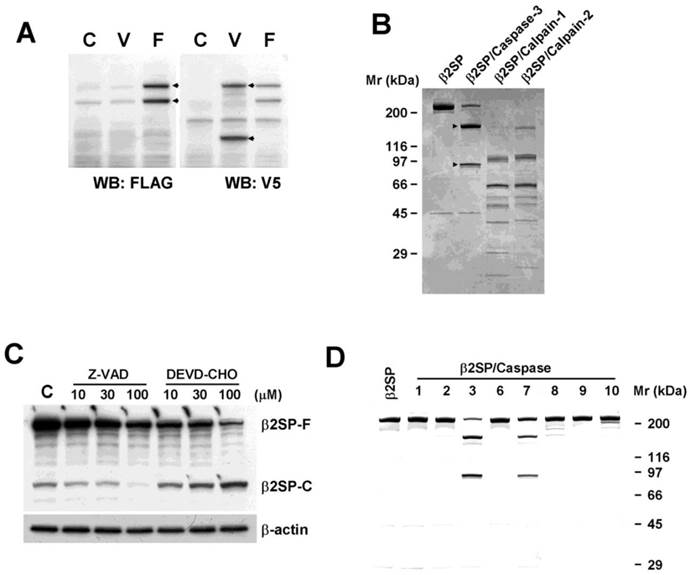
Based on data from our in vitro assay showing that β2-spectrin is a caspase-3 substrate, we further examined whether caspase-3 is the protease responsible for β2-spectrin cleavage upon acetaminophen treatment in Huh7 cells. Ac-DEVD-CHO, an inhibitor of caspase-3, was unable to prevent β2-spectrin fragmentation in Huh7 cells, but the addition of Z-VAD-FMK, an inhibitor of pan-caspase, blocked cleavage of β2-spectrin in a dose-dependent manner, suggesting mediation of cleavage by caspases other than caspase-3 (Fig. 2C). To identify the specific caspase responsible for acetaminophen-induced β2-spectrin cleavage, we performed an in vitro cleavage assay with purified recombinant β2-spectrin and the caspase family. As shown in Fig. 2D, both caspase-3 and -7 induced cleavage of β2-spectrin that yielded fragments exhibiting the same pattern as acetaminophen treatment, while other caspases did not induce fragmentation of β2-spectrin. We also identified that Z-VAD-FMK efficiently prevented the TGF-β-induced β2-spectrin cleavage in Huh7 cells and spontaneous β2-spectrin cleavage in COS7 (Supplementary Fig. 1B, and 1C). The findings suggest that β2-spectrin is cleaved by caspase-3 and -7 in acetaminophen-induced liver injury.
Prevention of β2-spectrin cleavage suppresses its function
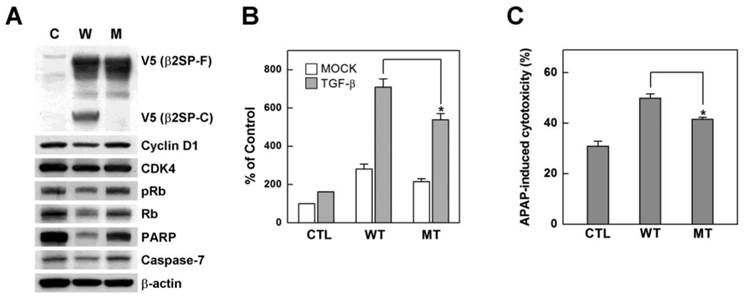
Based on data from our in vitro assay showing that β2-spectrin is a substrate of caspase-3 and caspase-7, we further examined the precise sites at which these caspases cleave β2-spectrin. Since the two enzymes display many similarities in their primary specificities and cleave DEVD peptides efficiently [18], we investigated whether β2-spectrin contains this sequence. The DEVD sequence in human β2-spectrin protein was located at positions 1454-1457 and conserved in mouse and chicken with similar sequences around DEVD. To ascertain whether DEVD is the caspase-3/7 cleavage site in β2-spectrin, we introduced a point mutation with a single amino acid substitution at D1457A. Next, both wild-type and mutant β2-spectrin were introduced into Huh7 cells, and the proteins analyzed via Western blot. Spontaneous cleavage of β2-spectrin was completely abrogated in the alanine-containing mutant protein, but not wild-type β2-spectrin, indicating that the caspase-3/7 cleavage site identified in β2-spectrin is correct and unique (Fig. 3A). Earlier studies by our group showed that upregulation of β2-spectrin decreases regulatory proteins of the cell cycle and apoptosis, and its deficiency leads to G1/S progression, which contributes to dysregulation of the cell cycle, cellular proliferation, oncogene overexpression, and formation of HCC [19]. To further determine whether β2-spectrin activity in cell cycle regulation is dependent on its cleavage, we examined the expression patterns of regulatory proteins of cell cycle and apoptosis with upregulation of D1457A mutant and wild type β2-spectrin. As shown in Fig. 3A, overexpression of wild-type β2-spectrin induced a dramatic decrease in Rb (24% to untransfected after β-actin normalization) and full-length PARP (50%). Uncleaved mutant β2-spectrin was associated with decreased levels of Rb (83%) and full-length PARP (74%), but not to the same extent as wild type. We subsequently compared TGF-β-dependent transcription activities with the 3TP promoter assay in Huh7 cells in three independent experiments. Introduction of β2-spectrin in Huh7 cells stimulated TGF-β-induced transcription activity to a significant extent. Cells with uncleaved D1457A mutant β2-spectrin exhibited significantly reduced TGF-β-induced transcription, compared to those with wild-type β2-spectrin (Fig. 3B). Furthermore, our analysis of acetaminophen-induced cytotoxicity showed that uncleaved mutant β2-spectrin (41.5%) induces a statistically significant decrease in growth suppression, compared to wild-type (49.9%) (Fig. 3C). These results indicate that caspase-3/7-dependent cleavage is required for β2-spectrin functions, including regulation of cell cycle, apoptosis, and transcription.
