Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2016; 12(4):381-388. doi:10.7150/ijbs.12947 This issue Cite
Research Paper
Targeting Oncogenes into a Defined Subset of Mammary Cells Demonstrates That the Initiating Oncogenic Mutation Defines the Resulting Tumor Phenotype
Kimberly R. Holloway1*, Vidya C. Sinha1,2*, Wen Bu1,2, Michael Toneff2, Jie Dong2, Yi Peng3,4, Yi Li1,2 ![]()
1. Lester & Sue Smith Breast Center, Baylor College of Medicine, Houston, TX, USA;
2. Department of Molecular and Cellular Biology, Baylor College of Medicine, Houston, TX, USA.
3. National Center for International Research of Biological Targeting Diagnosis and Therapy, Guangxi Medical University, Nanning, Guangxi 530021, China;
4. Guangxi Key Laboratory of Biological Targeting Diagnosis and Therapy Research, Guangxi Medical University, Nanning, Guangxi 530021, China.
* These two authors contributed equally to this work.
Received 2015-6-12; Accepted 2015-11-3; Published 2016-2-5
Abstract

Breast cancers exhibit high intertumoral heterogeneity in genetic alterations as well as histopathological and other phenotypic characteristics. The contribution of the initiating oncogenic mutation to tumor phenotype remains controversial, largely due to the technical difficulties in delivering genetic alterations into well-defined subsets of mammary epithelial cells. To examine how different initiating oncogenes drive tumor phenotype, we somatically delivered two oncogenes (ErbB2, PyMT) into a narrow and distinct subset of the mouse mammary epithelium defined by the expression of the progenitor marker keratin 6a (Krt6a), and compared the phenotypes of the resulting mammary tumors. While PyMT-induced tumors were well-differentiated and displayed glandular and papillary features, ErbB2-induced tumors were poorly differentiated and exhibited epithelial-to-mesenchymal transition as well as β-catenin activation. These in vivo data demonstrate that the initiating oncogene plays a key role in driving mammary tumor phenotype.
Keywords: Keratin 6a, PyMT, ErbB2, Breast Cancer, RCAS, tva
Introduction
The heterogeneity of breast cancer has been well-established by several key studies that have examined tumor histology, biomarkers, gene signature, and pathophysiology [1, 2]. While the exact determinants of tumor phenotype have yet to be defined, tumor heterogeneity may be explained (at least in part) by the nature of the initiating oncogenic mutation [3-6] and/or the cellular origin of the tumor (recently reviewed in [6-8]). Our group has previously reported that somatic induction of mammary tumors by distinct oncogenes (PyMT and ErbB2) from the MMTV-expressing population [9] gives rise to tumors of distinct histological phenotypes [9, 10]. Specifically, we found that the PyMT oncogene led to tumors of papillary/tubular appearance, while ErbB2-driven tumors exhibited a typical solid nodular neu-type [11] appearance.
While the above studies suggest that tumor heterogeneity can be driven by the initiating oncogene, a recurring caveat in these studies is that the mammary epithelial population to which the oncogenes were introduced is a broadly heterogeneous one. It is possible that the tumor heterogeneity observed was due not purely to the initiating oncogene but also to preferential transformation of different cell types by different oncogenes. As a result, we remain unable to distinguish between the contributions to tumor phenotype of cell-of-origin versus initiating oncogene.
To identify unique tumor features arising as a consequence of the initiating oncogene, we utilized the RCAS-tva strategy of mammary tumor induction previously described [9]. The oncogene of interest (either constitutively-activated ErbB2 or PyMT) was introduced into a narrow, well-defined population of mammary epithelial cells to ensure that the cell of origin for all tumors is as similar as possible. In this particular study, the cell of origin we utilized is a very small subpopulation of mammary epithelial cells expressing the cytokeratin 6a protein. The reasons for this choice were twofold: Firstly, this Krt6a-positive subpopulation is a well-defined population that comprises a very small fraction (<1%) of mammary luminal epithelial cells, presumably consisting of a single cell type. Secondly, the Krt6a-positive subpopulation has been recently reported by our group to mark progenitor cells of the mammary gland [12]. This is of particular interest due to the growing body of evidence that points toward stem and progenitor cells as being the origin of various cancers (reviewed in [6]).
