Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results
Discussion
Supplementary Material
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2016; 12(2):184-197. doi:10.7150/ijbs.13710 This issue Cite
Research Paper
Lysozyme Mutants Accumulate in Cells while Associated at their N-terminal Alpha-domain with the Endoplasmic Reticulum Chaperone GRP78/BiP
Yoshiki Kamada, Yusuke Nawata, Yasushi Sugimoto ![]()
Laboratory of Biochemistry and Bioscience, The United Graduate School of Agricultural Sciences, Kagoshima University, Kagoshima 890-0065, Japan
Received 2015-9-1; Accepted 2015-11-9; Published 2016-1-1
Abstract

Amyloidogenic human lysozyme variants deposit in cells and cause systemic amyloidosis. We recently observed that such lysozymes accumulate in the endoplasmic reticulum (ER) with the ER chaperone GRP78/BiP, accompanying the ER stress response. Here we investigated the region of lysozyme that is critical to its association with GRP78/BiP. In addition to the above-mentioned variants of lysozyme, we constructed lysozyme truncation or substitution mutants. These were co-expressed with GRP78/BiP (tagged with FLAG) in cultured human embryonic kidney cells, which were analyzed by western blotting and immunocytochemistry using anti-lysozyme and anti-FLAG antibodies. The amyloidogenic variants were confirmed to be strongly associated with GRP78/BiP as revealed by the co-immunoprecipitation assay, whereas N-terminal mutants pruned of 1-41 or 1-51 residues were found not to be associated with the chaperone. Single amino acid substitutions for the leucine array along the α-helices in the N-terminal region resulted in wild-type lysozyme remaining attached to GRP78/BiP. These mutations also tended to show lowered secretion ability. We conclude that the N-terminal α-helices region of the lysozyme is pivotal for its strong adhesion to GRP78/BiP. We suspect that wild-type lysozyme interacts with the GRP at this region as a step in the proper folding monitored by the ER chaperone.
Keywords: lysozyme, variants, aggregate, amyloidosis, GRP78/BiP, endoplasmic reticulum stress
Introduction
Not only neurodegenerative disorders such as Alzheimer's disease but also diabetes and non-neurogenic systemic diseases have become serious public health problems. These diseases are all attributed to the deposition of aberrant proteins in tissues [1-4]. Such proteins, accumulated both extracellularly and intracellularly, become dysfunctional and are thought to have different types of pathogenic influences. Malignant polypeptides are often formed by folding mistakes, which are enhanced by genetic mutations and various environmental factors such as salt, heat, oxidizing and toxic conditions, and molecular crowding [5-7]. Misfolded proteins are prone to expose their hydrophobic regions that are buried within the interior when the proteins are folded correctly. The exposed hydrophobic surfaces facilitate the oligomerization and aggregation of proteins [8,9]. In many cases, such protein molecule phenomena lead to the formation of fibers with a regular structure [10].
The mechanisms underlying the protein quality control (PQC) system, i.e., the cellular response that copes with aberrant proteins to maintain cell homeostasis have been extensively investigated [11-14]. A major part of the PQC system resides in the endoplasmic reticulum (ER), and is called ER-associated degradation (ERAD) [13]. The ER receives proteins that fail to undergo proper folding, includes them up to the step of ubiquitination and transfers them to further processes such as degradation by the proteasome system [15,16]. Sometimes, accumulated proteins in the cells are known to become a target of autophagy and to be degraded [16]. Abnormal proteins are occasionally sequestered to a specific cellular compartment to generate an intracellular inclusion body called the aggresome [17,18].
The most prominent factors in the PQC system are the molecular chaperones such as heat shock proteins (Hsp) that identify abnormal proteins, assist in their refolding, prevent their aggregation and help to restore the injured proteins [18]. It was reported that a decrease in the cell of molecular chaperones and the loss of their function induced folding failure and an increase in the cell of aggregate proteins [19].
A protein disorder in cells prompts an intracellular signal transduction pathway called the “unfolded protein response system.” This system, highly conserved from yeasts to humans, controls the homeostasis of the ER by activating the ER stress response to recover and protect cells from damage due to the aggregation of misfolded proteins [14,20]. In mammals, three independent pathways are used upon ER stress: the inositol-requiring enzyme-1 (IRE1) pathway [21], the double-stranded RNA-activated protein kinase-like ER kinase pathway, and the activating transcription factor 6 pathway [22].
There are two forms of IRE1: one in which IRE1α is expressed ubiquitously and the other in which IRE1β is expressed solely in the intestinal and lung epithelium [23,24]. In humans, IRE1α and IRE1β proteins are encoded by the ERN1 and ERN2 genes [25], respectively. IRE1α is bound to the ER chaperone GRP78/BiP under normal cell conditions. In an emergency, IRE1α is detached from GRP78/BiP and activates RNase activity, and this in turn processes immature XBP1 transcript to mature XBP1 mRNA. XBP1 (the XBP1 product) is a transcription factor that enhances the expression of GRP78/BiP (as well as that of GRP94) [26,27]. Thus, GRP78/BiP is one of the central players in the ER stress response [28].
Lysozyme, a small protein secreted from various tissues, is known for its bias toward amyloid fibril formation [29]. In humans, at least five naturally occurring single amino-acid mutants (I56T, F57I, I59T, W64R and D67H) of lysozyme accumulate in many organs such as liver, spleen and kidney, and they form amyloid fibrils, resulting in non-neuropathic systemic amyloidosis [30-32]. These mutants are therefore called amyloidogenic variants. The sites of mutations in the variants converge in or near the so-called amyloidogenic core region, which is important for amyloid fibril formation [33-35]. Our recent study using four of the five variants revealed that, when overexpressed in cultured human embryonic kidney (HEK) 293 cells, I56T, F57I, W64R and D67H were accumulated in the ER together with GRP78/BiP while enhancing the ER stress response via the IRE1-XBP1-GRP78/BiP pathway [36]. In the present study, using newly constructed prune mutants as well as the four amyloidogenic variants, we obtained results indicating that the N-terminal hydrophobic region of lysozymes is critical for the association with GRP78/BiP.
Materials and methods
Cells and chemicals
Cells of the HEK293 line, culture medium and supplements added to the medium, anti-GAPDH antibody, HRP-conjugated secondary antibody and Alexa Fluor 488- or 546-conjugated IgG were purchased as described in our previous study [36]. An anti-lysozyme antibody with a recognition site of 37-50 was obtained from Abcam (Cambridge, MA), and we created an anti-lysozyme antibody with a recognition site of 49-65 in our laboratory. The latter was used for N-terminal deletion mutants (see below). Anti-GRP78/BiP antibody was from Santa Cruz Biotechnology (Santa Cruz, CA); anti-FLAG antibody and anti-FLAG M2 Affinity Gel were from Sigma-Aldrich (St. Louis, MO). MG-132 (z-Leu-Leu-Leu-CHO) was purchased from Peptide Institute (Osaka, Japan), and other reagents were obtained from Wako Fine Chemicals (Osaka).
Construction of mutants and expression vectors
The cDNAs for wild-type human lysozyme and its amyloidogenic variants were prepared as described [36]. Deletion mutants and substitution mutants were newly produced from the wild-type cDNA using the inverse polymerase chain reaction (PCR) method with confirmation by sequencing. These sequences were introduced into the pEBMulti-neo expression vector (Wako). The expression vector of tagged GRP78/BiP was constructed as follows: Human GRP78/BiP cDNA was isolated from a HEK293 cDNA library prepared by reverse transcription-PCR (RT-PCR) of total RNA and introduced into the pBApo-EF1α-neo vector (Takara Bio, Shiga, Japan) using the In-Fusion HD cloning system (Takara Bio). To this vector was added the FLAG tag sequence (coding for DYKDDDDK) at the position behind the signal sequence of the GRP78/BiP gene.
