Impact Factor ISSN: 1449-2288
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Androgen receptor: Historical...
Early treatment for advanced PCa
Modern treatment for advanced PCa
Pre-receptor level treatment...
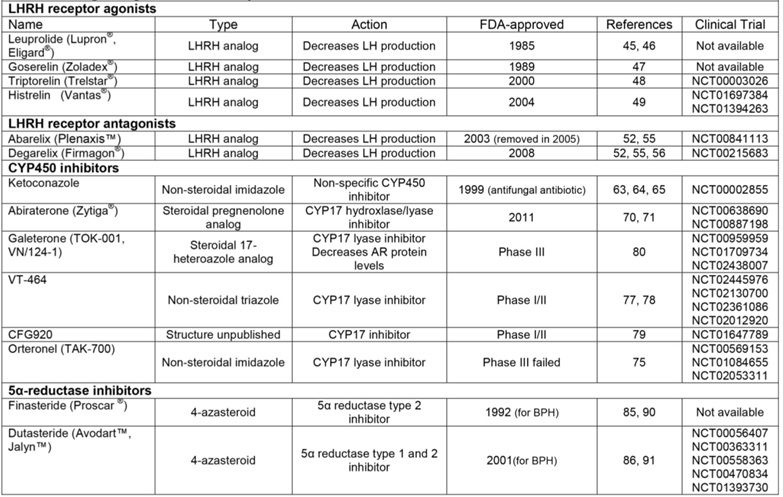
LHRH receptor agonists
LHRH receptor antagonists
CYP450 inhibitors
5α-reductase inhibitors
Receptor level treatment...
First generation AR antagonists
Second generation AR antagonists
Inhibitors of AR expression
Post-receptor level treatment...
Concluding remarks
Abbreviations
Acknowledgements
References
Author Biography

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2016; 12(4):409-426. doi:10.7150/ijbs.14090 This issue Cite
Review
Therapeutic Rationales, Progresses, Failures, and Future Directions for Advanced Prostate Cancer
Kristine M Wadosky, Shahriar Koochekpour ![]()
Departments of Cancer Genetics and Urology, Center for Genetics and Pharmacology, Roswell Park Cancer Institute, Elm and Carlton Streets, Buffalo, NY, USA.
Received 2015-10-10; Accepted 2015-11-15; Published 2016-2-6
Abstract

Patients with localized prostate cancer (PCa) have several therapeutic options with good prognosis. However, survival of patients with high-risk, advanced PCa is significantly less than patients with early-stage, organ-confined disease. Testosterone and other androgens have been directly linked to PCa progression since 1941. In this review, we chronicle the discoveries that led to modern therapeutic strategies for PCa. Specifically highlighted is the biology of androgen receptor (AR), the nuclear receptor transcription factor largely responsible for androgen-stimulated and castrate-recurrent (CR) PCa. Current PCa treatment paradigms can be classified into three distinct but interrelated categories: targeting AR at pre-receptor, receptor, or post-receptor signaling. The continuing challenge of disease relapse as CR and/or metastatic tumors, destined to occur within three years of the initial treatment, is also discussed. We conclude that the success of PCa therapies in the future depends on targeting molecular mechanisms underlying tumor recurrence that still may affect AR at pre-receptor, receptor, and post-receptor levels.
Keywords: Prostate cancer, androgen, AR, castrate-recurrent, treatment, FDA.
Introduction
Prostate cancer (PCa) is the most common non-dermatological cancer among men [1]. In 2015, it is expected that PCa will represent 26% of new cancer diagnoses for males in the United States [1]. Second only to lung cancer, it is estimated that 27540 deaths will be attributed to PCa in 2015 [1]. The advent of the prostate specific antigen (PSA) test has increased the rate of early diagnosis—consequently, the majority of tumors are discovered when they are low-grade [2, 3]. Also termed localized PCa, treatment choices for patients with these low-risk tumors are widespread. Therapeutic options include interstitial brachytherapy, external beam radiation, and radical prostatectomy [2, 3]. In addition, eligible patients can also undergo “active surveillance” or “watchful waiting”, where disease progression is monitored without any therapeutic intervention [4]. Identifying the best therapy for patients with indolent disease is a significant challenge in urologic oncology [2, 3]. Detailed description of low-risk, localized PCa and parameters used in disease management for these patients are reviewed elsewhere [2, 3].
While cases of low-grade PCa represent the largest portion of diagnoses, the 5-year survival rate in the United States for patients in these early stages approaches 100% [1]. However, for advanced PCa, defined as disease that has metastasized beyond the pelvic lymph nodes, the 5-year survival rate drops to 28% [1]. This staggering statistic represents a void in therapeutic efficacy and is undoubtedly a primary concern for clinicians, scientists, public health officials, and patients. Correspondingly, treatment of this high-risk class of prostatic malignancies is the major focus of this review article.
Nearly 75 years of work has been dedicated to understanding the pathophysiology of metastatic PCa. The foremost impact these studies have had is establishing the role of androgens, namely testosterone, in this disease [5]. Androgen receptor (AR), a transcription factor part of the nuclear receptor family, is the primary effector of testosterone and activates genes involved in growth, metabolism, and pro-survival signaling [6]. Since AR governs this malignant phenotype, the molecular biology affecting AR function has driven the pharmaceutical development of PCa therapies [7]. Indeed, excluding chemotherapy and immunotherapy, therapies for advanced PCa all center on blocking or interrupting AR-dependent signaling pathways.
In this review, we first describe AR biology, with special attention given to AR protein structure and regulation. Using a chronological framework, we go on to describe therapies used to treat advanced PCa that target androgen signaling. We categorize therapies via their relationship to AR; referring to each therapy as part of a pre-receptor, receptor, or post-receptor targeted strategy. For each subtype, we also describe new drugs under clinical development and examine their potential using the available data. Finally, we analyze the continued effort to overcome resistance to androgen-targeted therapies.
Androgen receptor: Historical background, structure, and function
The importance of androgens in the pathogenesis of PCa was first revealed by Drs. Charles Huggins and Clarence Hodges at the University of Chicago in 1941 [8]. They observed that bilateral orchiectomy reduced serum alkaline phosphatase, a clinical measure of PCa at the time. Conversely, they found that administration of testosterone increased serum levels of alkaline phosphatase, providing further evidence for the positive relationship between androgens and the expression of this enzyme [8]. This discovery earned Drs. Huggins and Hodges the Nobel Prize in 1966.
The study of the intracellular mechanism of androgen action in the prostate was initiated by Drs. Shutsung Liao and Senmaw Fang, also at the University of Chicago, in the late 1960s. This work first showed that dihydrotestosterone (DHT) is the predominant androgen in the prostate and went on to demonstrate that DHT is present in the nuclear fraction of the rat ventral prostate [9]. The finding that androgen hormones localize to the nucleus in prostatic cells was confirmed by another group that detected testosterone in the nuclear fraction of prostate homogenates [10]. Treatment of nuclear extracts with DNase, RNase, and phospholipase A did not affect DHT retention in the nuclear fraction—but protease treatment decreased DHT. These data suggested for the first time that DHT is complexed with a protein in prostatic nuclei [11].
Further elegant work showed that DHT can be displaced from protein fractions by the then new steroidal drug cyproterone—which was the first indication that “anti-androgen” activity occurs via interference with DHT binding to a putative protein receptor [11]. Years of work finally culminated in the cloning of AR in the late 1980s by Dr. Liao's group [12]. These results were simultaneously published by Dr. Elizabeth Wilson's group at the University of North Carolina at Chapel Hill, that also showed that the gene encoding AR is localized to the X chromosome [13].
AR is located at Xq11-12 and is about 90kb and contains 8 exons [14]. The AR mRNA transcript is 10.6 kb—with a 1.1 kb 5' untranslated region (UTR), followed by a 2.7 kb open reading frame (ORF), and a 6.8 kb 3' UTR [15] (Figure 1A and Figure 1B). AR is a 110 kDa protein with 919 amino acids and is a member of the steroid nuclear receptor transcription factor super family, sharing the same general structural and activation domains with other members [14, 16]. These domains include an N-terminal domain (NTD) that contains activation function domain 1 (AF-1), a DNA-binding domain (DBD), a hinge domain, and a ligand-binding domain (LBD) that contains AF-2 [14, 16] (Figure 1C).