We previously reported that PyMT-driven tumors arising from the Krt6a-positive population led to tumors of papillary appearance, resembling normal-like human breast cancer [12]. It remains unclear whether this phenotype is driven primarily by the PyMT oncogene or by the Krt6a-positive cell-of-origin. To address this question, we compare the effect of ErbB2 on the phenotype of tumors arising from the Krt6a-population and report that ErbB2-tumors arising from this Krt6a-positive subpopulation exhibit a tumor phenotype that is completely distinct from that of PyMT-tumors. These findings suggest that the initiating oncogene can indeed drive distinct tumors from the same cell of origin.
Results
PyMT gives rise to papillary tumors while ErbB2 gives rise to solid nodular tumors
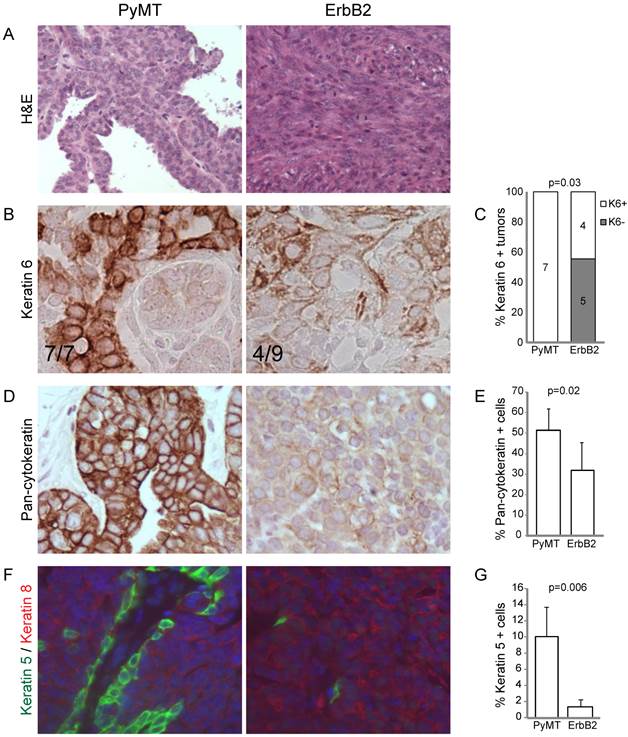
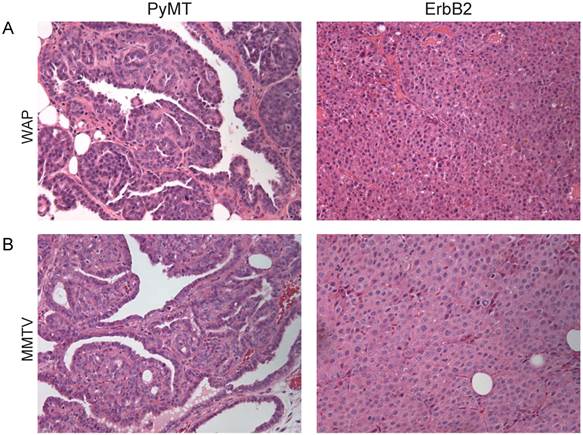
We previously reported that tumors induced from the Krt6a-positive population by the RCAS-PyMT oncogene were papillary and cystic in appearance, with ample spaces between the papillary fingers and resembled human normal-like breast cancer based on their molecular signature (Figure 1A, left panel; [12]). To determine whether these phenotypic features were determined primarily by cell-of-origin versus the PyMT oncogene, we evaluated the effect of a second oncogene, ErbB2, on the phenotype of tumors arising from the same cell population. Interestingly, ErbB2-induced tumors displayed vastly different features from PyMT-induced tumors. ErbB2 tumors exhibited dense (solid/nodular) epithelial architecture (Figure 1A, right panel), as is typical for ErbB2 tumors induced from the general mammary epithelium [11]. In addition, ErbB2-induced tumors arising from the Krt6a-positive population exhibited spindloid metaplasia (Figure 1A, right panel), which was absent in PyMT-induced tumors. To determine the extent to which these phenotypic differences are driven by the initiating oncogenes versus the cell-of-origin, we compared tumors induced by the PyMT or ErbB2 oncogenes from the small population of WAP-positive alveolar progenitors and found similar results; namely, PyMT tumors induced from the WAP-positive population appeared papillary/tubular, while ErbB2 tumors exhibited a typical solid nodular appearance (Figure 2A). We observed similar results when inducing tumors from the general MMTV-positive population (Figure 2B). Together, these findings reveal that, despite being targeted to the identical cell population within the mammary epithelium, different oncogenes can give rise to tumors of different histopathologic features. Furthermore, our findings suggest that the primary determinant of tumor histology, at least for our model system, is not the cell of origin but, rather, the initiating oncogene.