Cell culture, transfection and analyses
The cultivation and transfection of cells were performed essentially as described [36], with the following modifications. For the experiment using the proteasome inhibitor MG-132, transfected cells were cultured for 24 h in Dulbecco's Modified Eagle's Medium (DMEM) with fetal bovine serum (FBS), non-essential amino acids (NEAAs) and Antibiotic-Antimycotic solution (Life Technologies, Carlsbad, CA) in 5% CO2 at 37°C overnight. After the replacement of medium with Opti-MEM medium (Life Technologies), followed by additional incubation for 1 h, the cells were transfected with each of the expression vectors with lysozyme sequences using X-tremeGENE™ HP DNA Transfection Reagent (Roche, Mannheim, Germany) and incubated for 24 h. The culture media were then mixed with 5 µg MG-132 and further incubated for 12 h. The cells were collected and treated with NP-40 lysis buffer to obtain the lysates and pellets. These, with the culture media, were analyzed together by western blotting.
For the preparation of co-immunoprecipitated fractions, HEK293 cells were infected first with the vector with FLAG-tagged GRP78/BiP, followed by incubation for 24 h, and then infected with the lysozyme vector. After further incubation for 48 h, the cells were collected and homogenized in NP-40 lysis buffer, to which 10 U/ml hexokinase (Sigma Aldrich) plus 0.2 mM glucose were added in order to maintain the GRP78/BiP function [37,38]. Two types of NP-40 lysis buffer, with and without 1 mM ATP, were used in some experiments. The lysates were added to the anti-FLAG M2 Affinity Gel to recover FLAG-GRP78/BiP and allowed to react for 2 h with gentle rotation at 4°C. After being washed three times with phosphate-buffered saline (PBS), proteins were eluted with 2 × sodium dodecyl sulfate (SDS) sample buffer (0.25 M Tris-HCl buffer, pH 6.8, containing 20% 2-mercaptoethanol, 20% SDS and 20% sucrose). The eluants (co-immunoprecipitated lysozyme/GRP) and other fractions including the media were analyzed by Western blotting.
Immunocytochemistry was done using the cultured cells as described [36]. In brief, transfected cells were cultured on a collagen-coated cover glass for 72 h and treated with the first antibody followed by the secondary antibody and viewed under a confocal laser microscope. The Western blotting procedure, immunocytochemistry, PCR and approximate quantification of gel bands were as described [36]. The evaluation of XBP1 mRNA splicing was done by RT-PCR according as described [36,39].
Results
Association of amyloidogenic lysozyme variants and GRP78/BiP without degradation
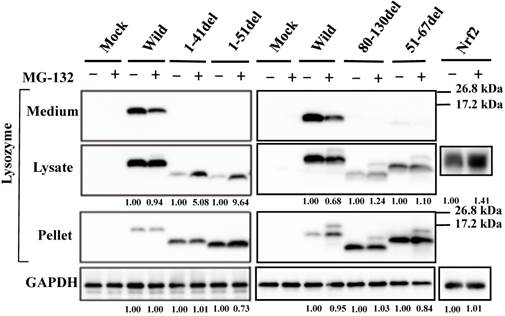
Our previous study demonstrated that the four amyloidogenic lysozyme variants I56T, F57I, W64R and D67H remain in the ER together with GRP78/BiP [36]. In the present study, to determine whether or not these variant proteins are affected by the intrinsic proteasomes, we overexpressed each of the variants and the wild-type lysozyme in HEK cells, which were further incubated with or without the proteasome inhibitor MG-132. The cell lysates, pellets and media were then subjected to Western blotting with the anti-lysozyme antibody. The results were in agreement with those of our previous experiments [36] in that the variants exhibited stronger bands in the pellet fractions compared to the wild type (Fig. 1, third panel). Regarding the results of the medium fractions (Fig. 1, top panel), the differences between the wild type and the variants were not marked unlike our earlier results in which clear differences were seen [36]; this may be because of the shortened cultivation time of the transfected cells in the MG-132 study (36 h instead of 72 h; see the Materials and methods section).
Effects of MG132 on the amyloidogenic lysozyme variants overexpressed in HEK293 cells. Wild, wild-type lysozyme; I56T etc., the four variants. Transfected cells were incubated for 24 h and further for 12 h with (+) or without (-) MG132, and fractionated into the lysates and pellets, which, together with the media, were subjected to western blotting for lysozyme. Mock, the cells transfected with vector alone; GAPDH, the GAPDH bands in the lysate fractions analyzed as an internal control; Nrf2 lanes, positive control by using NF-E2-related factor 2. Each of the numbers in the (+) MG132 lanes under the second and fourth panels represents the intensity ratio toward its (-) MG123 control.
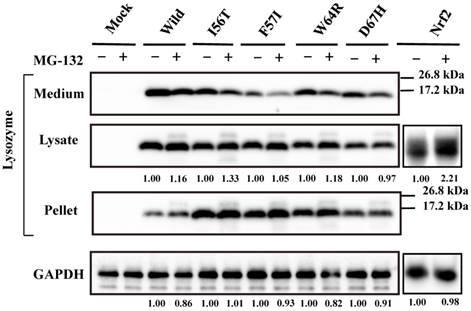
A new finding herein was that MG-132 produced little change in the band intensity compared to the respective control lanes for all specimens tested (Fig. 1; an alteration in the medium fraction of F57I was spontaneous). In the lysate fractions, the differences between with and without the inhibitor were only to a limited extent, from -3% to +18% (Fig. 1, second panel). We infer that the influence of the cellular degradation system on the expressed amyloidogenic lysozymes as well as the wild type was very small, if it existed at all. In other words, the lysozyme variants seemed to remain in the ER without entering the proteosome.
Our previous observation that the lysozyme variants were co-localized with GRP78/BiP raised the question of whether these variants were persistently associated or not with the chaperone. We therefore collected the lysate from cells wherein each vector of the lysozyme variants and that for FLAG-tagged GRP78/BiP had been co-transfected as described in Materials and methods. The products were subjected to co-immunoprecipitation with the anti-FLAG M2 Affinity Gel (referred to as “anti-FLAG gel” hereafter) and subjected to western blotting for lysozyme (Fig. 2A, second panel). Each of the variants afforded a marked band, whereas the wild-type specimen did not. We thus favor the idea that a stable association of lysozyme variants and GRP78/BiP had occurred. Other lanes including those marked as FLAG (blotting with anti-FLAG antibody) exhibited patterns as expected, indicating that the transfected genes were well expressed.
Binding assay of lysozyme and GRP78/BiP by co-immunoprecipitation. (A) HEK293 cells were incubated first for 24 h after being transfected with the vector for FLAG-tagged GRP78/BiP and then for 48 h after being transfected with the vector for the indicated lysozyme gene. Lysates were prepared in the presence of hexokinase and glucose as detailed in Materials and Methods, and treated with the anti-FLAG gel to obtain co-immunoprecipitated lysozyme-GRP fractions (IP:FLAG panels), which were analyzed by western blotting with anti-lysozyme antibody or the anti-FLAG antibody (marked as Lysozyme and FLAG, respectively). Lysates before immunoprecipitation were also subjected to western blotting (Lysate panels). (B) Experiments similar to those in A were repeated for I56T with or without adding 1 mM ATP in the lysis buffer. In this experiment, control series without tagged GRP78/BiP were also analyzed.