Structure of androgen receptor gene, transcript, and protein. (A) Relative lengths of the AR ORF and its flanking UTRs. (B) Exon structure of the AR transcript depicting the relative sizes of each exon. (C) AR protein functional domains including the NTD, DBD, hinge, and LBD and their respective coding exons. The relative locations of the AF-1, poly-glutamine (Gln), poly-glycine (Gly), NLS, NES, and AF-2 regions are indicated. Also shown are the AR protein domains to which post-translational modifications are known to occur. (D) Schematic representation of the spatial orientation of an AR homodimer complexed with a typical ARE. NTD: N-terminal domain; DBD: DNA-binding domain; LBD: Ligand-binding domain; AF-1: Activation function domain 1; NLS: Nuclear localization sequence; NES: Nuclear export signal; AF-2: Activation function domain 2; ARE: Androgen response element.
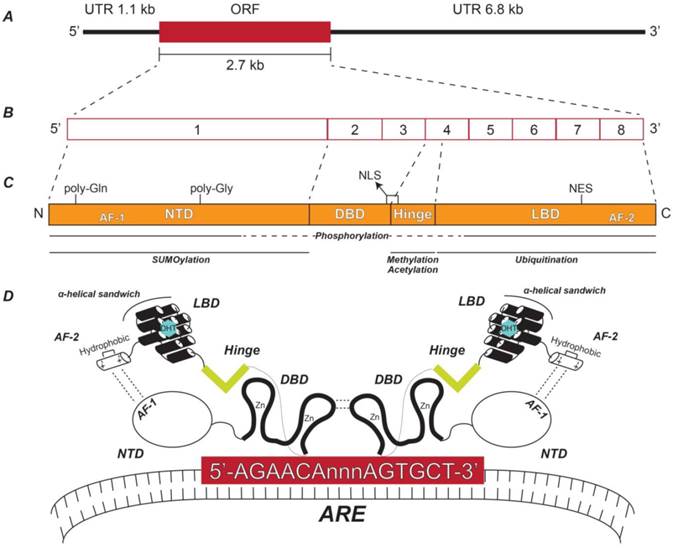
The AR NTD is encoded by exon 1 and contains two regions of variable-length with polymorphic CAG and GGC repeats, causing slight individual differences in the size of the AR NTD due to multiples of either glutamine or glycine [14] (Figure 1C). The NTD is flexible and disordered in solution—therefore its crystal structure is notoriously unsolved [16]. This struggle is considered a significant gap in the field, especially since the NTD is the largest of the AR domains and has the highest number of known post-translational modifications—including phosphorylation and small ubiquitin-like modified (SUMO)ylation [16-18] (Figure 1C) (for specific modified residues for all known AR post-translational modifications, see Reference [18]). Despite this setback, important elements of the NTD have been identified. Namely, the AF-1 is a highly effective transcriptional activator alone by virtue of its potent ability to recruit coactivators [16, 19, 20]. AF-1 is also involved in an intramolecular interaction with AF-2 in the LBD that is unique to AR among other nuclear receptors [21] (Figure 1D). This additional folding of the AR molecular structure recruits chromatin remodeling complexes, enabling AR to interact with androgen response element (ARE) DNA motifs in the promoter regions of androgen-regulated genes (ARGs) [22] (Figure 1D).
Initial identification of AREs occurred in the late 1990s [23-26] and years of work have identified the ideal ARE sequence to be 5'-AGAACAnnnAGAACA-3'; where three non-specific nucleotides provide space for the DBDs of two ARs to bind the flanking hexamers in a “head-to-head” dimer formation [16, 27] (Figure 1D). Like the DBDs for all nuclear receptors, the AR DBD contains two zinc finger domains encoded by AR exons 2 and 3 [14] (Figure 1C), where the second zinc finger domain acts as the scaffold for AR dimerization [27] (Figure 1D). In addition, phosphorylation of the DBD has been shown to promote proper nucleocytoplasmic shuttling of AR [18] (Figure 1C).
Immediately downstream of the DBD is the hinge domain, the smallest region of AR, encoded by exon 4 [16] (Figure 1C and Figure 1D). Despite being a short sequence lacking structure, the hinge domain plays an important role in AR function by harboring a portion of a ligand-dependent nuclear localization sequence (NLS) that spans the border between the DBD and the hinge region [28] (Figure 1C). In addition, the hinge domain can be subjected to post-translational modifications such as phosphorylation, methylation, or acetylation [18, 29, 30] (Figure 1C), which increase nuclear localization ability and transcriptional activity, further emphasizing the importance of this small region.
Lastly, exons 4-8 encode AR's LBD (Figure 1C), which is the region that binds DHT, testosterone, or other steroid hormones [14]. Like the DBD, all nuclear receptor LBDs share similar structure—where 11-13 α-helices form a three-layer anti-parallel α-helical sandwich [31]. For the AR LBD specifically, 11 helices make up the α-helical sandwich (Figure 1D), where 18 key amino acids compose the ligand-binding pocket [16]. One of these amino acids, Thr877, is of utmost importance, since this residue is frequently mutated to alanine (T877A) in PCa and in the LNCaP PCa cell line, the most commonly used cell line in PCa research [14, 16]. The LBD also contains a non-canonical nuclear export sequence (NES) that is independent of exportin 1-mediated export [32]. The AR LBD has been shown to be poly-ubiquitinated (Figure 1C), a modification that does not promote the degradation of AR, but instead allows AR to interact with certain co-activator proteins and bind the promoter regions of select ARGs [33]. While AR has been shown to undergo poly-ubiquitination-dependent degradation via the ubiquitin proteasome system [34, 35], the regions of AR where this modification occurs remain unknown.
The AR LBD also contains the C-terminal AF-2 (Figure 1C), which interacts with the AR AF-1, as discussed above [21] (Figure 1D). Interestingly, AF-2 has weak transcriptional activity alone and functions more efficiently in concert with AF-1 [16]. Like other nuclear receptors, AF-2 forms an α-helical structure [31]. Specifically, the AR AF-2 has a hydrophobic groove with regions of positive and negative charges on each side, allowing the binding of co-activator proteins [16] (Figure 1D).
The first step in AR activation in prostatic cells occurs with the diffusion of testosterone across the plasma membrane. Once testosterone is within the cell, 5α-reductase converts testosterone to DHT, a more potent AR ligand than testosterone [36] (Figure 2). Under basal conditions, AR is complexed in the cytoplasm with the protein chaperones heat shock protein 70 (HSP70) and 90 (Figure 2). Several other protein chaperones, including HSP27 and 40, p23, and Bag-1L, are also involved in maintaining AR in a ligand-responsive conformation and promoting AR's nuclear localization [37]. When DHT binds to AR, a conformational change exposes its NLS, causes the chaperone complex to dissociate, and enables AR's nuclear translocation [17, 37] (Figure 2). Androgen binding also triggers AR phosphorylation—a modification that promotes AR nuclear import [18] (Figure 2).
Activation of androgen regulated genes. Testosterone from the peripheral blood diffuses across the plasma membrane and is converted to DHT by 5α-reductase. DHT binding to AR induces HSP70 and 90 to dissociate from AR and phosphorylation of AR then promotes AR nuclear localization. In the nucleus, AR interacts with transcriptional co-regulators forming the transcriptional machinery complex that engages with AREs in the promoter regions of ARGs and triggers transcription. AR: androgen receptor; T: Testosterone; DHT: Dihydrotestosterone; HSP: Heat shock protein; ARE: Androgen response element.