RCAS-ErbB2 induces solid, poorly differentiated tumors with spindloid metaplasia. (A) Based on hematoxylin and eosin staining, histopathological comparison between mammary tumors arising from Krt6a-tva mice infected with RCAS-PyMT (107 IU, 5 weeks; n= 8) and RCAS-ErbB2 (107 IU, 5 weeks; n = 8). (B) Keratin 6 was measured via immunohistochemical staining of tumors induced by PyMT (n = 7) and ErbB2 (n = 8), with (C) quantitation of Krt6a-positivity, defined by presence of Krt6a-positive cells in the majority (≥3) of 5 random fields. (D) Pancytokeratin was measured via immunohistochemical staining of tumors induced by PyMT (n = 7) and ErbB2 (n = 6), with (E) quantitation of the mean percentage of pancytokeratin+ cells in PyMT and ErbB2 induced tumors. (F) Keratins 5 and 8 were measured by immunofluorescence staining of tumors induced by PyMT (n = 7) and ErbB2 (n=10), with (G) quantitation of the mean percentage of Krt5+ cells in PyMT and ErbB2 induced tumors.
RCAS-ErbB2 induces tumors are less differentiated compared to RCAS-PyMT induced tumors. Based on hematoxylin and eosin staining, histopathological comparison between mammary tumors arising from (A) WAP-tva and (B) MMTV-tva mice infected with RCAS-PyMT or RCAS-ErbB2.
ErbB2 tumors have decreased markers of epithelial differentiation versus PyMT tumors
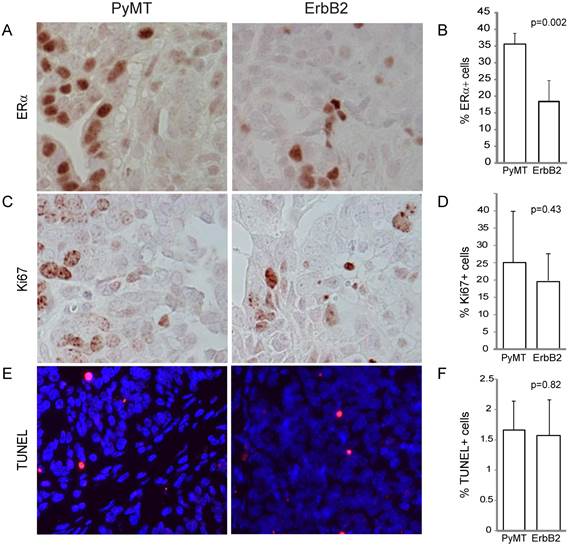
To gain further insight on the effect of different oncogenes on tumor pathology, we compared the effect of PyMT versus ErbB2 on common indices of tumor pathophysiology, focusing primarily on tumors induced from the Krt6a-positive population. We quantified the estrogen receptor (ER) expression level of both tumor types and found that while both ErbB2- and PyMT-induced tumors expressed ER in greater than 10% of tumor cells, PyMT-induced tumors exhibited a two-fold increased ER-positivity versus ErbB2-induced tumors (p=0.002; Figure 3A, B), possibly a corollary of the more-differentiated phenotype of PyMT tumors. Proliferation (assayed by Ki67 staining) was comparable in both groups (p=0.43; Figure 3C, D), as was apoptotic index (assayed by TUNEL, p=0.82; Figure 3E, F).