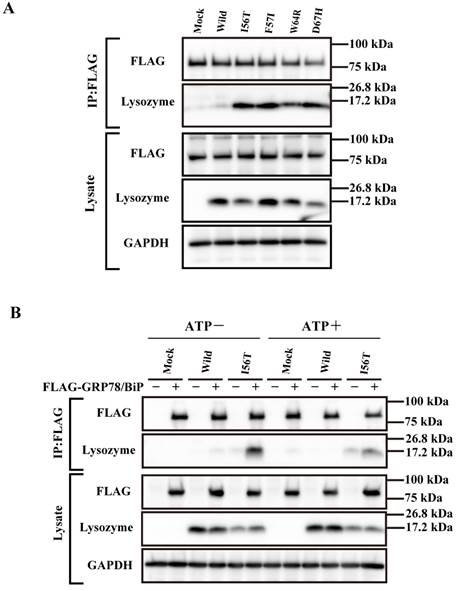
Next, we performed a similar co-expression study using one of the four amyloidogenic I56T variants in the presence or absence of ATP, since GRP is known to release the substrate when the amount of ATP is sufficient. As recognized in the western blots for lysozyme in the co-immunoprecipitated fractions obtained by the anti-FLAG gel, the l56T band was weaker in the ATP+ lanes than in the ATP- controls (Fig. 2B, second panel). Lysozyme signals were scarce in the other control series in which FLAG-GRP78/BiP vector was not added. It seemed that I56T and GRP78/BIP were once attached to each other and then detached by the addition of excess ATP.
Search for the region critical to the association with GRP78/BiP
What is the region that interacts with GRP78/BiP? To address this question, we constructed several types of lysozyme mutations, each with a deletion at the N-terminal, internal or C-terminal region as illustrated in Figure 3A, and we inserted the mutations into the expression vector (see Materials and Methods). The products were individually co-expressed with tagged GRP78/BiP as described above, and the synthesized proteins were subjected to western blotting for lysozyme.
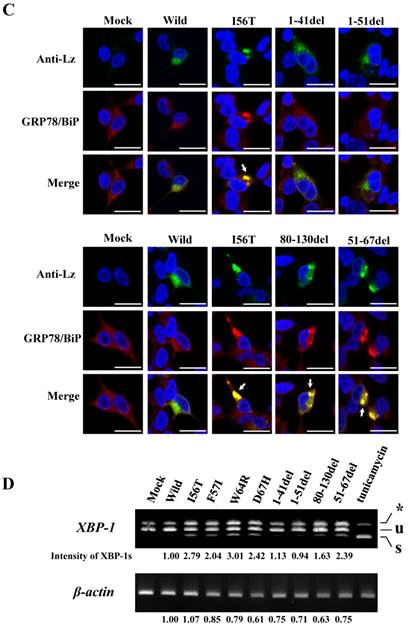
Search of possible GRP78/BiP-binding sites in lysozyme. (A) Schematic of prune mutants made from the wild type by truncating the N-terminal 1-41 and 1-51 residues, the C-terminal 80-130 residues, the internal 51-67 residues and the α1-helix (putative binding region to GRP78/BiP) 28-34 residues (the latter was used in the experiments shown in Fig. 5). The top image depicts the wild-type lysozyme. All of the constructs have the signal sequence of lysozyme. Broken line, deletion region; diagonal line, the core region of amyloid fibril formation; del, deleted residues. (B) Binding assay of the four derivatives (1-41del, 1-51del, 80-130del and 51-67del) to GRP78/BiP by co-immunoprecipitation. The mutants were each co-expressed with tagged in HEK293 cells (incubated for a total of 72 h) and fractionated into the lysates and pellets. The lysates were subjected to co-immunoprecipitation, and western blotting was done for lysozyme and FLAG. The cultivation media were also analyzed. Abbreviations are as in the legends to Figs. 1 and 2. (C) Co-localization of lysozyme and GRP78/BiP in HEK293 cells transfected with lysozyme (incubated for 72 h). The cells were fixed and double-labeled with Alexa 488-conjugated secondary IgG against anti-lysozyme antibody (green) and the anti-GRP78/BiP antibody (red), followed by confocal laser microscopy. Top panels, detection of lysozyme; middle panels, detection of GRP78/BiP; bottom panels, superimposed images. Scale bar: 20 μm. (D) RT-PCR of XBP1 for lysozyme expression. Transfected cells incubated for 72 h were used (see Materials and Methods). RT-PCR for XBP1 mRNA, where u, s, and * denote the unspliced, spliced, and combined forms. Bottom panel: internal control (β-actin). Numbers under each panel represent the s-band intensity ratios of the mutants to that of the wild type.
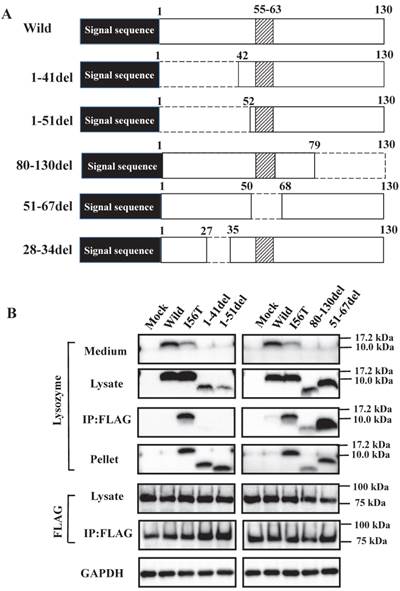
The bands in the cell lysate fractions were quite clear, although those of the N-terminal mutants 1-41del and 1-51del had slightly less intensity compared to the other specimens (Fig. 3B, second panels). All four of the deletion mutants tested here showed no signals in the culture media (Fig. 3B, top panels). The most noteworthy was the finding that the mutants pruned of the N-terminal region scarcely gave bands in the co-immunoprecipitated fractions obtained by the anti-FLAG gel (Fig. 3B, left third panel). This result implies that the N-terminal deletion mutants (1-41del and 1-51del) were without GRP78/BiP. The opposite situation was given by the C-terminal or core region pruned mutants (80-130del and 51-67del, respectively); these might have coexisted with GRP78/BiP as they exhibited definite bands in the co-immunoprecipitates (Fig. 3B, right third panel).
It should be noted here that the lysozyme proteins that lost N-terminal region gave intense signals in the pellet fractions (Fig. 3B, left fourth panel), although these proteins were believed to be free from GRP78/BiP. This can be attributed to self-aggregation into insoluble form.
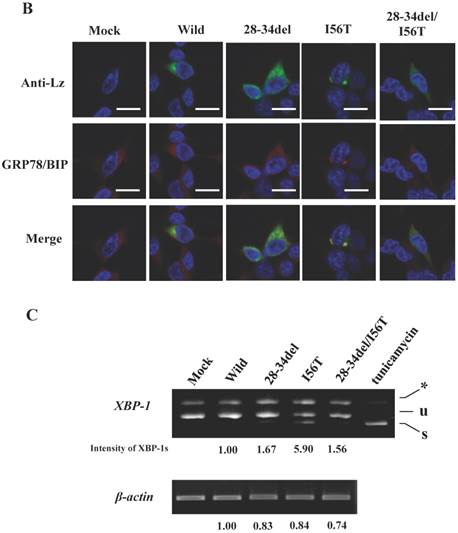
The same specimens were further subjected to immunocytochemical analyses. The intracellular localization positions of 51-67del and 80-130del as well as of those of I56T were in exact agreement with those of GRP78/BiP (Fig. 3C; arrows show fully merged regions). In contrast, the localization of the N-terminal 1-41 and 1-51 prune mutants as well as the wild-type lysozyme hardly matched that of GRP78/BiP (Fig. 3C, upper-half panels). These further verify the above results with respect to the association property toward GRP78/BiP.