Prevention of β2-spectrin cleavage suppresses its activities (A) Patterns of cell cycle and apoptosis regulatory proteins following transfection of wild-type (W) or uncleaved mutant (M) β2-spectrin in SNU-761 cells are shown. Expression of exogenous β2-spectrin was monitored using the V5 antibody, with β-actin as the loading control. (B) Following transient transfection with 3TP-Lux construct containing wild-type or mutant forms of β2-spectrin, Huh7 cells were incubated for 24 h in the absence or presence of 100 pM TGF-β and TGF-β-dependent transcriptional activity measured using the luciferase activity assay, as described in Materials and Methods. Data are expressed as mean values ± SE of three independent experiments. (C) Acetaminophen-induced cytotoxicities of wild-type and mutant β2-spectrin-transfected Huh7 cells were estimated with the MTT assay. Each number represents the mean ± SE of quadruplicate determinations. Statistically significant differences (P<0.05) are indicated by asterisks.
Cleaved fragments of β2-spectrin perform separate functions
Since prevention of β2-spectrin cleavage weakened the acetaminophen-induced physiological response, we further examined whether the cleaved fragments of β2-spectrin are biologically active or intermediates of degradation. To determine the functions of the caspase-3/7 cleavage products, V5-tagged N-and C-terminal fragments of β2-spectrin were cloned and transfected into Huh7 cells (Fig. 4A). Overexpression of all β2-spectrin fragments led to reduced levels of cell cycle regulatory proteins, including cyclin D1, CDK4, phosphorylated Rb and Rb, along with increased apoptosis signals with induction of cleaved caspase-7, indicating that both β2-spectrin fragments function in cell cycle arrest and apoptosis in a similar manner to full-length β2-spectrin.
Cleaved fragments of β2-spectrin exhibit individual functions. (A) Patterns of cell cycle and apoptosis regulatory proteins induced following transfection of various forms of β2-spectrin in Huh7 cells. β-Actin was used as the loading control. (B) 293T cells were transfected with FLAG-Smad3 and various forms of V5-β2-spectrin or empty vector. Cell lysates were immunoprecipitated with anti-V5 antibody-conjugated agarose and subjected to Western blotting using anti-V5 and anti-FLAG mouse IgG. The inputs represent 5% of protein extracts without immunoprecipitation. (C) Following transient transfection with various forms of β2-spectrin and the 3TP-Lux construct, Huh7 cells were incubated for 24 h in the absence or presence of 100 pM TGF-β, and TGF-β-dependent transcriptional activity measured using the luciferase assay, as described in Materials and Methods. Data are expressed as mean ± SD of three independent experiments. (D) V5-β2-spectrin and HA-53BP2-transfected 293T cells were immunoprecipitated with anti-V5 antibody-conjugated agarose, and analyzed with Western blotting using the indicated antibodies. (E) Representative histograms showing DNA content of Huh7 cells. Full-length or cleaved β2-spectrin-transfected Huh7 cells were treated with TGF-β (100 pM) for 1 day and analyzed via flow cytometry with PI staining. (F) Percentages of cell distribution in the sub-G1 phase are shown on the histogram. Data are expressed as mean ± SD of three independent experiments. *P<0.01.