RCAS-ErbB2-induced tumors consist of lower estrogen receptor alpha levels, while proliferation and apoptosis within PyMT- and ErbB2- induced tumors are similar. (A) Estrogen receptor was measured via immunohistochemical staining of tumors induced by PyMT (n = 8) and ErbB2 (n = 10), with (B) quantitation of mean percentage of ER+ cells in the tumors. (C) Ki67 immunohistochemical staining indicating proliferation was measured between PyMT (n = 7) and ErbB2 (n = 10) induced tumors, with (D) quantitation. (E) TUNEL staining indicating apoptosis was measured between PyMT (n = 8) and ErbB2 (n = 9) induced tumors, with (F) quantitation.
Next, we examined the degree of differentiation in tumors using a panel of cytokeratin markers. Since tumors were originally induced from Krt6a-expressing progenitor cells, we examined the degree to which Krt6a-positivity was retained in the subsequent tumor cells and found that ErbB2 tumors had significantly decreased Krt6a-positivity versus PyMT tumors (p=0.03; Figure 1B, C), suggesting a change in the differentiation status of tumor cells over the course of ErbB2-induced tumor initiation and progression. To explore this possibility, we examined tumors using pancytokeratin, luminal marker Krt8 and basal marker Krt5. We found that, compared to PyMT tumors, ErbB2 tumors had decreased overall pancytokeratin, based on both intensity of staining and frequency of positive staining (p=0.02; Figure 1D, E), suggesting altered epithelial status in ErbB2 tumors. Indeed, the bulk of cells in ErbB2 tumors were either cytokeratin-negative or Krt8-positive, with a stark loss of basal Krt5 expression (versus PyMT tumors, p=0.006, Figure 1F, G). In contrast, PyMT tumors exhibited luminal-basal expression similar to that of the differentiated mammary gland; namely monolayers of Krt5-positive cells underlying Krt8-positive luminal cells. Taken together, these data suggest that while the PyMT oncogene allows differentiation of Krt6 progenitor cells-of-origin to differentiate into both luminal and basal epithelial cells, the ErbB2 oncogene may limit or alter epithelial differentiation.
ErbB2 tumors exhibit increased EMT and Wnt signaling compared to PyMT tumors
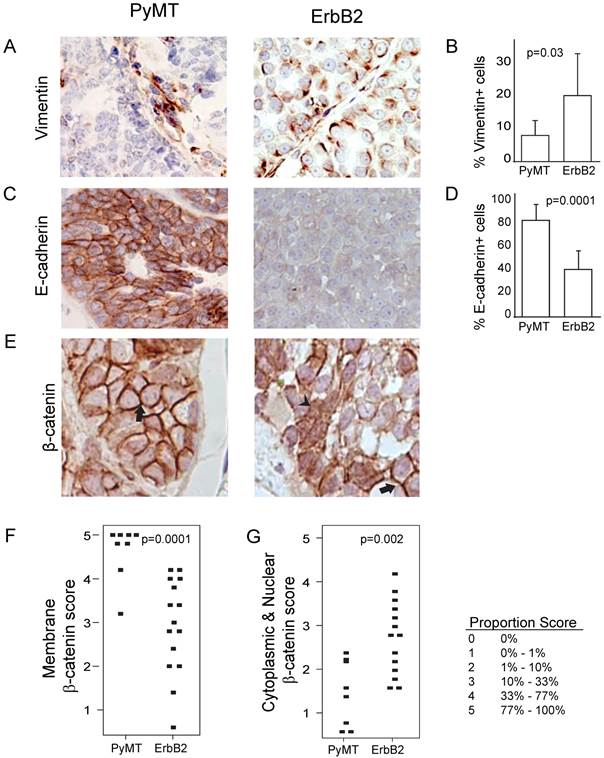
Because of the increased spindloid metaplasia and decreased cytokeratin expression in ErbB2-induced tumors (Figure 1), we next asked whether this phenotype was associated with increased epithelial-to-mesenchymal transition (EMT) events. We found that ErbB2 tumors exhibited increased vimentin (p=0.03; Figure 4A, B) and decreased E-cadherin (p=0.001, Figure 4C, D). Furthermore, we detected in ErbB2 tumors decreased membrane-bound β-catenin (p=0.0001; Figure 4E, F), consistent with induction of EMT. Since membrane-bound β-catenin was decreased in ErbB2-tumors, we wished to determine whether these tumors displayed a concomitant increase in nuclear β-catenin, suggestive of an active Wnt signaling pathway. Indeed, we observed significantly increased nuclear β-catenin in ErbB2 versus PyMT tumors (p=0.002; Figure 4G). Given the well-reported synergy between ErbB2 and Wnt signaling in promoting tumor progression [13-17], it is possible that the contrasting tumor phenotypes we observed in ErbB2 versus PyMT tumors (including loss of glandular architecture, altered epithelial status, and elevated EMT) are driven in part by differences in Wnt pathway activity.