The four amyloidogenic lysozyme variants were observed to elicit the splicing of XBP1 during the ER stress response [36]. We tested this point for the deletion mutants (Fig. 3D). The band of XBP-1s, the splicing product of XBP-1, was scarcely detected in the lanes for the N-terminal mutants (1-41del and 1-51del) as well as the lane for the wild-type lysozyme. However, the XBP-1s was observed in the lanes for the core region mutant and the C-terminal region mutant (51-67del and 80-130del, respectively) as well as in the lanes for the four amyloidogenic mutants. These data, together with those of the immunocytochemical study, coincided with the above-mentioned western blot patterns. We therefore conclude that, for the persistent association with GRP78/BiP, the N-terminal region of lysozyme is essential but its core and C-terminal regions are not.
To determine whether the current prune derivatives especially those deleted at the N-terminal 1-41 and 1-51 residues that were free from GRP78/BiP in the cells were affected by proteasomes or not, we treated the transfected cells with the proteasome inhibitor MG-132 followed by western blotting for lysozyme. As shown in the left second panel of Figure 4, the presence of the inhibitor caused a marked increase in band intensity for the N-terminal mutants, indicating that the proteins received the degradative activity. Little or no effects of MG-132 on the 80-130 and 51-67 deletion mutants were observed (Fig. 4, right second panel); these mutants have been shown to coexist with GRP78/BiP in the ER, and were thus detected in the pellet fractions (Fig. 4, right third panel). The banding patterns of the left third panel of Figure 4 might be attributed to self-aggregation as mentioned above (see Fig. 3B, left fourth panel).
Effects of MG132 on the truncated mutants of lysozyme overexpressed in HEK293 cells. 1-41del, etc., the deletion mutants shown in Fig. 3A. Transfected cells were incubated with (+) or without (-) MG132, fractionated and subjected to western blotting for lysozyme. Each number in the (+) MG132 lanes under the second and fourth panels shows the intensity ratio toward its (-) MG123 control. Details are as described in the Fig. 1 legend.
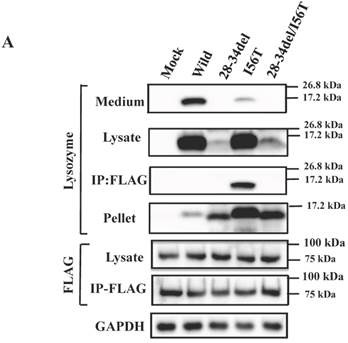
The preceding data indicated that the N-terminal region of lysozyme is imperative for the adhesion of GRP78/BIP. GRP78/BiP has been shown to interact with a redundant heptapeptide motif, Hy(W/X)HyXHyXHy, in which alternating large hydrophobic or aromatic amino acids (Hy) provide high affinity to the substrate domain of GRP78/BiP [40,41]. After searching for a sequence resembling the motif in the N-terminal region of lysozyme, we noticed a WMCLAKW heptapeptide at the 28-34 residues. We thus designed a prune mutation in which these seven amino acids were absent (see Fig. 3A, bottom), using both wild-type lysozyme and the I56T variant as starting materials. The prepared two constructs (28-34del and 28-34del/I56T) were used in co-expression experiments for lysozyme and tagged GRP78/BiP as described above.
The results clearly showed that, although the western blots for lysozyme were weak in the lysates, the two prune proteins were absent in the immunoprecipitated fractions eluted from the anti-FLAG gel (Fig. 5A, second and third panels, respectively). This indicated that the mutants were free from GRP78/BiP. The same conclusion was reached in the immunocytochemical study (Fig. 5B), where the two mutants and wild-type lysozyme gave equivocal patterns in contrast to I56T, which was reconfirmed to coexist with GRP78/BiP in narrow regions (also see Fig. 3C). Moreover, these deletion mutants barely promoted the splicing of XBP-1 (Fig. 5C), an important link of the IRE1-XBP1-GRP78/BiP pathway. The two mutants also resembled the above-mentioned four prune mutants in that these were not secreted into the media (Fig. 5A, top panel; cf. Figs. 3B and 4).
Biding assay to GRP78/BiP of lysozyme truncated at 28-34 residues of the wild type (28-34del) or of I56T (28-34del/I56T). (A) These were co-expressed with tagged GRP78/BiP in HEK293 cells, which were fractionated. The lysates were subjected to co-immunoprecipitation and western blotting was done for lysozyme and FLAG. See the Fig. 2B legend for details. (B) Localization of lysozyme proteins and GRP78/BiP. The co-transfected cells were fixed, double-labeled and carried to confocal laser microscopy (see the legend to Fig. 3C). Top panels: Detection of lysozyme. Middle panels: Detection of GRP78/BiP. Bottom panels: Superimposed images. Scale bar: 20 μm. (C) RT-PCR of XBP1 for lysozyme expression in the above-described transfected cells. RT-PCR for XBP1 mRNA, where u, s and * denote unspliced, spliced and combined form. See the Fig. 3D legend for further descriptions about this experiment.
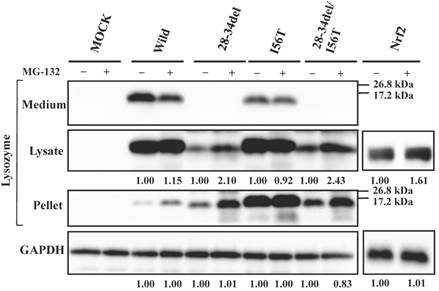
Since 28-34del and 28-34del/I56T were free from GRP78/BiP, these were expected to be sensitive to proteasome systems. We confirmed that this was the case by the expression experiments for lysozyme using MG-132 (Fig. 6). The band intensities were higher in the case of MG-132+ compared to the MG-132- control in the lysate fractions. In an additional co-expression study, the western blot bands of the deletion mutant proteins in the immunoprecipitated fractions obtained by the anti-FLAG gel did not differ between with and without MG-132 treatment (data not shown). All of these findings suggested that the N-terminal 28-34 residues of lysozyme are critical for the affinity to the ER chaperone.
Effects of MG132 on the core region-pruned lysozymes overexpressed in HEK293 cells. 28-34del, deletion mutant from the wild type; 28-34del/I56T, deletion mutant from I56T. Transfected cells were incubated with (+) or without (-) MG132, and fractionated and subjected to western blotting for lysozyme. See the Fig. 4 legend for details.
Effects of N-terminal hydrophobic residues of lysozyme on GRP78/BiP association
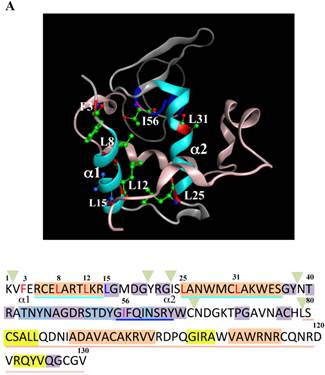
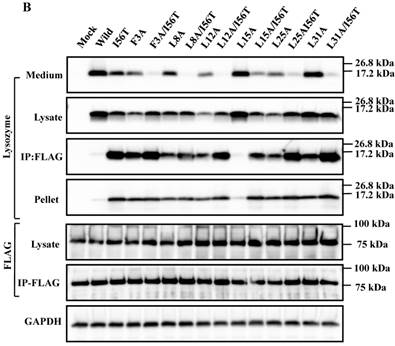
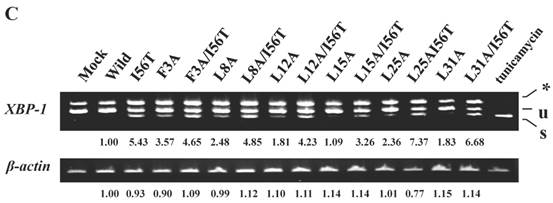
The 28-34 heptapeptide described above nearly equals the α2-helix (25-35 residues) of the lysozyme's N-terminal region (Fig. 7A). The α2-helix is paralleled by the α1-helix, and the two helices possess an array of hydrophobic amino acids (F3, L8, L12, L15, L25 and L31) with approximately identical intervals. I56 lies as if it is a member of the array. We tentatively assumed that such a structure works for the stable co-localization with GRP78/BIP. We therefore changed each of these aromatic amino acids to alanine in both the wild-type lysozyme and the I56T variant. The resulting mutant series were individually co-expressed with the vector for tagged GRP78/BiP, and the products were analyzed by western blotting. In the co-immunoprecipitated fractions obtained by the anti-FLAG gel, the mutants made from the wild type displayed very clear lysozyme bands (Fig. 7B, third panel, lanes for F3A, etc.), indicating that these were changed to have enhanced adhesion bias to GRP78/BiP in the cells. L15A was an exception, as it produced no signals similarly to the wild type. The I56T-based double substitution mutants were still like the original I56T (Fig. 7B, third panel, lanes for F3A/I56T, etc.). All of the current mutant specimens that were shown to be coexisting with GRP78/BiP exhibited enhanced XBP1 splicing (Fig. 7C), again indicating that the attachment of GRP accompanies the ER stress response via the IRE1-XBP pathway. In addition, the introduced mutations tended to exhibit impaired secretion ability as seen by lowered protein banding in the medium fractions compared to the controls (Fig. 7B, top panel).