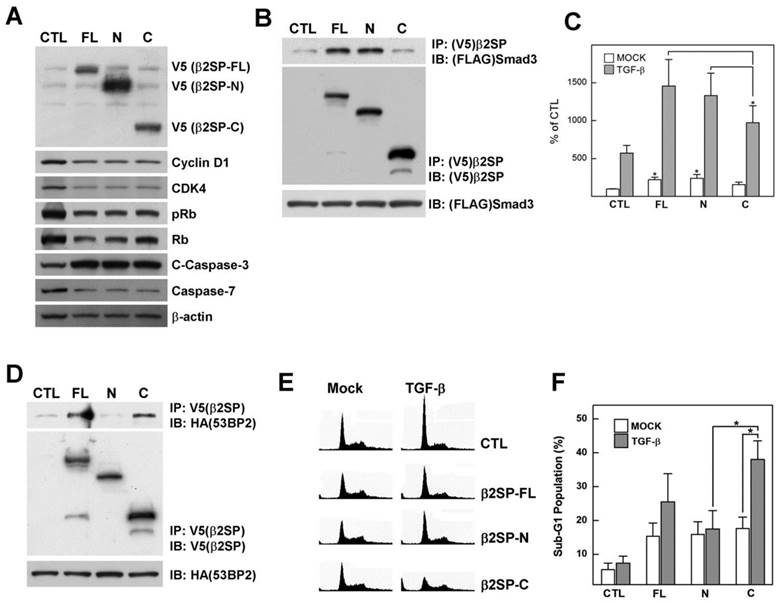
We previously reported that β2-spectrin interacts with Smad3, which regulates binding of Smad3 and CDK4 for cell cycle progression [19]. Based on this finding, we examined whether Smad3 selectively interacts with any of the β2-spectrin fragments. Surprisingly, the N-terminal fragment of β2-spectrin (β2SP-N) interacted with Smad3 to a similar extent as full-length β2-spectrin (β2SP-FL), while the C-terminal fragment (β2SP-C) did not bind Smad3 (Fig. 4B). We further investigated whether b2SP-N plays a specific role in TGF-β-dependent transcription activity via interactions with Smad proteins. The 3-TP lux promoter assay was performed with several forms of β2-spectrin in the absence or presence of TGF-β, and the extent of luciferase activity was measured. Our data showed significant induction of TGF-β-dependent transcription by β2SP-N, similar to β2SP-FL, compared to β2SP-C (Fig. 4C).
53BP2 was initially identified as a p53-binding protein in a yeast two-hybrid screen using p53 as bait. The protein plays a central role in regulation of apoptosis and cell growth [20]. Recently, we identified 53BP2 as a β2-spectrin interacting protein using proteomic analysis. Here, we continued to examine the interactions between β2-spectrin fragments and 53BP2 via immunoprecipitation of V5 epitope-tagged β2-spectrin and HA epitope-tagged 53BP2 expressed in 293T cells. In the immunoprecipitation assay, both β2SP-FL and β2SP-C interacted with 53BP2, but not β2SP-N (Fig. 4D). We additionally examined whether 53BP2-binding β2-spectrin proteins influence cell cycle and apoptosis. Specifically, Huh7 cells were transfected with several forms of β2-spectrin, followed by treatment with TGF-β, and cell cycle phases analyzed via fluorescence-activated cell sorting (FACS) with propidium iodide staining. Introduction of different forms of β2-spectrin increased the population of cells in the DNA fragmented sub-G1 phase to similar extents (Fig. 4E, and 4F). Interestingly, the apoptotic sub-G1 populations with the transfection of β2SP-C exhibited the statically significant increment upon from 17.6% to 38.0% following TGF-β treatment (100 pM), while transfection of β2SP-FL in the presence of TGF-β induced an increase in the number of cells in this phase from 15.3% to 25.5%. Simultaneously, we did not detect significant accumulation of cells in the sub-G1 phase in β2SP-N- or empty vector-transfected cells exposed to TGF-β. Thus, cleaved fragments of β2-spectrin are physiologically active, and the N- and C-terminal fragments retain their discrete interaction partners and functions in transcriptional regulation and apoptosis, respectively.