ErbB2 tumors exhibit increased EMT and Wnt signaling compared to PyMT tumors. (A, B) Vimentin expression via immunohistochemistry staining in PyMT (n = 7) and ErbB2 (n = 8) induced tumors. (C, D) E-cadherin expression via immunohistochemistry staining in PyMT (n = 7) and ErbB2 (n = 8) induced tumors. (E) β-catenin immunohistochemical staining was utilized to measure membrane-bound β-catenin (arrow) and nuclear β-catenin (arrowhead) between PyMT (n = 8) and ErbB2 (n = 15) induced tumors. Proportion based on Allred scoring of (F) membrane-bound and (G) nuclear β-catenin per tumor is represented via dot plot. Allred scoring system is as indicated.
Discussion
Our findings suggest that tumor phenotype may be determined primarily not by the cell-of-origin but instead by the initiating oncogene. We found that, regardless of the Krt6a or WAP status of the cell-of-origin, the PyMT oncogene gave rise to papillary, cystic tumors whereas the ErbB2 oncogene gave rise to solid, nodular tumors. Our findings are consistent with previous studies reporting that mouse mammary tumors induced by transgenic PyMT displayed acinar/glandular/papillary features more frequently than those induced by transgenic ErbB2 (~34% versus 10%, [18]).
Our findings do not rule out a role for the cell-of-origin in influencing tumor phenotype. While introduction of PyMT or ErbB2 into the general MMTV-tva expressing population (the majority of which is terminally differentiated) also leads to papillary or nodular phenotypes, respectively (Figure 2B, [9, 10]), tumors induced from the highly diverse MMTV-expressing population tend to exhibit a greater degree of inter-tumoral heterogeneity, in agreement with previous work showing that targeting the PyMT oncogene into various cell types (including differentiated cell types) produces a range of mammary tumor subtypes [19]. Such findings suggest that the cell-of-origin does play some role in determining tumor phenotype.
Although both PyMT and ErbB2 activate both Ras and PI3K signaling pathways, differences in the pathophysiology of PyMT versus ErbB2-induced tumors strongly suggest that the spectrum of pathways dysregulated by each oncogene must be only partially overlapping. Previous studies profiling gene expression of mouse mammary tumors revealed that PyMT-induced tumors closely clustered with but remained distinct from ErbB2-induced tumors [4, 5, 20]. It is likely that the unique pathways activated by each oncogene play, in aggregate, a determinant role in shaping tumor initiation and pathophysiology.