Mutations at the hydrophobic amino acids in the α-helices region of lysozyme. (A) Six sites of single amino acid substitutions introduced here are expressed by red color plus one character/number in the 3D diagram (top) and by red alphabet in the sequence (bottom) of human lysozyme. I56 is colored magenta in both. The α1 and α2-helices (5-14 and 25-34, respectively) are colored and underlined by cyan in the 3D structure and the sequence, respectively. The C-terminal region (80-130) is colored flesh in the 3D diagram, and the core sequence of amyloid fibril formation (55-63) is colored blue in the sequence. Solid squares show the second structure (PDB: 1JWR, http://www.rcsb.org/pdb/explore/remediatedSequence.do?structureId=1JWR). Light brown, alpha- helix; yellow, 310-helix; light purple, turn; grey, beta- strand; green triangle, beta-bridge. (B) Binding assay of single-residue mutants to GRP78/BiP by co-immunoprecipitation. HEK293 cells were co-transfected with indicated lysozyme sequence and FLAG-tagged GRP78/BiP. F3A and F3A/I56T etc., the single-site substitution mutants made of the wild type and I56T, respectively. Co-immunoprecipitated fractions (IP:FLAG panels) were analyzed by western blotting with anti-lysozyme antibody or the anti-FLAG antibody. Further details are as described in the Fig. 2A legend. (C) RT-PCR of XBP1 for lysozyme expression in the cells transfected with the substitution mutants. Experiments were performed as described in the Fig. 3D legend. RT-PCR for XBP1 mRNA, where u, s and * denote unspliced, spliced and combined form.
Effects of I56 mutations on the association property with GRP78/BiP
I56, the mutation site of the variant I56T, is in the amyloidogenic core region. Here, I56 of wild-type lysozyme was replaced into different amino acids, and the products were individually co-expressed with tagged GRP78/BiP as described above. Substitution to leucine, valine and methionine of the locus did not extensively alter the wild-type lysozyme's negative associability toward GRP78/BiP or the positive secreting ability to the media. This was evidenced by the weak or no western blots for lysozyme in the co-immunoprecipitated fractions and strong blots in the medium fractions, respectively (Fig. 8A, third and first panels; I56L, I56V, I56M and Wild lanes).
Binding assay of mutations at the 56th residue in the amyloidogenic core region of lysozyme and GRP78/BiP. I56A, etc.: the single amino acid substitution mutants made of the wild type. (A) Co-immunoprecipitated fractions (IP:FLAG panels) were analyzed by western blotting with anti-lysozyme antibody or the anti-FLAG antibody (marked as Lysozyme and FLAG, respectively). Lysates before immunoprecipitation were also subjected to western blotting (Lysate panels). See the Fig. 2A legend for other descriptions. (B) Localization of selected lysozyme proteins in the GRP78/BiP-positive regions. Details of these procedures and abbreviations are as described in the Fig. 3C legend.
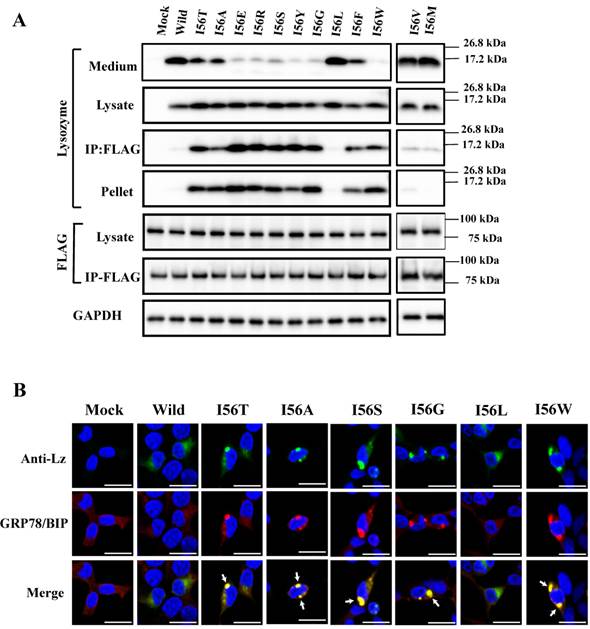
In contrast, the substitution to one of eight amino acids (alanine, glutamic acid, etc.) exploited in the present study changed the wild-type lysozyme into the I56T type, which displayed marked bands in the co-immunoprecipitated fractions (Fig. 8A, third panel; the lanes from I56T to I56W except for the I56L lane), indicating that these remain associated with GRP78/BiP in the cells. Whether these mutations were secreted into the media or not depended on the substituted amino acid (Fig. 8A, top panel).
The results obtained here for the I56 mutants are summarized by rough degrees in Table 1. Some of the I56 mutants were optically observed for the co-localization in the cytoplasm by the immunocytochemical method after co-expression with the GRP78/BiP vector. In accord with the western blots, I56L exhibited a pattern like that of the wild-type lysozyme whereas the patterns of I56A, I56S, I56G and I56W resembled that of I56T (Fig. 8B).
Comparison of associability with GRP78/BiP, in silico-predicted thermal stability and other properties among wild-type and I56-mutated human lysozyme.
| Lysozyme | Secretion to medium | Pelleting | Association to GRP78/BiP | Thermal stability | Property of amino acid 56 |
|---|---|---|---|---|---|
| Wild type 56I | +++ | ± | - | +++ | Hydrophobic |
| I56L* | +++ | - | - | ++ | = |
| I56V* | +++ | - | ± | + | = |
| I56M* | +++ | - | ± | + | = |
| I56A | ++ | +++ | +++ | - - - | = |
| I56G | - | +++ | +++ | - - - - | = |
| I56F | ++ | +++ | +++ | + | = |
| I56W | - | +++ | +++ | - - | = |
| I56T** | ++ | +++ | +++ | - - - | Hydrophilic |
| I56E | - | +++ | +++ | - - - | = |
| I56R | - | +++ | +++ | - - | = |
| I56S | - | +++ | +++ | - - - | = |
| I56Y | - | +++ | +++ | ++ | = |
I56L etc., lysozyme mutants denoted by intrinsic and substituted amino acids framing the residue number; *, those with wild-type like properties; **, that belongs to the so-called amyloidogenic variants. = is used as ditto mark. Columns 2, 3 and 4 are from the first, fourth and third panels, respectively, of Fig. 8A. Relative thermal stability was predicted by computer simulation (see text). The score shows rough degree, where +, intensive or stable; ±, subtle; -, negative or unstable. Also unused I56X mutants were simulated for the stability. When X was hydrophobic amino acid, most of the scores were ++ or +, and when X was hydrophilic one, most of the scores were - - - or - -. I67Y and I56C gave the score of ++.