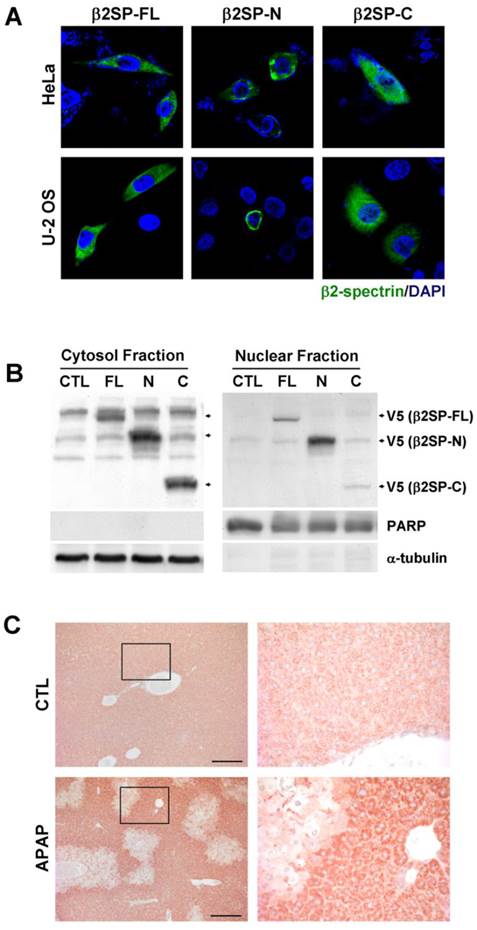
Differences in β2-spectrin fragment localization
We previously showed that nuclear co-localization of β2-spectrin and Smad4 is enhanced with strengthened interactions in the presence of TGF-β [21]. Accordingly, we examined the intracellular distribution of fragments of β2-spectrin cleaved by acetaminophen and subjected to TGF-β treatment. Analysis of the subcellular localization of β2-spectrin in HeLa, and U-2 OS cells revealed the predominant presence of β2SP-FL and β2SP-C with dispersed cytoplasmic distribution (Fig. 5A). In contrast, β2SP-N was concentrated around the nuclear membrane, consistent with earlier observations in gastrointestinal cells [21]. Subcellular fractionation further disclosed higher levels of β2SP-N than β2SP-C in the nuclear fraction, whereas similar levels of both fragments were observed in the cytoplasmic fraction (Fig. 5B). In addition, we performed the immunohistochemical staining to the mice liver sections using the antibody raised against C-terminus of β2-spectrin (2101-2189) in the absence or presence of acetaminophen administration. As shown in Fig. 5C, localization of β2-spectrin in normal liver was detected in cytosol with some granular shape. However, after treatment of acetaminophen, necrotic area of liver exhibited the disappearance of β2-spectrin while surrounding areas of necrotic region exhibited the increased accumulation of β2-spectrin in cytosol with diffused pattern. These results suggest that cleavage of β2-spectrin facilitates the redistribution of β2-spectrin fragments during the process of liver damage by acetaminophen.
Differences in β2-spectrin fragment localization. (A) HeLa and U-2 OS cells were transfected with V5-tagged full-length, N-terminal and C-terminal β2-spectrin, and immunostained with an antibody against the V5-epitope. (B) Huh7 cells were transfected with various forms of V5-tagged β2-spectrin. Total cell lysates were additionally separated into cytosol and nuclear fractions, and subjected to immunoblot analysis using an anti-V5 antibody to determine the subcellular localization of fragments. PARP and α-tubulin were used as the nuclear and cytosol localization markers, respectively. (C) Four-month old male C57BL/6 mice were injected with acetaminophen (500 mg/kg, ip), and their livers were collected at 24 h day later. The left panels show the liver sections stained with the antibody raised against C-terminus (2101-2189) of β2-spectrin. Right panels are magnified images of the boxed areas. Scale bar: 200 μm.
Downregulation of β2-spectrin impairs liver damage response to acetaminophen
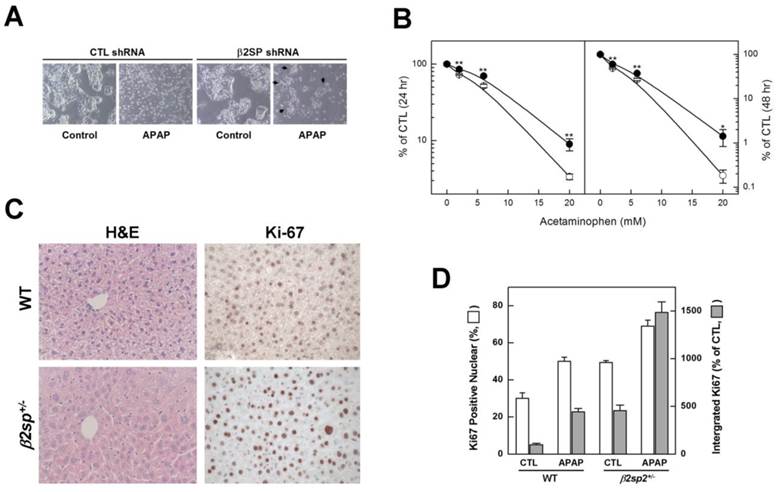
To establish the effect of deficiency of β2-spectrin on acetaminophen-induced damage response in liver, we eliminated endogenous β2-spectrin via transduction of HepG2 cells with β2-spectrin shRNA, and examined cytotoxicity in response to acetaminophen (5 mM). As seen in Fig. 6A, the majority of normal HepG2 cells could not survive without typical attachment, whereas β2-spectrin knockdown HepG2 cells were less dense than their untreated counterparts, but some attached cells were identified. Comparison of the survival of HepG2 cells at various concentrations of acetaminophen revealed that those with β2-spectrin knockdown were significantly more resistant to acetaminophen than mock transduced cells (Fig. 6B). To further determine whether deficiency of β2-spectrin alters liver damage induced by acetaminophen, we investigated the liver status of 4M old β2sp+/- mice, compared to age-matched wild-type controls administered acetaminophen (200 mg/kg, ip). At 3 days after acetaminophen injection, we observed no differences in liver damage between wild-type and β2sp+/- mice, based on H&E staining and collagen deposition. However, at 9 days after injection, H&E staining around the hepatic vein revealed that liver from β2sp+/- mice retained the typical structure while that from wild-type mice exhibited extensive regions with hepatic steatosis (Fig. 6C). Moreover, in immunohistochemical analyses of the midzonal area using antibodies against Ki-67, markers of proliferating cells were more easily identified in liver tissues of β2sp+/- mice than their matched wild-type counterparts. Ki-67-positive nuclei in regenerating livers were calculated as 50% and 69% in wild type and β2sp+/- mice, respectively, and accumulation of signal intensities was more than 3.4 times higher in mutant mice (Fig. 6D). Our data suggest that β2-spectrin is required for acetaminophen-mediated liver damage, and its deficiency leads to resistance to drug-induced tissue toxicity.