ErB2-induced tumors harbor a high percentage of nuclear β-catenin in comparison to PyMT-induced tumors (Figure 4). The relationship between ErbB2 and Wnt signaling has been well-reported and are presumed to synergize to promote tumor formation and progression [13-17]; however the functional relationship between Wnt and ErbB2 signaling pathways in the setting of tumorigenesis may be more complex than previously envisioned. ErbB2 and Wnt signaling in tumors are clearly not analogous: Comparison of tumors induced by MMTV-Wnt versus MMTV-ErbB2 has shown that, in stark contrast to solid, poorly-differentiated appearance of ErbB2-induced tumors, Wnt-induced tumors are of a highly glandular nature [18]. Furthermore, our group has previously reported that mammary epithelial cells with active canonical Wnt signaling appear to resist transformation by ErbB2 [13].
One aspect of tumor pathophysiology at which Wnt and ErbB2 signaling may converge is in mediating EMT. We found that the ErbB2 oncogene alters epithelial differentiation and shifts tumor cells toward a more mesenchymal cell type. Many cells in ErbB2-induced tumors expressed the mesenchymal marker vimentin while retaining the round/cuboidal morphology typical of an epithelial cell (Figure 4A, right panel), suggesting that these cells may be in some intermediate state between epithelial and mesenchymal. The concomitant increase in EMT and Wnt in our ErbB2-induced tumors is consistent with previous findings that tumors driven by transgenic ErbB2 engages β-catenin signaling and that the transcriptional signature of these tumors were enriched for genes involved in mesenchymal development [16]. Taken together, these findings lend support to a potential functional relationship between Wnt signaling and the induction of EMT in ErbB2-induced tumors. It would be interesting to study the effect(s) of blockade of Wnt signaling on EMT in ErbB2-induced tumors in order to precisely define the role of Wnt signaling as either a consequence or a cause of EMT in the setting of ErbB2-mediated tumorigenesis. Furthermore, it remains necessary to define the phenotype of Wnt-driven tumors induced from distinct, well-defined cells-of-origin, such as Krt6a-positive or WAP-positive cells, to truly delineate the contributions of Wnt versus ErbB2 to mammary tumor pathophysiology.
In summary, we have shown that ErbB2 tumors induced from the Krt6a-positive progenitor subset of the mammary epithelial compartment are characterized by an undifferentiated histopathology, and a high degree of epithelial-to-mesenchymal transition. In contrast, PyMT tumors induced from the same cell population are, as previously published [12], more differentiated and exhibit minimal EMT markers. This in vivo study demonstrates that different oncogenes introduced into the same mammary epithelial cell type can, indeed, give rise to tumors of highly contrasting phenotypes, and underscores the need to develop clinically applicable methods to accurately identify driver mutations in a patient-specific manner.
Methods
Transgenic Mice and Animal Care. Animal care and procedures were approved by the Institutional Animal Care and Use Committee of Baylor College of Medicine and were in accordance with the procedures detailed in the Guide for Care and Use of Laboratory Animals (NIH publication 85-23). Generation of Krt6a-tva [12] and WAP-tva [21] mouse lines have been described previously.
Virus Preparation and Delivery to the Mammary Gland. RCAS virus preparation and mammary intraductal infection have been previously described [9]. RCAS-PyMT [22] (107 IU) or RCAS-ErbB2 [9] (107 IU) was delivered into the numbers 2, 3, and 4 mammary gland through intraductal injection into pubertal (5 weeks of age) mice. A tracking dye (0.1% bromophenol blue) was used to determine injection success.