These findings indicate that the character of the 56th amino acid of lysozyme affects the GRP78/BiP-lysozyme relationship, and this may mirror the folding state of the lysozyme proteins and in turn the difference in thermal stability. To investigate the latter point, we performed 'molecular modeling and simulation' to predict relative heat stability data of the single amino acid-substituted lysozymes by using the Molecular Operating Environment (MOE) program (Chemical Computing Group, Montreal, Canada). The results are expressed as a coarse evaluation and listed in parallel with the other properties (Table 1). The mutants that remain associated with GRP78/BiP in the cells were shown to be unstable (I56R, I56E, I56T, I56S, I56A, I56W and I56G), and the reverse was also likely (wild type, I56L, I56V and I56M). There were exceptions: I56Y and I56F showed rather higher marks for stability despite their association with the GRP. The pelleting tendency (i.e., insolubility) was in parallel with the associability toward GRP78/BiP (for exceptions see Figs. 3B and 5A). Secretion is the intrinsic behavior of wild-type lysozyme, and the mutants with negative scores for secretion tended to have low stability, although I56T, I56Y and I56A were exceptions. The wild-type lysozyme and the mutants I56L, I56L and I56M, which has the above properties that were similar to those of the wild type as summarized in Table 1, have a hydrophobic amino acid at the 56th residue; this might have significance related to the possible normal folding and secretion of lysozyme.
Discussion
We have reported the intracellular coexistence of the well-known amyloidogenic lysozyme variants I56T, F57I, W64R and D67H with GRP78/BiP, which is an ER-specific member of the HSP70 family molecular chaperones, while showing the occurrence of ER stress response [36]. In the present study, we further revealed that not all but many of the newly constructed lysozyme mutants were found to be associated with GRP78/BiP. Here, the strong adhesion was ascertained by co-immunoprecipitation of the mutants and the tagged GRP78/BiP by the anti-tag antibody. An accompanying ER stress response was also observed. Generally, aberrant proteins such as those misfolded or unfolded will meet their fate of aggregation or degradation in cells, as described in the Introduction. It is worth noting that the lysozyme variants and mutants that bound to GRP78/BiP in the cells did not undergo the degradation by the proteasome system. When GRP78/BiP was knocked down by RNAi, lysozyme variants in the pellets were greatly reduced (unpublished data). This supports the idea that the relevant proteins were carried to the degradation system under the insufficiency of GRP78/BiP.
The induction of aggresome formation as well as that of apoptosis was shown to be negative in the four amyloidogenic variants [36]. By contrast, the mutations pruned at the N-terminal did not match the descriptions; these were not bound with GRP78/BiP but tended to undergo aggregation and degradation (see Fig. 4). In addition, the secretion from the cells was partly or completely impaired in many of the newly constructed mutants. The results are summarized in Tables 1 and 2, in which the data are stated by rough degrees. The latter table also includes some of the results obtained in our previous work [36].
Comparison of associability with GRP78/BiP and other properties among wild-type, amyloidogenic variants, deletion mutants and substitution mutants of human lysozyme.
| Lysozyme | Degradation | Secretion to medium | Pelleting | Association to GRP78/BiP |
|---|---|---|---|---|
| Wild type | - | +++ | ± | - |
| I56T** | - | ++ | +++ | +++ |
| F57I** | - | ++ | +++ | +++ |
| W64R** | - | ++ | +++ | +++ |
| D67H** | - | ++ | +++ | +++ |
| 1-41del | +++ | - | +++*** | - |
| 1-51del | +++ | - | +++*** | - |
| 28-34del | + | - | +++*** | - |
| 28-34del/I56T | + | - | +++*** | - |
| 51-67del | ± | - | +++ | +++ |
| 80-130del | ± | - | +++ | ++ |
| F3A | n.d. | + | ++ | ++ |
| F3A/I56T | n.d. | - | ++ | +++ |
| L8A | n.d. | ++ | ++ | ++ |
| L8A/I56T | n.d. | - | ++ | +++ |
| L12A | n.d. | ++ | ++ | ++ |
| L12A/I56T | n.d. | - | ++ | +++ |
| L15A* | n.d. | +++ | - | - |
| L15A/I56T | n.d. | - | ++ | +++ |
| F25A | n.d. | + | ++ | ++ |
| F25A/I56T | n.d. | - | ++ | +++ |
| L31A | n.d. | ++ | ++ | ++ |
| L31A/I56T | n.d. | - | ++ | ++ |
I56T etc., lysozyme mutants denoted by intrinsic and substituted amino acids framing the residue number; F3A/I56T etc., those received two changes; *, that with wild-type like properties; **, the so-called amyloidogenic variants; ***, observations attributed to self-aggregation (see text). Columns 2 to 5 are from Figs.1 to 7 of the present study and from the data in our previous article [2015-BBA]. The score shows rough degree, where +, intensive; ±, subtle; -, negative. n.d., not determined.
The amyloidogenic variants in which the sites of single amino acid mutations are around the amyloidogenic core region (55-63 residues, see Fig. 3A) at the β-domain produce no regularity of amino acid features, e.g., in hydrophobicity. The steric structure of I56T revealed little difference in comparison to the wild type [PDB; 3FE0 and 1LOZ], whereas the D67H variant has greatly changed its configurational features [42]. Thus, the implication of the difference of steric structure in the core region for the association propensity with GRP78/BiP was unclear. In the association with the chaperone, partially folded state of lysozyme variants might have involvement. Such problems are accessible via the secondary structure prediction of the variants, although these remain to be elucidated.
The fate of lysozyme variants in the cells may not be determined only by the type of amino acids at the critical sites. For instance, the production of the human lysozyme variants F57I and W64R in the yeast Pichia pastoris was 1/300 and 1/30 less than that of wild-type lysozyme, respectively [43]. As the mRNA levels and the protein secretion of the variants were comparable to those of the wild type, the reduction of protein was ascribed to the yeast PQC system. Such observations were reproducible for I56T in the experiments performed with the yeast system during our study (Supplementary Figure S1). In this case, the reduction degree of protein production was less than 1/100 of the wild type. These variant human proteins have been shown to exist without degradation in human cells. The discrepancy may be explained by the notion that protein folding depends on the cellular environment, which might differ from species to species.
In the present study, we focused on the search for the site of lysozyme that is indispensable to the stable association with GRP78/BiP. Deletion mutants showed that it was the N-terminal region of lysozyme. We further recognized in the N-terminal region of lysozyme a heptapeptide stretch that we speculate is the motif possessing high affinity to GRP78/BiP. In support of this speculation, lysozyme mutants pruned of these seven amino acids was found free from the GRP78/BiP. This heptapeptide resides in the α2-helix, which, in concert with the α1-helix, makes a regular array of hydrophobic/aromatic amino acids (see Fig. 7A). Replacing one of these amino acids with alanine often altered the wild-type lysozyme into those like the amyloidogenic variants, steadily associated with GRP78/BiP (Table 1). We infer that the position of the helices α1 and α2 might be an entrance for GRP78/BiP, which monitors the proper folding of normal lysozyme. Mutations such as I56T, i.e., hydrophobic/hydrophilic substitution, would probably prevent the dissociation of protein from the ER chaperone. It is of interest why the L15A mutant had wild-type like properties, i.e., not associable with GRP78/BiP and secreted into the medium. The L15 residue is at the bottom position between the helices α1 and α2 as seen in Fig. 7A and probably with less implications in the association with the chaperone.