Impairment of acetaminophen-induced damage response upon β2-spectrin downregulation. (A) Following transduction with β2-spectrin shRNA or empty construct containing lentivirus, HepG2 cells were incubated in the absence or presence of 5 mM acetaminophen for 48 h. The arrows indicate surviving colonies in the presence of acetaminophen upon downregulation of β2-spectrin shRNA. (B) Survival of HepG2 cells at the indicated concentrations of acetaminophen was estimated with the MTT assay after transduction with empty or β2-spectrin shRNA-containing lentivirus. The numbers represent mean values ± SD, with significant differences indicated by asterisks. **P<0.01; *P<0.05. (C, D) Four-month old male mice with spnb2+/+, and spnb2+/- were injected with acetaminophen (200 mg/kg, ip), and livers collected 9 days later. The left panels show the morphology of liver sections stained with H&E. Proliferation was evaluated immunohistochemically in wild-type and mutant livers using an antibody against Ki-67 (right panels). Relative amounts of Ki-67-positive nuclear and integrated signal intensities are shown on the histogram.
Discussion
Acetaminophen is one of most widely used analgesics and antipyretics worldwide but the main cause of drug-induced hepatotoxicity. Acetaminophen administered at therapeutic doses is predominantly metabolized in liver to sulfate and glucuronide metabolites, and excreted in urine. However, at excess doses, the sulfate and glucuronide pathways become saturated. Thus, an increased proportion of acetaminophen is converted to NAPQI by cytochrome P450, which is detoxified by glutathione to water-soluble non-toxic cysteine and mercapturic acid. With NAPQI accumulation, depletion of glutathione causes oxidant stress that may trigger signaling pathways involved in mitochondrial toxicity, ultimately leading to lethal cell injury [10]. Even at regular doses, long-term use of acetaminophen may lead to the development of chronic hepatic necrosis and fibrosis with portal abnormalities [22]. In addition, hepatotoxicity in response to acetaminophen frequently occurs in patients with chronic liver disease or alcoholics, signifying that acetaminophen-induced acute liver failure is a cumulative effect [23] [24].
The TGF-β signaling pathway plays a critical role in diverse cell functions, including inhibition of differentiation, cell growth, apoptosis, adhesion, migration, and extracellular matrix (ECM) deposition. In an acute liver injury model, treatment of B6C3F1 mice with hepatotoxic doses (600 mg/kg) of acetaminophen produced a marked increase in TGF-β1 mRNA expression in liver, but not spleen [25]. The results of our study showed that in addition to transcriptional induction, phosphorylation of TGF-β receptor II, Smad2, and Smad3 is significantly induced by acetaminophen treatment of Huh7 cells, signifying that liver damage induced by high doses of acetaminophen is mediated via the TGF-β signaling pathway. Under toxic conditions, liver fibrosis is the typical response to tissue damage, and accumulation of excessive ECM protein is caused by increased synthesis and deposition of newly formed components, eventually leading to cirrhosis and many complications, including portal hypertension, liver failure, and hepatocellular carcinoma [26]. High levels of TGF-β are frequently observed in hepatic fibrosis, suggesting that TGF-β is a mediator of fibrosis and other chronic liver diseases [27]. Moreover, activated TGF-β is suggested to cause fibrotic disease in several tissues by enhancing ECM synthesis and inhibiting of ECM degradation through suppression of matrix-degrading enzymes [28] [29].