Tissue Processing and Immunostaining. Mammary tumors were removed and fixed in 4% paraformaldehyde overnight at 4°C. Fixed tissues were paraffin-embedded, and 3-µm sections were generated and placed on slides for immunostaining, hematoxylin and eosin staining, or TUNEL staining. For immunohistochemical staining, sections were deparaffinized and rehydrated in a graded xylene/ethanol series and then stained using the Vector ABC and M.O.M. immunostaining kits (Vector Laboratories, Burlingame, CA) according to the manufacturer's instructions. For immunofluorescent staining, deparaffinized slides were incubated with the primary antibodies overnight at 4°C. Slides were incubated with fluorophore-conjugated secondary antibodies for 30 minutes at room temperature and counterstained with 4',6-diamidino-2-phenylindole (DAPI) to identify nuclei. Primary antibodies used in these studies include IgG against keratin 5 (Covance; PRB-160P), keratin 6 (Covance; PRB-169P), keratin 8 (TROMA-I; MS-977-PI), pancytokeratin (Santa Cruz; sc-8018), E-cadherin (Cell Signaling; 3195), β-catenin (BDBiosciences; 610153), vimentin (Santa Cruz; sc-7557), estrogen receptor α (Santa Cruz; sc-542), and Ki-67 (Santa Cruz; sc-7846). TUNEL assay was performed using the TUNEL Apoptosis Detection Kit (Millipore; 17-141) according to manufacturer's instructions.
Image Processing and Quantitation. Images were visualized and quantitated using freeware ImageJ [23] or Adobe® Photoshop® software (version CS5 or later). Unless specified, staining was quantified by counting percentage of positive-staining cells per tumor, with the mean and standard error reported. Statistical analysis was performed in R [24] and the R Commander package [25], using Student's t-test and Mann-Whitney U test for normal and non-normal data, respectively, and Fisher's Exact test for count data.
Abbreviations
Krt5 - Keratin 5, Krt6 - Keratin 6, Krt8 - Keratin 8, MMTV - Mouse mammary tumor virus, PyMT - Polyoma Middle T antigen, RCAS - Replication-competent avian sarcoma-leukosis virus LTR splice acceptor, WAP - Whey Acidic Protein
Acknowledgements
The authors thank Dr. Michael Lewis, Dr. Jeffrey Rosen, and Ms. Claire Faust for provision of reagents and technical assistance. This work was supported in part by funds from NIH CA124820 (to Y.L) and U54CA149196 (to Y. L; PI: Stephan Wong) as well as by the resources from the Dan L. Duncan Cancer Center (P30CA125123) and the Sue & Lester Breast Center (P50-CA058183). K.H. was supported by NIH training award T32AG000183, and V.S. was supported by the Cancer Prevention Research Institute of Texas training award RP101499.
Authors' contributions
Experimental approach was designed by W.B, M.T, Y.L. Virus was generated and injected into mice by W.B and M.T. Tumors were collected by K.H and V.S. Tumor sections were stained by K.H, V.S, Y.P, and J.D. Analysis and interpretation of tumor data, and writing of manuscript was performed by K.H, V.S, with guidance from Y.L.
Competing interests
The authors declare no competing interests.