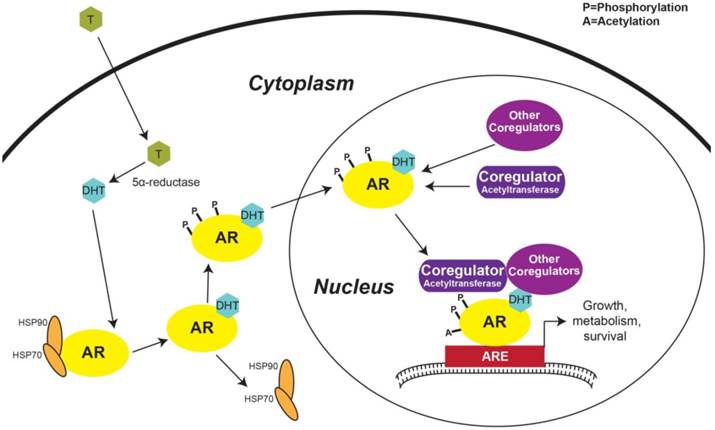
Once in the nucleus, activated AR interacts with various proteins, including molecular scaffolds, histone modifiers, chromatin remodelers, members of the ubiquitination and SUMOylation pathways, other transcription factors, and components of the transcriptional machinery [36]. AR co-regulators are defined as any protein that enhances or inhibits AR's transcriptional activity [36]. Notable proteins shown to activate AR transcriptional activity include members of the p160 co-activator family, including steroid receptor co-activator (SRC)-1, SRC-3, and transcriptional intermediary factor (TIF), p300, CREB-binding protein (CBP), Tat interacting protein 60 kDa (Tip60), and AR-associated (ARA)-54 and -70, many of which are acetyltransferases [36] (Figure 2). Not only histone acetylation, but AR acetylation in the hinge region (Figure 1C) [29, 30] has been shown to be important during the formation of the AR transcriptional complex—since mutation of AR acetylation sites decreases induction of ARGs [38]. This inhibitory mechanism occurs via the recruitment of nuclear receptor co-repressor (NCoR) to hypoacetylated AR [36, 37]. NCoR is a member of the group of AR inhibitory co-factors which also includes silencing mediator for retinoid and thyroid hormone receptors (SMRT) [36].
The active AR transcriptional complex binds gene promoter regions containing AREs (Figure 2), which induces a transcriptional profile that causes the primary effect that androgens have on prostatic cells [14]. The most well-known AR target gene is PSA, which is among several other ARGs that code for seminal proteins, including kallikren 2 and prostatic acid phosphatase, and other prostate-specific proteins, including transmembrane protease, serine 2 (TMPRSS2) [6]. AR also targets genes involved in general cell processes, including growth, proliferation, and survival (Figure 2). AR activates transcription of the growth factor Wnt-induced secreted protein-2 (WISP-2), insulin-like growth factor receptor (IGF1-R), and mitogen activated kinase 2 kinase 4 (MAP2K4) [6, 39]. Many cell cycle genes have recently been shown to be activated by AR, including cyclin-dependent kinase (CDK)-6, CDK2-associated protein 2 (CDK2AP2), and E2F1 [39]. ARGs involved in apoptosis include caspase-2 and Mdm2, where AR inhibits and activates their transcription, respectively [6]. The advent of Next-Generation DNA sequencing, improvement of the chromatin immunoprecipitation technique, and increased computational capacity has recently enabled researchers to compile a comprehensive list of all AR-binding sites and target genes in the human genome [39]. Many of the ARGs that were also within 25 kb of an AR-binding site include those associated with metabolism, such as pyruvate dehydrogenase complex component X (PDHX) and fatty acid synthase (FASN) [39]. Altogether, AR's transcriptional effect on the prostatic epithelial cell is to increase growth, proliferation, and metabolism, while inhibiting apoptosis—emphasizing the importance that AR plays in oncogenic signaling in the prostate.
Early treatment for advanced PCa
Localized PCa accounts for 93% of cases [1]. These patients have several options (i. e. interstitial brachytherapy, external beam radiation, and radical prostatectomy) and good prognosis since these are mostly curative therapies [40]. A minority of patients present with locally-advanced or metastatic PCa, defined as tumors that have spread to the pelvic lymph nodes or other more-distant organs, respectively. The growth of these tumors is largely dependent on androgen hormones [40]. Diagnosis with this high-risk PCa has a far worse prognosis since curative therapy has yet to be developed. The central role of androgens in advanced PCa has led to treatment strategies to limit the production and downstream effects of testosterone [40]. As described in the previous section, Huggins and Hodges were the first to show that bilateral orchiectomy alleviates symptoms of metastatic PCa—proving that removal of the source of testosterone is an effective therapy [8]. This initial study by Huggins and Hodges also concluded that estrogen treatment performed similarly to orchiectomy [8]. This was the first time chemical castration, widely in use today, was utilized in the treatment of metastatic PCa. A large-scale study carried out by the Veterans Administration Co-operative Urological Research Group in the 1960s determined that the therapeutic effect of estrogen was comparable to orchiectomy [5, 41]. These data led to the use of estrogen in combination with other therapies or as a non-surgical monotherapy for several years. Unfortunately, significant cardiovascular side effects were associated with estrogen therapy and despite substantial work to determine an optimal dosing regimen, estrogen was abandoned in the early 1980s [5]. The failure of estrogen treatment for metastatic PCa brought on the age of modern therapies.
Modern treatment for advanced PCa
The initial step in the generation of testosterone is the production of luteinizing hormone releasing hormone (LHRH) (also known as gonadotropin-releasing hormone [GnRH]) by the hypothalamus (Figure 3). LHRH stimulates the production of luteinizing hormone (LH) by the anterior pituitary, which releases LH into the circulation. In response to LH, Leydig cells in the testes synthesize testosterone (Figure 3) [42]. At the molecular level, testosterone freely diffuses across the cell membrane and 5α-reductase converts testosterone to DHT (Figure 2 and Figure 3), a more potent ligand for AR, as discussed in the previous section [36]. DHT-dependent activation of AR induces a prostate-specific transcriptional program, the main purpose of which is production of seminal proteins, including PSA [26]. Consequently, PSA is used as a biomarker for PCa as well as a measure of treatment effectiveness in patients with active disease [26].
Physiological and anatomical targets of FDA-approved drugs for prostate cancer. LHRH produced by the hypothalamus stimulates production of LH by the anterior pituitary. LH then activates the production of testosterone by the testes. Gonadal testosterone, along with testosterone produced by the adrenal glands and the prostate itself, activates transcription of ARGs. Currently approved therapies for advanced PCa target multiple nodes along this physiological pathway. LHRH: Luteinizing hormone releasing hormone; LH: Luteinizing hormone; CYP17: Cytochrome P450-C17; T: Testosterone; DHT: Dihydrotestosterone; DHEA: Dehydroepiandrosterone; AR: androgen receptor; ARE: Androgen response element.
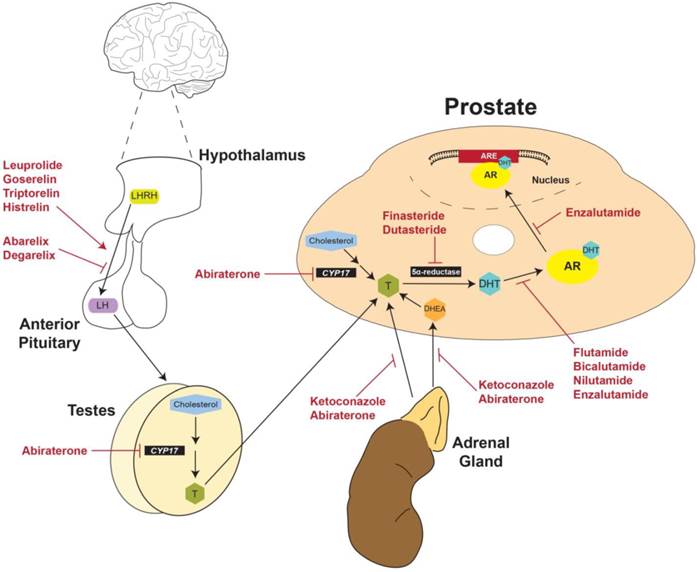
Strategies for current treatment regimens and development of new therapies for PCa could be divided into three categories: pre-receptor, receptor, and post-receptor [43]. The goal of pre-receptor level therapies is to interrupt the biosynthesis of testosterone and decrease plasma testosterone to < 0.5 ng/mL (50 ng/dL), the level clinically-defined as castration [5, 26]. At the receptor level, the goal of treatment is to suppress AR expression or activity and thereby inhibit downstream effects of testosterone at the molecular level. Finally, post-receptor level therapies aim to destabilize AR protein by inhibiting the interaction of AR with HSP chaperones, disrupt the binding of AR and coregulators/cofactors, or inhibit the active AR transcriptional complex from binding DNA promotor elements [43].