Previous studies by our group demonstrated that β2-spectrin is a mediator of TGF-β signaling in growth control of liver cells from β2sp heterozygous mutant mice to hepatocellular cancer development [6] [30] [31]. Moreover, β2-spectrin has been identified as a Smad3/4 interacting protein that plays a critical role in localizing Smads and facilitates the tumor suppressor function of TGF-β/Smad signaling. Deficiency of β2-spectrin protein has been shown to result in mislocalization of Smad3 and Smad4, as well as loss of the TGF-β-dependent transcriptional response. These defects are rescued upon restoration of β2-spectrin [3]. In terms of its contribution to TGF-β signaling, we showed here that β2-spectrin is not only a scaffolding protein that interacts with Smad proteins but also a target molecule of TGF-β for transmission and amplification of signals to generate the physiological response in liver damage. Acetaminophen treatment induced phosphorylation of TGF-β receptor II and Smad2/3, and simultaneously, fragmentation of β2-spectrin in mouse liver and Huh7 cell cultures. In addition, treatment of cultured Huh7 cells with TGF-β dramatically induced cleavage of β2-spectrin from 2 h. Indeed, proteolysis of spectrin has been observed under conditions other than liver damage, including injury and aging of brain and development of lens, which also require extensive cytoskeletal reorganization and apoptosis [15, 32, 33]. Depending on the diverse proteases and physiological demands, different regulatory processes for β2-spectrin proteolysis have been identified. Recent reports have shown that cleavage of β2-spectrin is catalyzed by cathepsins and two endopeptidases, calpain and caspase-3 [16] [34]. In contrast to the exopeptidase activity of cathepsin, cleavage by endopeptidases, calpain and caspase, generates relatively large and long-lived active substrate fragments. Under conditions of brain injury, the 260 kDa β2-spectrin is degraded into four major fragments (110/85 kDa by calpain-2 and 108/80 kDa by caspase-3) [34]. On the other hand, analyses of the β-spectrin status during fludarabine/mitoxantrone/dexamethasone-induced apoptosis in Jurkat T cells revealed proteolytic degradation by caspase-8, with the predominant appearance of a specific 55 kDa fragment containing the actin-binding domain [35]. To provide an insight into the relationship between acetaminophen-induced liver damage and β2-spectrin cleavage, we investigated the protease responsible for β2-spectrin cleavage in acetaminophen-induced liver damage in cell cultures in vitro. β2-Spectrin was cleaved by caspase-3 and caspase-7 at a single position into 160 and 80 kDa fragments. Importantly, β2-spectrin was cleaved at high doses of acetaminophen (10 mM). At the same acetaminophen dose, we observed a dramatic increase in apoptosis, based on cleavage of PARP and caspase-7, while TGF-β receptor II and Smad2/3 phosphorylation was begun to detect at lower concentrations of acetaminophen (3 mM). Lethal liver injury by acetaminophen is mediated via oxidant stress from depletion of glutathione which, in turn, triggers the signaling pathway involved in mitochondrial toxicity, leading to cytochrome c release. Cytochrome c from mitochondria acts in combination with caspase-9, processing procaspase-3/7 to active cleaved caspase-3/7 [36]. Based on these results, we suggest that proteolysis of β2-spectrin is mediated through the mitochondrial pathway by caspase-3/7 in acetaminophen-induced liver damage.
Interestingly, two different proteolytic modulations occur in a tetrameric spectrin protein complex consisting of two α-spectrins and β-spectrins that lead to distinct physiological responses: one enhancing skeletal plasticity without destroying the cytoskeletal network, characterized by cleavage of α-spectrin and preservation of the β-spectrin component, and an alternative response closely correlated with cell death and characterized by the proteolysis of both types of spectrins and complete dissolution of the spectrin skeleton [17]. Our data clearly revealed that cleavage of β2-spectrin in acetaminophen-induced liver damage is closely related to cell death. Firstly, the dramatic appearance of cleaved caspase-7 and PARP corresponded to β2-spectrin cleavage following acetaminophen treatment in Huh7 cells. Next, in the presence of TGF-β, marked increase in the apoptotic sub-G1 population was detected following the introduction of the C-terminal cleavage fragment, which also interacts with apoptosis-regulating 53BP2 protein. In addition, prevention of β2-spectrin cleavage led to reduction of PARP and caspase-7 cleavage and a significant decrease in acetaminophen-induced cytotoxicity. Cleavage of β2-spectrin facilitated relocalization of the proteolytic fragments with transcription and apoptosis functions in the nuclear and cytosol compartments, respectively. Finally, downregulation of β2-spectrin using specific shRNA in cultured cells and heterozygous mutant mice (β2sp+/-) significantly reduced suppression of cell growth and induced cell regeneration. Our results collectively imply that cleavage of β2-spectrin is required for determination of cell death in acetaminophen-induced liver damage, and inhibition of cleavage or downregulation of β2-spectrin alters the regulation of cell growth and death triggered by acetaminophen. In view of the present findings, we propose that β2-spectrin plays a critical role as a mediator and effector molecule in liver damage by modulating the cytoskeletal network and survival. The absence or downregulation of β2-spectrin leads to resistance to liver damage and enhancement of regeneration, suggesting that loss of β2-spectrin is triggered upon liver damage and this protein is a primary target during the selection process for growth advantages. Our study clearly supports the hypothesis that the deleterious effects of β2-spectrin serve as a double-edged sword that suppresses damage to the liver early in life but may later lead to hepatocellular cancer formation.