References
1. Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA. et al. Molecular portraits of human breast tumours. Nature. 2000;406:747-52
2. Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61-70
3. Cardiff RD, Sinn E, Muller W, Leder P. Transgenic oncogene mice. Tumor phenotype predicts genotype. The American journal of pathology. 1991;139:495-501
4. Desai KV, Xiao N, Wang W, Gangi L, Greene J, Powell JI. et al. Initiating oncogenic event determines gene-expression patterns of human breast cancer models. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:6967-72
5. Herschkowitz JI, Simin K, Weigman VJ, Mikaelian I, Usary J, Hu Z. et al. Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome biology. 2007;8:R76
6. Visvader JE. Cells of origin in cancer. Nature. 2011;469:314-22
7. Blanpain C. Tracing the cellular origin of cancer. Nature cell biology. 2013;15:126-34
8. Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature. 2013;501:328-37
9. Du Z, Podsypanina K, Huang S, McGrath A, Toneff MJ, Bogoslovskaia E. et al. Introduction of oncogenes into mammary glands in vivo with an avian retroviral vector initiates and promotes carcinogenesis in mouse models. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:17396-401
10. Toneff MJ, Du Z, Dong J, Huang J, Sinai P, Forman J. et al. Somatic expression of PyMT or activated ErbB2 induces estrogen-independent mammary tumorigenesis. Neoplasia. 2010;12:718-26
11. Cardiff RD, Anver MR, Gusterson BA, Hennighausen L, Jensen RA, Merino MJ. et al. The mammary pathology of genetically engineered mice: the consensus report and recommendations from the Annapolis meeting. Oncogene. 2000;19:968-88
12. Bu W, Chen J, Morrison GD, Huang S, Creighton CJ, Huang J. et al. Keratin 6a marks mammary bipotential progenitor cells that can give rise to a unique tumor model resembling human normal-like breast cancer. Oncogene. 2011;30:4399-409
13. Bu W, Zhang X, Dai H, Huang S, Li Y. Mammary cells with active Wnt signaling resist ErbB2-induced tumorigenesis. PloS one. 2013;8:e78720
14. Podsypanina K, Li Y, Varmus HE. Evolution of somatic mutations in mammary tumors in transgenic mice is influenced by the inherited genotype. BMC medicine. 2004;2:24
15. Radu M, Semenova G, Kosoff R, Chernoff J. PAK signalling during the development and progression of cancer. Nature reviews Cancer. 2014;14:13-25
16. Schade B, Lesurf R, Sanguin-Gendreau V, Bui T, Deblois G, O'Toole SA. et al. beta-Catenin signaling is a critical event in ErbB2-mediated mammary tumor progression. Cancer research. 2013;73:4474-87
17. Schlange T, Matsuda Y, Lienhard S, Huber A, Hynes NE. Autocrine WNT signaling contributes to breast cancer cell proliferation via the canonical WNT pathway and EGFR transactivation. Breast cancer research: BCR. 2007;9:R63
18. Rosner A, Miyoshi K, Landesman-Bollag E, Xu X, Seldin DC, Moser AR. et al. Pathway pathology: histological differences between ErbB/Ras and Wnt pathway transgenic mammary tumors. The American journal of pathology. 2002;161:1087-97
19. Smith BA, Shelton DN, Kieffer C, Milash B, Usary J, Perou CM. et al. Targeting the PyMT Oncogene to Diverse Mammary Cell Populations Enhances Tumor Heterogeneity and Generates Rare Breast Cancer Subtypes. Genes & cancer. 2012;3:550-63
20. Pfefferle AD, Herschkowitz JI, Usary J, Harrell JC, Spike BT, Adams JR. et al. Transcriptomic classification of genetically engineered mouse models of breast cancer identifies human subtype counterparts. Genome biology. 2013;14:R125
21. Haricharan S, Hein SM, Dong J, Toneff MJ, Aina OH, Rao PH. et al. Contribution of an alveolar cell of origin to the high-grade malignant phenotype of pregnancy-associated breast cancer. Oncogene. 2014;33:5729-39
22. Holland EC, Li Y, Celestino J, Dai C, Schaefer L, Sawaya RA. et al. Astrocytes give rise to oligodendrogliomas and astrocytomas after gene transfer of polyoma virus middle T antigen in vivo. The American journal of pathology. 2000;157:1031-7
23. Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nature methods. 2012;9:671-5
24. R Core Team. R: A Language and Environment for Statistical Computing. 2014.
25. Fox J. The R Commander: A Basic Statistics Graphical User Interface to R. Journal of Statistical Software. 2005;14:1-42
Author contact
![]() Corresponding author: Breast Center, BCM600, One Baylor Plaza, Houston, TX 77030. Phone: 713-798-3963. Fax: 713-798-1659. Email: liyiedu.
Corresponding author: Breast Center, BCM600, One Baylor Plaza, Houston, TX 77030. Phone: 713-798-3963. Fax: 713-798-1659. Email: liyiedu.