Pre-receptor level treatment strategies
Following the landmark studies by Huggins and Hodges [8], interfering with testosterone production by either bilateral orchiectomy or estrogen treatment was standard therapy for metastatic PCa [5]. Despite the initial success of these therapies, the psychological and cosmetic impacts of orchiectomy and cardiovascular side effects of estrogen treatment initiated the development of new pre-receptor level therapies to limit testosterone production or the conversion of testosterone to DHT.
LHRH receptor agonists
Discovery of the LHRH amino acid sequence [44] introduced a novel treatment strategy for metastatic PCa [5, 45, 46]. The premise of this therapy is founded on a negative feedback loop induced in response to excess stimulation of LHRH receptors with exogenous LHRH analogs in the anterior pituitary. Since production of LHRH by the hypothalamus is pulsatile, constant stimulation of LHRH receptors leads to receptor desensitization and eventually triggers a permanent decrease in receptor expression, creating negative feedback [45, 46]. Consequently, future production of LHRH by the hypothalamus does not stimulate LH production by the anterior pituitary and thereby indirectly inhibits downstream gonadal testosterone production [45, 46] (Figure 3).
The first LHRH analog to be developed was leuprolide, a nonapeptide which was FDA-approved in 1985 (Table 1, Figure 3) [45]. Shortly after the release of leuprolide, another LHRH analog was approved by the FDA called goserelin [47] (Table 1, Figure 3). Additional LHRH analogs include triptorelin and histrelin, both of which were FDA-approved in the early 2000s (Table 1, Figure 3) [48, 49]. Meta-analysis of ten randomized clinical trials found that treatment with LHRH agonists is equivalent to orchiectomy in survival, clinical outcomes (testosterone and PSA levels), and time to disease progression [42, 50]. This analysis and others have found no significant differences between leuprolide and goserelin in overall survival, however no trials have directly compared the efficacy of different LHRH agonists [50, 51]. Consequently, choice of LHRH agonist in a treatment paradigm depends on the clinician's and patient's preference of drug dosage and delivery [5, 45, 47-49]
Drugs Targeted at Pre-receptor Level.
LHRH receptor antagonists
A significant side effect of LHRH agonists is naturally an initial surge in LH and testosterone that occurs for about 1-2 weeks after being administered [52]. To avoid initial hormonal flares and decrease the delay to castration, development of LHRH analogs with receptor antagonist activity began in the 1990s [53, 54]. One of these analogs, abarelix, was FDA-approved in 2003 as the first of its class [52] (Table 1, Figure 3). However, once abarelix was in clinic, immediate-onset systemic allergic reactions emerged as a significant adverse response and it was removed from the US market in 2005 [55]. In the early 2000s, development of other LHRH analogs produced degarelix, a new LHRH antagonist approved by the FDA in 2008 (Table 1, Figure 3) [55]. There have been several clinical trials comparing the efficacy of LHRH receptor agonists and antagonists. Most considerable of these studies is Klotz et al. [56], which compared leuprolide with degarelix. The results of this study showed that 81% of patients who received leuprolide experienced an initial flare in testosterone, as expected. But during the same time period, > 95% of patients who received degarelix experienced reduction in circulating testosterone levels to ≤ 0.5 ng/mL within only 3 days [52, 56]. However, leuprolide required 28 days of treatment to achieve the same reduced testosterone level [52, 56]. Following 28 days, suppression of testosterone was equivalent between degarelix and leuprolide until the completion of the one-year study [52, 56]. Altogether, clinical data has proven that the LHRH antagonist degarelix is at least as effective, if not more so, than LHRH agonists for patients with metastatic PCa. Some would argue that for patients with symptomatic disease who want to avoid initial testosterone surges and subsequent micro-surges, degarelix is superior to LHRH agonists. Surely, in patients at high risk for progression, data suggest that choosing degarelix over leuprolide could increase overall survival [52, 55, 56].
CYP450 inhibitors
The majority of testosterone (> 90%) is synthesized in the testes and the remaining is produced in the adrenal gland and peripheral tissues (including the prostate) [57]. Activation of adrenal production of androgens is much like the hypothalamus-pituitary-gonadal axis. The release of corticotropin-releasing hormone (CRH) by the hypothalamus induces the release of adrenocorticotropic hormone (ACTH) from the anterior pituitary. ACTH then activates synthesis of testosterone and dehydroepiandrosterone (DHEA), another androgen, by the adrenal gland. In the prostate, testosterone is produced largely from adrenal DHEA or other steroid precursors [57] (Figure 3). LHRH analogs do not affect the production of CRH and ACTH and therefore only suppress production of testosterone by the testes [57, 58]. This limitation of LHRH-targeted therapies poses a significant clinical problem, since even small amounts of testosterone or DHT can activate AR and fuel PCa growth, especially when produced locally by the prostate or at sites of metastasis [59-61]. It has been shown that adrenal and intratumoral androgens can contribute to disease progression, an advanced stage clinically-referred to as castrate-resistant or castrate-recurrent (CR) PCa, following treatment with LHRH analogs [59-61]. About 80% of patients will progress to CR-PCa within 24 months of starting LHRH-directed treatment [40]. Since CR-PCa is fatal after 9-12 months if left untreated, effective therapies for this subset of patients are vital [40].
The central enzyme in testosterone biosynthesis across all organ systems is cytochrome P450-C17 (CYP17); therefore, a single agent that inhibits CYP17 could suppress gonadal, adrenal, and intratumoral androgen production and decrease overall testosterone levels even after treatment with LHRH agonists/antagonists [57, 62]. Early in its use as an antifungal antibiotic, it was found that ketoconazole could inhibit steroidogenesis by virtue of its ability to antagonize CYP450 enzymes [63] (Table 1, Figure 3). This discovery triggered the off-label use of ketoconazole at high doses for the treatment of CR-PCa. Ketoconazole proved to be efficacious in decreasing testosterone and DHEA, but as a pan-CYP450 inhibitor, it also decreased production of corticosteroids by the adrenal gland. Consequently, hydrocortisone co-treatment was required with ketoconazole [64, 65]. Since at the time the only FDA-approved treatment for CR-PCa was chemotherapy, ketoconazole was considered a success even with the need for corticosteroid replacement [40]. Nevertheless, the off-target effects of ketoconazole initiated the development of therapies that specifically target CYP17.
Work on the development of CYP17-specific inhibitors in the 1990s led to the discovery of abiraterone, a derivative of the steroid-hormone precursor pregnenolone that irreversibly inhibits both hydrolyase and lyase activities of CYP17 [66, 67]. Early clinical studies using abiraterone to treat CR-PCa determined that the dose required to repress testosterone also alters the levels of multiple corticosteroids, since they also require CYP17 for their synthesis [68, 69]. To correct this imbalance, patients were given prednisone along with abiraterone in phase III clinical trials [70, 71]. Abiraterone with prednisone was FDA-approved in 2011 specifically for treatment of CR-PCa and was the first agent to significantly increase survival of these most advanced patients, but only by 4.6 months (Table 1, Figure 3) [70-72]. Nevertheless, abiraterone is currently in use for CR-PCa patients that have received prior chemotherapy and those that are chemotherapy-naïve [70, 71].
While the specificity of abiraterone for CYP17 has been demonstrated in vitro, additional analysis has shown that it inhibits multiple hepatic CYP enzymes [57]. Indeed, liver dysfunction and drug-sensitivity are significant side effects of abiraterone [57]. In addition, abiraterone also inhibits CYP11B1 and 2, essential enzymes in the synthesis of mineralocorticoids and glucocorticoids; these off-target effects of abiraterone explains the changes in corticosteroids observed in clinical trials and the need for prednisone [57]. Finally, inhibition of the hydrolyase activity of CYP17 shifts the steroidogenesis pathway toward mineralcorticoid synthesis. Consequently, mineralocorticoid excess is common in patients receiving abiraterone and is not fully corrected by prednisone [57].