Supplementary Material
Supplementary Figure S1.
Abbreviations
β2SP, β2-spectrin; APAP, acetaminophen; TGF-β, transforming growth factor-β.
Acknowledgements
This study was supported by National Cancer Center of Korea NCC-1310660, Korea Healthcare Technology R & D Project HI11C1289, and National Research Foundation of Korea 2014R1A2A1A11049935.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Viel A, Branton D. Spectrin: on the path from structure to function. Curr Opin Cell Biol. 1996;8:49-55
2. Bennett V. Spectrin-based membrane skeleton: a multipotential adaptor between plasma membrane and cytoplasm. Physiol Rev. 1990;70:1029-65
3. Tang Y, Katuri V, Dillner A, Mishra B, Deng CX, Mishra L. Disruption of transforming growth factor-beta signaling in ELF beta-spectrin-deficient mice. Science. 2003;299:574-7
4. Thenappan A, Li Y, Kitisin K, Rashid A, Shetty K, Johnson L. et al. Role of transforming growth factor beta signaling and expansion of progenitor cells in regenerating liver. Hepatology. 2010;51:1373-82
5. Mishra B, Tang Y, Katuri V, Fleury T, Said AH, Rashid A. et al. Loss of cooperative function of transforming growth factor-beta signaling proteins, smad3 with embryonic liver fodrin, a beta-spectrin, in primary biliary cirrhosis. Liver Int. 2004;24:637-45
6. Kitisin K, Ganesan N, Tang Y, Jogunoori W, Volpe EA, Kim SS. et al. Disruption of transforming growth factor-beta signaling through beta-spectrin ELF leads to hepatocellular cancer through cyclin D1 activation. Oncogene. 2007;26:7103-10
7. Yao ZX, Jogunoori W, Choufani S, Rashid A, Blake T, Yao W. et al. Epigenetic silencing of beta-spectrin, a TGF-beta signaling/scaffolding protein in a human cancer stem cell disorder: Beckwith-Wiedemann syndrome. J Biol Chem. 2010;285:36112-20
8. Lee WM. Acetaminophen and the U.S. Acute Liver Failure Study Group: lowering the risks of hepatic failure. Hepatology. 2004;40:6-9
9. Larson AM, Polson J, Fontana RJ, Davern TJ, Lalani E, Hynan LS. et al. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology. 2005;42:1364-72
10. Chen C, Hennig GE, Manautou JE. Hepatobiliary excretion of acetaminophen glutathione conjugate and its derivatives in transport-deficient (TR-) hyperbilirubinemic rats. Drug Metab Dispos. 2003;31:798-804
11. Kass GE, Macanas-Pirard P, Lee PC, Hinton RH. The role of apoptosis in acetaminophen-induced injury. Ann N Y Acad Sci. 2003;1010:557-9
12. Lim JA, Baek HJ, Jang MS, Choi EK, Lee YM, Lee SJ. et al. Loss of beta2-spectrin prevents cardiomyocyte differentiation and heart development. Cardiovasc Res. 2014;101:39-47
13. Baek HJ, Lim SC, Kitisin K, Jogunoori W, Tang Y, Marshall MB. et al. Hepatocellular cancer arises from loss of transforming growth factor beta signaling adaptor protein embryonic liver fodrin through abnormal angiogenesis. Hepatology. 2008;48:1128-37
14. Kitisin K, Pishvaian MJ, Johnson LB, Mishra L. Liver stem cells and molecular signaling pathways in hepatocellular carcinoma. Gastrointest Cancer Res. 2007;1:S13-21
15. Pineda JA, Wang KK, Hayes RL. Biomarkers of proteolytic damage following traumatic brain injury. Brain Pathol. 2004;14:202-9
16. Wang KK, Posmantur R, Nath R, McGinnis K, Whitton M, Talanian RV. et al. Simultaneous degradation of alphaII- and betaII-spectrin by caspase 3 (CPP32) in apoptotic cells. J Biol Chem. 1998;273:22490-7
17. Glantz SB, Cianci CD, Iyer R, Pradhan D, Wang KK, Morrow JS. Sequential degradation of alphaII and betaII spectrin by calpain in glutamate or maitotoxin-stimulated cells. Biochemistry. 2007;46:502-13
18. Pop C, Salvesen GS. Human caspases: activation, specificity, and regulation. J Biol Chem. 2009;284:21777-81