Reports from other laboratories have indicated that amylotrophic lateral sclerosis-causing mutants of human copper/zinc superoxide dismutase-1 (SOD1) with altered folding patterns make aggregates inside the ER in cultured cells or transgenic mice, wherein these are bound to GRP78 accompanied by the induced ER stress response [44-46]. Interestingly, SOD1 is a cytoplasmic enzyme but its mutations seem to stimulate the ER stress. In addition, a human amyloidogenic mutant of transthyretin was shown to be stably bound to GRP78 in Escherichia coli [47] and in mammalian cells [48], but dissociated from it by the addition of ATP. When GRP78 was down-regulated the mutant protein was degraded, implying that GRP78 protects the aggregate-prone proteins from degradation in the cells.
Transthyretin's binding peptide to GRP78 was confirmed to be 88-103 residues comprising the β-strand F, which has been suggested as a possible elongation region of amyloid fibrils [47,49]. There is a similarity in the situations of these two enzymes and lysozyme. The strong affinity of the ER chaperone to abnormal proteins might arrest their secretion and in turn lower the circulation of aberrant proteins in vivo. This might at least partly explain why it takes a long period of time for systemic amyloidosis to develop in vivo.
The subsequent step of our experiment was to make a mutant series in which the isoleucine at the 56th residue in the core region of the α-domain was replaced with another amino acid. When substituted with a hydrophobic amino acid, some of the products remained as the normal type, exhibiting dissociation from GRP78/BiP, along with a marked secretion property and no pellet formation (3 of 7 specimens; Table 1). In contrast, substitution with a hydrophilic amino acid changed lysozyme I56T-like with propensities essentially in contrast to those of the wild type (all 4 samples; Table 1). It is possible that the lowering in hydrophobicity at the α-helical domain brings about insufficient or abnormal folding, and this in turn causes the strong retention of lysozymes with GRP78/BiP. As described, some mutants were exceptional to the rule, e.g., I56A and I56T showed secretion ability but were associated with GRP78/BiP. Whether these inconsistent results may be explained by the features of folding state is unclear without additional experiments.
Such lysozyme mutants that have rigid affinity to GRP78/BiP were considered to have lowered thermal stability. This notion was supported by the computational study to calculate the relative stability indices for the I56 mutants (Table 1). The results of in silico simulation of thermostability were reported to be in agreement with biochemical values (43). The wild-type lysozyme has the highest thermal stability among the specimens tested, and this may indicate that the wild-type protein undergoes proper folding, which is considered to be a key to complete the steric structure of the β-domain containing the amyloidogenic core region. However, how and to what extent the latter process is monitored by GRP78/BiP remains to be determined.
On the whole, a significant finding of the present study is that the site of mutant lysozyme necessary for the association with GRP78/BiP was the N-terminal region, where an array of hydrophobic amino acids in the α1 and α2-helices domain was shown to be crucial. Interestingly, the α2-helix is superimposed with the putative heptapeptide motif, reminiscent of the common degenerate GRP-biding sequences, although definite conclusions can only be drawn after the identification of the bound peptide region in the complex.
There have been many studies of the involvement of the amyloidogenic lysozyme variants in diseases since the first report in 1993 about the presence of I56T and D67H in humans [30]. Such variants are single-point mutations and are inherited in an autosomal dominant manner [32]. Even wild-type lysozyme is intrinsically amyloidogenic as mentioned above; however, incompletely folded states of wild-type protein are very rare in the human body [29], probably because proteins with aberrant structures are alleviated by the PQC system under healthy conditions. Lysozyme variants incline to the amyloid fibril formation despite such a mitigation process [31].
At present, there is no specific therapy for amyloidosis; patients are managed only symptomatically [30]. Further research is thus needed to elucidate the more detailed structural features of lysozyme its mutants in particular. In 2014, nine pathogenic mutations of human lysozyme were reported [50]; these include the newer members D68G, T70N, W82R and W112R. The mutants artificially prepared in the present study may also be of use in the future, if they have a differential amyloidogenic nature which has not been confirmed thus far.
Supplementary Material
Supplementary Figure S1.
Abbreviations
ER, endoplasmic reticulum; ERAD, ER-associated degradation; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; GRP78/BiP, glucose regulated protein 78/binding immunoglobulin protein; Hsp, heat shock protein; IRE1, inositol-requiring enzyme-1; Nrf2, NF-E2-related factor 2 (nuclear factor erythroid 2-related factor 2); PCR, polymerase chain reaction; PQC system, protein quality control system. XBP1, X-box binding protein 1, XBP1u, unspliced XBP1 transcript; XBP1s, spliced XBP1 transcript.
Acknowledgements
This work was supported in part by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (#25450476).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Blennow K, de Leon MJ, Zetterberg H. Alzheimer's disease. Lancet. 2006;368:387-403
2. Lynch MA. The impact of neuroimmune changes on development of amyloid pathology; relevance to Alzheimer's disease. Immunology. 2014;141:292-301
3. El-Kaissi S, Sherbeeni S. Pharmacological management of type 2 diabetes mellitus: an update. Curr Diabetes Rev. 2011;7:392-405
4. Pinney JH, Hawkins PN. Amyloidosis. Ann Clin Biochem. 2012;49:229-41
5. Valastyan JS, Lindquist S. Mechanisms of protein-folding diseases at a glance. Dis Model Mech. 2014;7:9-14
6. Gregersen N, Bolund L, Bross P. Protein misfolding, aggregation, and degradation in disease. Mol Biotechnol. 2005;31:141-50
7. Chatani E, Imamura H, Yamamoto N, Kato M. M. Kato, Stepwise organization of the β-structure identifies key regions essential for the propagation and cytotoxicity of insulin amyloid fibrils. J Biol Chem. 2014;289:10399-410
8. Gsponer J, Vendruscolo M. Theoretical approaches to protein aggregation. Protein Pept Lett. 2006;13:287-93
9. Lindgren M, Hammarström P. Amyloid oligomers: spectroscopic characterization of amyloidogenic protein states. FEBS J. 2010;277:1380-88
10. Kajava AV, Steven AC. Beta-rolls, beta-helices, and other beta-solenoid proteins. Adv Protein Chem. 2006;73:55-96
11. Chakrabarti A, Chen AW, Varner JD. A review of the mammalian unfolded protein response. Biotechnol Bioeng. 2011;108:2777-93
12. Tyedmers A, Mogk B, Bukau B. Cellular strategies for controlling protein aggregation. Nat Rev Mol Cell Biol. 2010;11:777-88
13. Ruggiano A, Foresti O, Carvalho P. Quality control: ER-associated degradation: protein quality control and beyond. J Cell Biol. 2014;204:869-79