The promiscuity of abiraterone has demonstrated the need for a more specific CYP17 inhibitor. One area of drug development has focused on agents that explicitly inhibit CYP17 lyase function, a strategy which could in theory specifically target androgen synthesis while retaining CYP17 hydrolyase activity and avoiding changes in mineralocorticoid synthesis [40]. One of these new drugs is orteronel (TAK-700) (Table 1), which is an imidazole-derivative that inhibits CYP17 lyase activity about 5 times more specifically than hydroxylase activity and has less profound off-target effects on hepatic CYP450 enzymes than abiraterone [73, 74]. While patients that received orteronel had significant radiologic improvement in phase III clinical trials, orteronel failed to increase overall survival in CR-PCa patients and use of this drug has been suspended for the treatment of PCa [75, 76]. Another drug that has higher inhibitory activity towards CYP17 lyase is VT-464, a non-steroidal triazole derivative [77] (Table 1). Preclinical studies have shown that VT-464 treatment decreases AR activation in vitro to a greater extent than abiraterone and intratumoral testosterone to similar levels as abiraterone [78]. As of this writing, VT-464 is in phase I/II clinical trials, the results of which have yet to be published (NCT02445976, NCT02130700, NCT02361086, NCT02012920, https://clinicaltrials.gov/). CFG920 is another drug that is in phase I/II clinical trials (NCT01647789, https://clinicaltrials.gov/) (Table 1) [79]. While it is reported to be a CYP17 inhibitor with anti-tumor effects in PCa [79], no structural, functional, or pre-clinical data have been published for CFG920 as of this writing.
Finally, the most encouraging of the novel CYP17 inhibitors in development is galeterone (TOK-001), a steroidal 17-heteroazole analog [80]. Pre-clinical studies have shown that not only does galeterone inhibit CYP17 [81], but also inhibits AR transcriptional activity [81, 82] and promotes the proteasomal degradation of both AR and its ligand-independent variants AR-V7 and ARv567es [83]. Phase I clinical trials have been completed and phase II trials are still ongoing for galeterone in CR-PCa patients [80]. A phase III clinical trial has started recruiting CR-PCa patients that specifically express AR-V7 to study the efficacy of galeterone (NCT02438007, https://clinicaltrials.gov/). While the complete results for the clinical trials have yet to be published in full, galeterone was effective in decreasing PSA levels and promoting radiologic tumor regression in CR-PCa patients without off target effects on corticosteroids [80]. Consequently, galeterone is used as a monotherapy without prednisone [80]. Considering the importance of both androgen production and AR signaling in progression of CR-PCa, use of galeterone as a multi-targeting therapy could provide significant benefit. Noteworthy is galeterone-dependent decrease in expression of AR variants, which play a key role in development of resistance to therapy in CR-PCa.
5α-reductase inhibitors
Decades of research has shown that androgens and the downstream signaling pathways they activate are drivers of PCa pathogenesis. This well-defined cause-and-effect relationship makes PCa unique among cancers and has led to the theory that PCa could not just be treated, but prevented by interfering with androgen-dependent signaling. 5α-reductase inhibitors limit the production of intracellular DHT by competitively binding to the enzyme that chemically reduces testosterone to DHT (Figure 3) [84]. Finasteride and dutasteride are 4-aza-steroidal 5α-reductase inhibitors that are FDA-approved for the treatment of benign prostatic hyperplasia (BPH) [85, 86] (Table 1), a non-neoplastic enlargement of the prostate caused by proliferation of epithelial and/or stromal cells [87]. Their ability to decrease DHT at the tissue level made finasteride and dutasteride potential candidates for PCa prevention, especially since 5α-reductase isoforms 1 and 2 are elevated in PCa [88]. Two clinical trials were performed in the early 2000s to determine if long term treatment with finasteride or dutasteride can prevent PCa either in healthy men or those at risk of developing PCa, respectively [89-91] (NCT00056407 [dutasteride trial], https://clinicaltrials.gov/). The results from both trials showed that PCa incidence was significantly decreased in the treatment groups compared to the placebo groups [89-91]. However, conclusions from these trials also determined that patients treated with finasteride or dutasteride who were diagnosed with PCa had higher-grade tumors (Gleason score ≥ 7 or ≥ 8, respectively) compared to patients who received placebo [89-91]. As a result of these data, neither finasteride nor dutasteride are FDA-approved for the prevention of PCa [92].
There has been controversy over whether 5α-reductase inhibitors truly increase severity of prostatic neoplasms or if implicit biases within these trials contributed to these results. For instance, 46.3% of patients in finasteride PCa prevention study were not included in the final analysis for various reasons (e.g. death, declining end-of-study biopsy) [89, 90]. The primary function of finasteride and dutasteride in the treatment of BPH is to decrease prostate size [93]. Since having a smaller prostate is associated with the increased sensitivity of screening methods (e.g. digital rectal examination and biopsy), the increased number of high grade tumors in the treatment groups compared to the placebo groups in both studies could be a result of increased detection [89]. Indeed, a recent retrospective study using a cohort of patients diagnosed with PCa between 1999-2009 showed that there were no difference in PCa-specific mortality between patients that received long-term treatment with a 5α-reductase inhibitor and those that did not [94]. Since higher grade tumors are associated with higher PCa-specific morality [95], the results of this retrospective study refute those of the PCa prevention clinical trials completed for finasteride and dutasteride [90, 91].
Despite the continuing debate about use of 5α-reductase inhibitors for patients prior to PCa diagnosis, work has continued to determine the efficacy of this class of drugs in slowing progression in patients already diagnosed with PCa. Specifically, studies have focused on dutasteride because it inhibits both type 1 and 2 isoforms of 5α-reductase and has been shown to decrease serum DHT to a greater extent than finasteride [93]. Results from a phase IV clinical trial have shown that dutasteride decreases incidence of histopathological progression compared to active surveillance in patients with low-grade PCa [96] (NCT00363311, https://clinicaltrials.gov/). For patients who underwent radical prostatectomy or radiation therapy for localized PCa, results from a phase II clinical trial have shown that two years of treatment with dutasteride delayed biochemical progression, as measured by time to PSA doubling [97] (NCT00558363, https://clinicaltrials.gov/). The use of dutasteride in combination with other therapies for advanced PCa has not been as successful. In a recent phase IV clinical trial, the addition of dutasteride to bicalutamide for patients with progressive non-metastatic PCa did not delay further progression compared to patients that received only bicalutamide [98] (NCT00470834, https://clinicaltrials.gov/). The effectiveness of dutasteride in combination with abiraterone in increasing time to progression is currently being studied in a phase II clinical trial (NCT01393730, https://clinicaltrials.gov/) [40], the results of which have yet to be published at the time of this writing. Altogether, while the true clinical benefit of 5α-reductase inhibitors for PCa still remains unclear, continuing work shows that potential exists for dutasteride as a preventive therapy for patients with existing low-grade PCa.
Receptor level treatment strategies
The therapies described thus far include drugs used in androgen deprivation therapy (ADT), the clinical definition of treatment that decreases testosterone/DHT (Table 1) [99]. A large limitation of ADT is that the majority of these compounds do not efficiently block the functional endpoint of AR-dependent signaling. Despite targeting gonadal, adrenal, and intratumoral androgen production (Figure. 3), low androgen levels can still have proliferative effects on tumor cells [59, 61]. Receptor level treatment strategies for PCa aim to decrease or inhibit AR expression and activity. In the first study that alluded to the existence of AR, Liao and Fang used cyproterone, a DHT-derivative known at the time to have “anti-androgenic” properties, to inhibit DHT's interaction with the protein fraction of prostatic nuclear lysates [11]. This experiment eventually led to the development of PCa therapeutics that interfere with AR- testosterone/DHT complex formation by competitively binding the AR LBD (Figure 1 and Figure 3). Indeed, cyproterone has been shown to be anti-tumorigenic in PCa [100], but was never approved in the United States because it is associated with hepatotoxicity and other serious off-target effects [7]. Similar safety profiles were observed for megestrol and medroxyprogesterone, other steroidal AR antagonists (also known as “anti-androgens”) that were never FDA-approved for treatment of PCa [7, 101-103].