19. Baek HJ, Pishvaian MJ, Tang Y, Kim TH, Yang S, Zouhairi ME. et al. Transforming growth factor-beta adaptor, beta2-spectrin, modulates cyclin dependent kinase 4 to reduce development of hepatocellular cancer. Hepatology. 2011;53:1676-84
20. Sullivan A, Lu X. ASPP: a new family of oncogenes and tumour suppressor genes. Br J Cancer. 2007;96:196-200
21. Katuri V, Tang Y, Marshall B, Rashid A, Jogunoori W, Volpe EA. et al. Inactivation of ELF/TGF-beta signaling in human gastrointestinal cancer. Oncogene. 2005;24:8012-24
22. Bonkowsky HL, Mudge GH, McMurtry RJ. Chronic hepatic inflammation and fibrosis due to low doses of paracetamol. Lancet. 1978;1:1016-8
23. O'Dell JR, Zetterman RK, Burnett DA. Centrilobular hepatic fibrosis following acetaminophen-induced hepatic necrosis in an alcoholic. JAMA. 1986;255:2636-7
24. Marino G, Lewis JH. Drug-induced liver disease. Curr Opin Gastroenterol. 2001;17:232-41
25. Jeon YJ, Han SH, Yang KH, Kaminski NE. Induction of liver-associated transforming growth factor beta 1 (TGF-beta 1) mRNA expression by carbon tetrachloride leads to the inhibition of T helper 2 cell-associated lymphokines. Toxicol Appl Pharmacol. 1997;144:27-35
26. Wells RG. The role of matrix stiffness in hepatic stellate cell activation and liver fibrosis. J Clin Gastroenterol. 2005;39:S158-61
27. Jonsson JR, Clouston AD, Ando Y, Kelemen LI, Horn MJ, Adamson MD. et al. Angiotensin-converting enzyme inhibition attenuates the progression of rat hepatic fibrosis. Gastroenterology. 2001;121:148-55
28. Leask A, Abraham DJ. TGF-beta signaling and the fibrotic response. FASEB J. 2004;18:816-27
29. Ogawa K, Chen F, Kuang C, Chen Y. Suppression of matrix metalloproteinase-9 transcription by transforming growth factor-beta is mediated by a nuclear factor-kappaB site. Biochem J. 2004;381:413-22
30. Tang Y, Katuri V, Srinivasan R, Fogt F, Redman R, Anand G. et al. Transforming growth factor-beta suppresses nonmetastatic colon cancer through Smad4 and adaptor protein ELF at an early stage of tumorigenesis. Cancer Res. 2005;65:4228-37
31. Katuri V, Tang Y, Li C, Jogunoori W, Deng CX, Rashid A. et al. Critical interactions between TGF-beta signaling/ELF, and E-cadherin/beta-catenin mediated tumor suppression. Oncogene. 2006;25:1871-86
32. Yan XX, Jeromin A. Spectrin Breakdown Products (SBDPs) as Potential Biomarkers for Neurodegenerative Diseases. Curr Transl Geriatr Exp Gerontol Rep. 2012;1:85-93
33. Lee A, Morrow JS, Fowler VM. Caspase remodeling of the spectrin membrane skeleton during lens development and aging. J Biol Chem. 2001;276:20735-42
34. Kobeissy FH, Liu MC, Yang Z, Zhang Z, Zheng W, Glushakova O. et al. Degradation of betaII-Spectrin Protein by Calpain-2 and Caspase-3 Under Neurotoxic and Traumatic Brain Injury Conditions. Mol Neurobiol. 2014
35. Toporkiewicz M, Grzybek M, Meissner J, Michalczyk I, Dubielecka PM, Korycka J. et al. Release of an ~55kDa fragment containing the actin-binding domain of beta-spectrin by caspase-8 during FND-induced apoptosis depends on the presence of protein 4.1. Arch Biochem Biophys. 2013;535:205-13
36. Li P, Nijhawan D, Wang X. Mitochondrial activation of apoptosis. Cell. 2004;116:S57-9 2 p following S9
Author contact
![]() Corresponding author: Sang Soo Kim, Radiation Medicine Branch, National Cancer Center, Goyang, Gyeonggi, 410-769, Korea. Tel: 8231-920-2491, Fax: 8231-920-2494, E-mail: sangsookimre.kr
Corresponding author: Sang Soo Kim, Radiation Medicine Branch, National Cancer Center, Goyang, Gyeonggi, 410-769, Korea. Tel: 8231-920-2491, Fax: 8231-920-2494, E-mail: sangsookimre.kr