14. FU XL, Gao DS. Endoplasmic reticulum proteins quality control and the unfolded protein response: the regulative mechanism of organisms against stress injuries. Biofactors. 2014;40:569-85
15. Hoozemans JM, Scheper W. Endoplasmic reticulum: The unfolded protein response is tangled in neurodegeneration. Int J Biochem Cell Biol. 2012;44:1295-98
16. Yang L, Zhao D, Ren J, Yang J. Endoplasmic reticulum stress and protein quality control in diabetic cardiomyopathy. Biochim Biophys Acta. 2015;1852:209-18
17. Kopito R. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000;10:524-30
18. Wolff S, Weissman JS, Dillin A. Differential scales of protein quality control. Cell. 2014;157:52-64
19. Gregeren N, Bross P. Protein misfolding and cellular stress: an overview. Methods Mol Biol. 2010;648:3-23
20. Wu H, Ng BS, Thibault G. Endoplasmic reticulum stress response in yeast and humans. Biosci Rep. 2014;34(4):e00118
21. Chen T, Brandizzi F. IRE1: ER stress sensor and cell fate executor. Trends Cell Biol. 2013;23:547-55
22. Yoshida H, Uemura A, Mori K. pXBP1(U), a negative regulator of the unfolded protein response activator pXBP1(S), targets ATF6 but not ATF4 in proteasome-mediated degradation. Cell Struct Funct. 2009;34:1-10
23. Sano R, Reed JC. ER stress-induced cell death mechanisms. Biochim Biophys Acta. 2013;1833:3460-70
24. Martino MB, Jones L, Brighton B, Ehre C, Abdulah L, Davis CW, Ron D, O'Neal WK, Ribeiro CMP. The ER stress transducer IRE1β is required for airway epithelial mucin production. Mucosal Immunology. 2013;6:639-54
25. Imagawa Y, A. Hosoda A, Sasaka S, Tsuru A, Kohno K. RNase domains determine the functional difference between IRE1alpha and IRE1beta. FEBS Lett. 2008;582:656-60
26. Clarke R, Cook KL, Hu R, Facey CO, Tavassoly I, Schwartz JL, Baumann WT, Tyson JJ, Xuan J, Wang Y, Wärri A, Shajahan AN. Endoplasmic reticulum stress, the unfolded protein response, autophagy, and the integrated regulation of breast cancer cell fate. Cancer Res. 2012;72:1321-31
27. Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881-91
28. Tsai YL, Zhang Y, Tseng CC, Stanciauskas R, Pinaud F, Lee AS. Characterization and mechanism of stress-induced translocation of 78-kilodalton glucose-regulated protein (GRP78) to the cell surface. J Biol Chem. 2015;290:8049-64
29. Morozova-Roche LA, Zurdo J, Spencer A, Noppe W, Receveur V, Archer DB, Joniau M, Dobson CM. Amyloid fibril formation and seeding by wild-type human lysozyme and its disease-related mutational variants. J Struct Biol. 2000;130:339-51
30. Pepys MB, Hawkins PN, Booth DR, Vigushin DM, Tennent GA, Soutar AK, Totty N, Nguyen O, Blake CC, Terry CJ. et al. Human lysozyme gene mutations cause hereditary systemic amyloidosis. Nature. 1993;362:553-57
31. Canet D, Sunde M, Last AM, Miranker A, Spencer A, Robinson CV, Dobson CM. Mechanistic studies of the folding of human lysozyme and the origin of amyloidogenic behavior in its disease-related variants. Biochemistry. 1999;38:6419-27
32. Sattianayagam PT, Gibbs SD, Rowczenio D, Pinney JH, Wechalekar AD, Gilbertson JA, Hawkins PN, Lachmann HJ, Gillmore JD. Hereditary lysozyme amyloidosis — phenotypic heterogeneity and the role of solid organ transplantation. J Int Med. 2012;272:36-44
33. Frare E, Mossuto MF, Polverino de Laureto P, Dumoulin M, Dobson CM, Fontana A. Identification of the core structure of lysozyme amyloid fibrils by proteolysis. J Mol Biol. 2006;361:551-61
34. Tokunaga Y, Sakakibara Y, Kamada Y, Watanabe K, Sugimoto Y. Analysis of core region from egg white lysozyme forming amyloid fibrils. Int. J Biol Sci. 2013;9:219-27
35. Tokunaga Y, Matsumoto M, Sugimoto Y. Amyloid fibril formation from a 9 amino acid peptide, 55th-63rd residues of human lysozyme. Int. J Biol Macromol. 2015;80:208-16
36. Kamada Y, Kusakabe T, Sugimoto Y. Amyloidogenic lysozymes accumulate in the endoplasmic reticulum accompanied by the augmentation of ER stress signals. Biochim Biophy Acta. 2015;1850:1107-19
37. Hurtley SM, Bole DG, Hoover-Litty H, Helenius A, Copeland CS. Interactions of misfolded influenza virus hemagglutinin with binding protein (BiP). J Cell Biol. 1989;108:2117-26
38. Gething MJ. Role and regulation of the ER chaperone BiP. Semin. Cell Dev Biol. 1999;10:465-72
39. Maiuolo J, Bulotta S, Verderio C, Benfante R, Borgese N. Selective activation of the transcription factor ATF6 mediates endoplasmic reticulum proliferation triggered by a membrane protein. Proc Natl Acad Sci. USA. 2011;108:7832-37
40. Blond-Elguindi S, Cwirla SE, Dower WJ, Lipshutz RJ, Sprang SR, Sambrook JF, Gething MJ. Affinity panning of a library of peptides displayed on bacteriophages reveals the binding specificity of BiP. Cell. 1993;75:717-28
41. Knarr G, Modrow S, Todd A, Gething MJ, Bunchner J. BiP-binding sequences in HIV gp160, Implications for the binding specificity of bip. J Biol Chem. 1999;274:29850-57
42. Chamberlain AK, Receveur V, Spencer A, Redfield C, Dobson CM. Characterization of the structure and dynamics of amyloidogenic variants of human lysozyme by NMR spectroscopy. Protein Sci. 2001;10:2525-30
43. Kumita JR, Johnson RJ, Alcocer MJ, Dumoulin M, Holmqvist F, McCammon MG, Robinson CV, Archer DB, Dobson CM. Impact of the native-state stability of human lysozyme variants on protein secretion by Pichia pastoris. FEBS J. 2006;273:711-20
44. Tobisawa S, Hozumi Y, Arawaka S, Koyama S, Wada M, Nagai M, Aoki M, Itoyama Y, Goto K, Kato T. Mutant SOD1 linked to familial amyotrophic lateral sclerosis, but not wild-type SOD1, induces ER stress in COS7 cells and transgenic mice. Biochem Biophys Res Commun. 2003;303:496-03
45. Wate R, Ito H, Zhang JH, Ohnishi S, Nakano S, Kusaka H. Expression of an endoplasmic reticulum-resident chaperone, glucose-regulated stress protein 78, in the spinal cord of a mouse model of amyotrophic lateral sclerosis. Acta Neuropathol. 2005;110:557-62
46. Kikuchi H, Almer G, Yamashita S, Guégan C, Nagai M, Xu Z, Sosunov AA, McKhann GM 2nd, Przedborski S. Spinal cord endoplasmic reticulum stress associated with a microsomal accumulation of mutant superoxide dismutase-1 in an ALS model. Proc Natl Acad Sci USA. 2006;103:6025-30
47. Sörgjerd K, Ghafouri B, Jonsson BH, Kelly JW, Blond SY, Hammarström P. Retention of misfolded mutant transthyretin by the chaperone BiP/GRP78 mitigates amyloidogenesis. J Mol Biol. 2006;356:469-82
48. Susuki S, Sato T, Miyata M, Momohara M, Suiko MA, Shuto T, Ando Y, Kai H. The endoplasmic reticulum-associated degradation of transthyretin variants is negatively regulated by BiP in mammalian cells. J Biol Chem. 2009;284:8312-21
49. Suzuki Y, Minami M, Suzuki M, Abe K, Zenno S, Tsujimoto M, Matsumoto K, Minami Y. The Hsp90 inhibitor geldanamycin abrogates colocalization of eIF4E and eIF4E-transporter into stress granules and association of eIF4E with eIF4G. J Biol Chem. 2009;284:35597-604
50. Jean E, Ebbo M, Valleix S, Benarous L, Heyries L, Grados A, Bernit E, Grateau G, Papo T, Granel B, Daniel L, Harlé JR, Schleinitz N1. A new family with hereditary lysozyme amyloidosis with gastritis and inflammatory bowel disease as prevailing symptoms. BMC Gastroenterol. 2014;14:156-61
Author contact
![]() Corresponding author: Yasushi Sugimoto: yasushiagri.kagoshima-u.ac.jp Tel: 81+ 99-285-8781 Fax: 81+99-285-8799
Corresponding author: Yasushi Sugimoto: yasushiagri.kagoshima-u.ac.jp Tel: 81+ 99-285-8781 Fax: 81+99-285-8799