First generation AR antagonists
While steroidal AR antagonists showed efficacy compared to estrogen in the treatment of locally advanced or metastatic PCa [7, 100-103], their off-target effects on other steroid receptors have limited their use. This drawback emphasized the need to develop AR antagonists that lack the steroidal carbon backbone. The earliest compound to be synthesized with these characteristics was flutamide, the AR antagonistic activity of which was comparable to cyproterone and other steroidal AR antagonists in pre-clinical studies [104, 105] (Figure 3). Also at this time, the theory of “complete androgen blockade” or “combined androgen blockade” (CAB), essentially treatment with a combination of pre-receptor and receptor level targeting agents, was introduced by Dr. Fernand Labrie's laboratory in 1982 [106]. Together, these discoveries led to clinical studies to determine the effectiveness of combination therapy with leuprolide and flutamide versus leuprolide alone for patients with metastatic PCa [107]. Results of a phase III trial showed that CAB increased progression-free survival, where improvement of symptomatic disease was most pronounced during the first 12 weeks of treatment [107]. In 1989, flutamide was FDA-approved specifically for combination therapy with LHRH analogs for treatment of metastatic PCa as part of the CAB treatment regimen [7] (Table 2). In addition, two other non-steroidal AR antagonists that derive their structure from flutamide, nilutamide and bicalutamide, were also FDA-approved for combination therapy with LHRH agonists in the mid-1990s [7] (Table 2). Together, these AR antagonists comprise the group currently known as first-generation “anti-androgens.”
Drugs Targeted at Receptor Level.
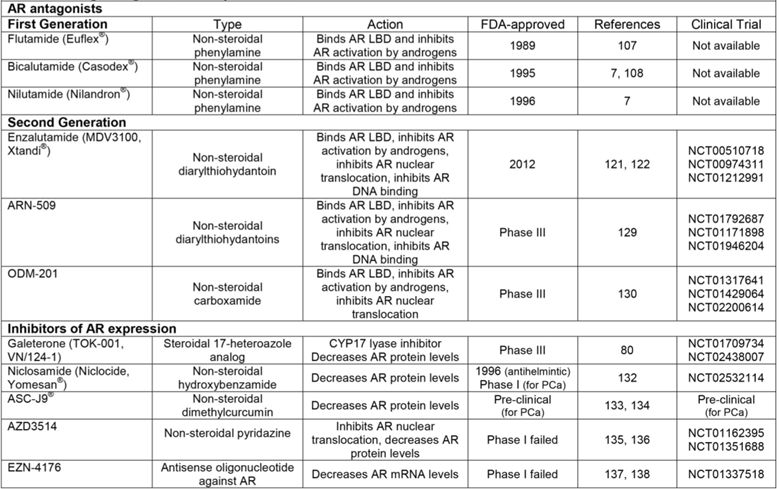
Of the first-generation AR antagonists, bicalutamide is the most widely studied and commonly used [7]. Indeed, clinical trials have shown that when in combination with an LHRH agonist, bicalutamide is superior to flutamide in time-to-treatment failure and better tolerated [108]. Numerous studies have been conducted to determine the underlying mechanisms driving bicalutamide-dependent AR inhibition. Bicalutamide slows AR nuclear localization [109], destabilizes the AR-DNA interaction specifically inhibiting AR's interaction with enhancer regions [110, 111], promotes the interaction of AR with co-repressors [111, 112], limits recruitment of AR co-activators [113], and inhibits histone acetylation at ARGs [111]. But despite lengthening the time to treatment failure for patients with metastatic PCa, CAB with first generation AR antagonists only delays development of CR-PCa [114]. Resistance to AR antagonists primarily occurs via the agonistic activity of these compounds towards AR mutants and the increased expression of ligand-independent AR splice variants [115, 116].
Second generation AR antagonists
To address the continuing challenge of therapy resistance in PCa, a comprehensive approach was conducted to identify more potent AR antagonists. In this context, the AR inhibitory activity of 200 derivatives of RU59063, an AR super-agonist, was assessed specifically in the presence of increased AR expression (a setting that approximates the CR-PCa disease state since AR protein levels are elevated in recurrent tumors and cell lines) [117-119]. This study identified enzalutamide, a second generation AR antagonist that has a four-fold increased binding affinity for AR compared to bicalutamide [118]. Other activities of enzalutamide that make it superior to first generation AR antagonists include inhibition of AR nuclear translocation and AR-DNA interaction [118] (Figure 3). The results of this seminal work brought enzalutamide to clinical trials. Phase I-II trials showed that enzalutamide as a monotherapy was well-tolerated, decreased PSA levels, and stabilized symptoms of both soft tissue and skeletal disease at all studied doses in CR-PCa patients [120] (NCT00510718, https://clinicaltrials.gov/). The first phase III clinical trial studied the effect of enzalutamide on overall survival in patients with CR-PCa who progressed following chemotherapy treatment with docetaxel [114, 121] (NCT00974311, https://clinicaltrials.gov/). At interim analysis, enzalutamide significantly increased overall survival over placebo (18.4 versus 13.6 months) and improved all secondary endpoints [121]. These data led to FDA-approval of enzalutamide in 2012 specifically for patients with progressive CR-PCa following chemotherapy [114] (Table 2). Another ongoing phase III trial is studying the effect of enzalutamide in chemotherapy-naïve patients with metastatic CR-PCa [122] (NCT01212991, https://clinicaltrials.gov/). Preliminary results of this study showed that enzalutamide decreased risk of progression and death as well as delayed time to initiation of chemotherapy compared to placebo [122]. Consequently, the FDA expanded its approval for enzalutamide to include chemotherapy-naïve CR-PCa patients [114]. In addition, further analysis of the pre-chemotherapy study determined that enzalutamide also improved quality of life as measured by responses to multiple standard clinical questionnaires [123].
The aim of evaluating potential second generation AR antagonists in an in vitro CR-PCa setting was to identify compounds that are impervious to known resistance mechanisms [118]. Regardless, patients who initially respond to enzalutamide still become resistant to therapy and progress following treatment [120]. Several resistance mechanisms have been determined for enzalutamide, including expression or overexpression of AR splice variants and mutant AR and alteration of other signaling pathways [124-127]. Further work on developing additional second generation AR antagonists identified ARN-509, a compound that is of the same structural class as enzalutamide [128] (Table 2). The activity of ARN-509 is also similar to enzalutamide in that ARN-509 specifically inhibits the nuclear localization and DNA binding of AR [128]. When the anti-tumor activity of ARN-509 was compared to enzalutamide in a preclinical human CR-PCa xenograft model, ARN-509 decreased tumor volume to equivalent levels, but required a lower dose than enzalutamide [128]. A phase I clinical study to determine the anti-tumor activity of ARN-509 in progressive metastatic CR-PCa showed that ARN-509 was able to decrease PSA levels as well as evidence of soft tissue disease in these patients [129] (NCT01792687, https://clinicaltrials.gov/). Results of a phase II study in metastatic CR-PCa have yet to be published (NCT01171898 https://clinicaltrials.gov/). In addition, a phase III clinical trial to study the efficacy of ARN-509 in patients with non-metastatic CR-PCa is currently recruiting (NCT01946204, https://clinicaltrials.gov/) (Table 2).
Even though ARN-509 is still in development, a potential mechanism of ARN-509 resistance has already been identified. Specifically, both enzalutamide and ARN-509 have been shown to act as agonists for the AR-F876L mutant [124]. In order to address the antagonist-to-agonist phenomenon occurring as a result of mutant AR expression, another second generation AR antagonist called ODM-201 was developed. ODM-201 is a carboxamide that also inhibits AR nuclear localization, but binds to the LBD and inhibits AR to a greater extent than both enzalutamide and ARN-509 [130] (Table 2). In addition, ODM-201 inhibits AR-F876L to a similar extent as wildtype AR [130]. Results of a phase I-II clinical trial for ODM-201 in progressive metastatic CR-PCa showed that ODM-201 decreased PSA levels by ≥ 50% at all doses by 12 weeks [131]. As of this writing, a phase III clinical trial is recruiting to study the efficacy of ODM-201 in high-risk non-metastatic CR-PCa (NCT02200614, https://clinicaltrials.gov/) (Table 2).
Inhibitors of AR expression
Resistance to AR LBD-targeted antagonists continues to develop as a result of cancer cells retaining dependency on AR for survival. As a result of this reliance on AR, researchers in PCa therapeutics have begun work on identifying compounds that decrease AR expression. The compound of this class that has progressed furthest in clinical development is galaterone (described above), a CYP17 lyase-specific inhibitor that also decreases expression of AR, AR-V7 and ARv567es [80-83]. A new phase III trial is currently recruiting to compare the anti-tumor effects of galaterone to enzalutamide specifically in men with metastatic CR-PCa expressing AR-V7 (NCT02438007, https://clinicaltrials.gov/) (Table 1 and Table 2). Since AR-V7 expression has been demonstrated to a greater extent in circulating tumor cells from patients who are resistant to enzalutamide [125], galaterone could be superior to enzalutamide by virtue of its ability to promote AR-V7 degradation.
Other compounds that inhibit AR expression have also been identified. For instance, niclosamide, an FDA-approved antihelmintic, promotes degradation of AR-V7 [132]. Importantly, preclinical studies have shown that niclosamide is effective in decreasing tumor growth of human xenografts derived from an enzalutamide-resistant cell line [132]. These results suggest that by triggering degradation of AR-V7, niclosamide could be effective for patients that progress following enzalutamide or prevent enzalutamide resistance altogether. As of this writing, a phase I clinical trial to test the efficacy of niclosamide in combination with enzalutamide for patients with AR-V7-positive CR-PCa has been received, but has yet to begin recruiting (NCT02532114, https://clinicaltrials.gov/) (Table 2). Work by Dr. Chawnshang Chang's lab group has identified the curcumin ASC-J9®, another compound capable of promoting AR degradation [133]. Mechanistically, ASC-J9® induces degradation of both AR and AR-V7 by interrupting AR's interaction with coregulators ARA55 and 70 [134]. While ASC-J9® has completed phase II clinical trials as a topical cream for treatment of acne vulgaris, it remains in preclinical development for PCa (Table 2).
While niclosamide and ASC-J9® have yet to be tested in the clinic, two other agents that showed promise in pre-clinical studies in their ability to decrease AR expression were unsuccessful in early clinical development. AZD3514 decreases AR expression in vitro; specifically, this pyridazine compound is capable of decreasing expression of full-length AR and inhibiting AR nuclear translocation [135]. Results from two parallel phase I clinical trials studying the efficacy of AZD3514 in patients with CR-PCa have recently been published [136] (NCT01162395, NCT01351688, https://clinicaltrials.gov/). Only 13% of patients treated with AZD3514 had a ≥ 50% decline in PSA and 17% of patients had decreased clinical indicators of soft tissue disease [136]. These marginal outcomes, as well as tolerability issues related to nausea and vomiting, have caused further clinical development of AZD3514 to be abandoned [136] (Table 2). Another drug that initially showed promise in preclinical studies but failed during phase I clinical trials is EZN-4176 (Table 2). As a locked antisense oligonucleotide, EZN-4176 is efficient in decreasing both AR transcript and protein levels in PCa cell lines and tumor xenografts [137]. Despite this positive biochemical profile, results from a phase I clinical trial in patients with progressive metastatic PCa showed that significant but reversible liver dysfunction, as measured by elevated serum aspartate transaminase (AST) and alanine aminotransferase (ALT), occurred as a result of EZN-4176 treatment [138] (NCT01337518, https://clinicaltrials.gov/). Since further dose escalation was limited, only one patient had a ≥ 50% decline in PSA with a tolerable dose of EZN-4176 [138]. These adverse events and partial anti-tumor effects discontinued further clinical development of EZN-4176. Despite the setbacks of AZD3514 and EZN-4176, degradation of AR as a therapeutic strategy for PCa remains viable with the latest clinical trials for galaterone and niclosamide. Indeed, these drugs may become the first in a new class recently referred to as selective androgen-receptor down-regulators (SARDs) or androgen receptor degradation enhancers (ARD enhancers). The long-term effectiveness of decreased AR expression against therapy resistance in PCa has yet to be determined.
Post-receptor level treatment strategies
Considering the near inevitability of developing resistance to available therapies, new treatment strategies for advanced PCa are always being explored. Many of these drugs neither target the T/DHT biosynthesis pathway nor AR directly, but instead target other proteins involved in AR stability and AR-dependent gene activation. Accordingly, these strategies are considered to be post-receptor targeted therapies. The multitude of possible targets, complexity of AR-transcriptional machinery, and intricacy of the molecular pathways that regulate ARGs has posed a significant barrier to clinical development of post-receptor level therapies for PCa. Consequently, no post-receptor level therapies are currently FDA-approved.
One group of co-chaperones that has been identified as a viable target in PCa is HSPs [139], proteins essential for AR protein stability, ligand interaction, nuclear localization, and DNA binding [37], as discussed earlier in this review. While two separate HSP90 inhibitors, 17-AAG and IPI-504, failed in phase II clinical trials [140, 141], onalespib and ganetespib have recently completed phase II clinical trials in patients with CR-PCa. As of this writing, results from the onalespib (NCT01685268) and ganetespib trials (NCT01270880, https://clinicaltrials.gov/) have yet to be published (Table 3). HSP27 is another co-chaperone that has been targeted in PCa therapeutics with the antisense oligonucleotide OGX-427 [142]. A phase II trials testing the effectiveness of OGX-427 with prednisone in patients with CR-PCa has been completed (NCT01120470, https://clinicaltrials.gov/), but results have yet to be published. In addition, another phase II trial is still recruiting to determine if adding OGX-427 for patients already receiving abiraterone can have a greater anti-tumor effect than abiraterone alone (NCT01681433, https://clinicaltrials.gov/) (Table 3).
Drugs Targeted at Post-receptor level.
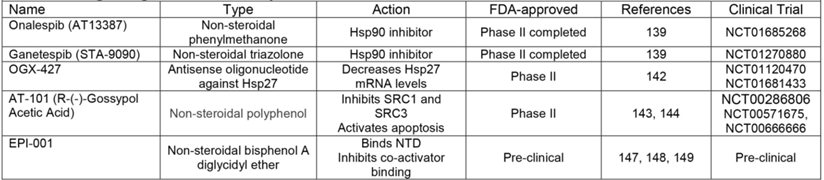
Another post-receptor treatment strategy includes targeting co-factors which are part of the active AR transcriptional complex. One agent that has been shown to inhibit SRC-1 and -3 is AT-101 [143]; in addition, this molecule also sensitizes PCa cells to apoptosis [144]. AT-101 showed minimal anti-tumor activity in a phase I/II trials in patients with CR-PCa [145] (NCT00286806, https://clinicaltrials.gov/), which led to additional phase II trials to determine the effectiveness of AT-101 either in combination with docetaxel or CAB. Despite the ability of AT-101 to promote apoptosis in PCa cell lines, AT-101 provided no additional anti-tumor activity when combined with docetaxel compared to docetaxel alone in phase II trials [146] (NCT00571675, https://clinicaltrials.gov/). The effectiveness of AT-101 in enhancing the anti-tumor effect of CAB has been studied in a phase II trial in patients with newly diagnosed metastatic PCa who are CAB-naïve (NCT00666666, https://clinicaltrials.gov/) (Table 3), but no results have published. Finally, one post-receptor agent that does directly target AR is EPI-001, an ether compound that binds the AF-1 domain in the AR N-terminus (the region that is most involved in co-activator recruitment) (Figure 1) [147-149]. While EPI-001 remains in preclinical development, its ability to inhibit AR transcriptional activity both in the presence and absence of androgens represents its specific potential to target ligand independent AR splice variants [147-149]. Altogether, while post-receptor treatment approaches signify the newest hypotheses surrounding possible therapeutics for CR-PCa, these strategies have yet to demonstrate their validity, superiority, or success in pre-clinical and clinical studies.
Concluding remarks
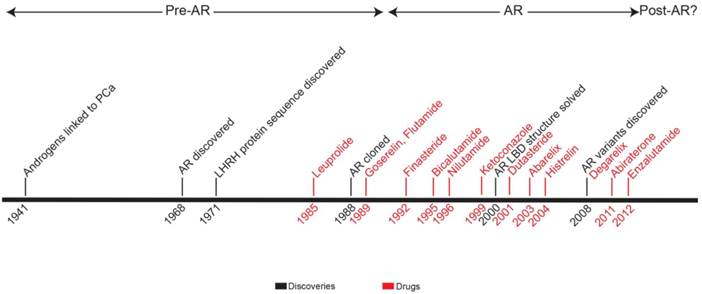
In this review, we present a timeline of therapeutic strategies for primary and advanced PCa therapeutic development. Since the 1940s, the field has built a solid model of PCa biology centered on androgen and AR (Figure 4). Treatment strategies take advantage of the positive relationship that exists among androgen, AR activation, and disease progression. By targeting the pre-receptor, receptor, and post-receptor nodes in androgen-AR signaling pathway, progress has been made in increasing the survival of patients with high-risk, locally-advanced, or metastatic PCa. However, if the extreme success of therapy for low-risk localized PCa is used as a standard, treatment efficacy for advanced PCa is lacking. Newly approved therapies for CR-PCa, abiraterone and enzalutamide, prolong life in these patients by just a few months [70, 71, 121, 122]—with the treatment of abiraterone providing the largest survival benefit, 8.2 months on average, for chemotherapy-naïve CR-PCa patients [71].
Timeline representing development of therapeutic strategies for prostate cancer. Years when key discoveries were made (black) and indicated drugs (red) were FDA-approved are shown. Based on major publications and clinical advances, the chronology is broken up into three eras: pre-AR, AR, and post-AR.
An additional treatment option (other than chemotherapy) for advanced PCa includes cell-based immunotherapy, with the FDA approval of Sipuleucel-T in 2010 [79]. As part of this treatment, a patient's hematopoietic progenitor cells are isolated and combined with a prostatic acid phosphatase-granulocyte-macrophage colony-stimulating factor fusion protein to produce antigen presenting cells. When reintroduced into the patients, these cells activate CD8+ T lymphocytes, which are then primed against cells expressing prostatic acid phosphatase—presumably PCa cells [150]. The theory behind this therapeutic strategy is promising—PCa cells are targeted for cell death directly, bypassing the need to alter intracellular signaling and inhibiting development of resistance mechanisms. However, in the three phase III trials performed for Sipuleucel-T, one showed an increased median survival of only 4.1 months [150], another only 4.5 months [151], and the last showed no significant increase in survival [152]. Therefore, the performance of Sipuleucel-T appears to be less than abiraterone and enzalutamide [70, 71, 121, 122]. In addition, time to disease progression was unchanged in patients who received Sipuleucel-T therapy compared to placebo in these clinical trials [152].
The drugs available to advanced PCa patients do not satisfy the needs of this population, especially since ADT substantially impacts quality of life [153] and can have adverse effects on cardiovascular [154] and musculoskeletal health [155] in an increasingly aging patient population. In addition, nearly all of these patients are destined to recur regardless of treatment regimens. This aggressive prostate malignancy perseveres largely as a result of reliance on AR and downstream signaling. Indeed, AR gene amplification, increased AR protein expression, emergence of AR mutants, expression of ligand-independent AR splice variants, and alteration in the signaling pathways and co-regulators that affect AR have all been documented in CR-PCa [116]. The role of AR at the nexus of underlying mechanisms for recurrence highlights the promise of agents that inhibit AR expression. Of these drugs, the biological profile of galeterone, in its ability to inhibit both CYP17 lyase and expression of AR, AR-V7, and ARv567es, is the most positive example. While galeterone has progressed furthest in clinical development, this multi-targeting agent has yet to prove its long-term therapeutic efficacy. The ultimate test for agents that inhibit AR expression is whether the loss of AR can eventually be circumvented by these recurrent tumors. In conclusion, despite significant progress in using drugs to manipulate androgen and AR-dependent signaling in high-risk PCa, a curative treatment strategy remains to be envisioned or discovered. Clinical trials testing novel agents continue, but the seemingly inevitability of recurrence for advanced PCa patients remains to be fully addressed by current therapies.
Abbreviations
ADT: Androgen-deprivation therapy; AF-1: Activation function domain 1; AF-2: Activation function domain 2; ALT: alanine aminotransferase; AR: Androgen receptor; ARG: Androgen-regulated gene; ARA: AR-associated; ARE: Androgen response element; AST: aspartate transaminase; BPH: Benign prostatic hyperplasia; CAB: Complete androgen blockade or combined androgen blockade; CDK6: Cyclin-dependent kinase 6; CDK2AP2: CDK2-associated protein 2; CGP: CREB-binding protein; CR: Castrate-resistant or castrate-recurrent; CYP17: Cytochrome P450-C17; DBD: DNA-binding domain; DHEA: Dehydroepiandrosterone; DHT: Dihydrotestosterone; GnRH: Gonadotropin-releasing hormone; HSP: Heat shock protein; IGF1-R: Insulin-like growth factor 1 receptor; LBD: Ligand-binding domain; LHRH: Luteinizing hormone releasing hormone; LH: Luteinizing hormone; MAP2K4: Mitogen activated kinase 2 kinase 4; NCoR: Nuclear receptor co-repressor; NES: Nuclear export signal; NLS: Nuclear localization sequence; NTD: N-terminal domain; PSA: Prostate specific antigen; SMRT: Silencing mediator for retinoid and thyroid hormone receptors; SRC: Steroid receptor co-activator; SUMO: Small ubiquitin-like modifier; Tip60: Tat interacting protein 60kDa; TIF: Transcriptional intermediary factor; WISP-2: Wnt-induced secreted protein-2.
Acknowledgements
This work was supported by the National Cancer Institute (R21CA181152 and R21CA183892 to S.K. and P30 CA016056 to Roswell Park Cancer Institute). The authors take sole responsibility for the content which may or may not represent the official views of the National Institutes of Health or National Cancer Institute.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Cancer Facts & Figures 2015. In: Society AC, editor. Atlanta: American Cancer Society. 2015
2. Thompson I, Thrasher JB, Aus G, Burnett AL, Canby-Hagino ED, Cookson MS. et al. Guideline for the management of clinically localized prostate cancer: 2007 update. The Journal of urology. 2007;177:2106-31
3. Kollmeier MA, Zelefsky MJ. How to select the optimal therapy for early-stage prostate cancer. Critical reviews in oncology/hematology. 2012;84(Suppl 1):e6-e15
4. Klotz L, Emberton M. Management of low risk prostate cancer-active surveillance and focal therapy. Nature reviews Clinical oncology. 2014;11:324-34
5. McLeod DG. Hormonal therapy: historical perspective to future directions. Urology. 2003;61:3-7
6. Dehm SM, Tindall DJ. Molecular regulation of androgen action in prostate cancer. Journal of cellular biochemistry. 2006;99:333-44
7. Helsen C, Van den Broeck T, Voet A, Prekovic S, Van Poppel H, Joniau S. et al. Androgen receptor antagonists for prostate cancer therapy. Endocrine-related cancer. 2014;21:T105-18
8. Huggins C, Hodges C. Studies on Prostatic Cancer. I. The Effect of Castration, of Estrogen and of Androgen Injection on Serum Phosphatases in Metastatic Carcinoma of the Prostate. Cancer Research. 1941;1:293-7
9. Anderson KM, Liao S. Selective retention of dihydrotestosterone by prostatic nuclei. Nature. 1968;219:277-9
10. Bruchovsky N, Wilson JD. The intranuclear binding of testosterone and 5-alpha-androstan-17-beta-ol-3-one by rat prostate. The Journal of biological chemistry. 1968;243:5953-60
11. Liao S, Fang S. Receptor-proteins for androgens and the mode of action of androgens on gene transcription in ventral prostate. Vitamins and hormones. 1969;27:17-90
12. Chang CS, Kokontis J, Liao ST. Molecular cloning of human and rat complementary DNA encoding androgen receptors. Science. 1988;240:324-6
13. Lubahn DB, Joseph DR, Sullivan PM, Willard HF, French FS, Wilson EM. Cloning of human androgen receptor complementary DNA and localization to the X chromosome. Science. 1988;240:327-30
14. Koochekpour S. Androgen receptor signaling and mutations in prostate cancer. Asian journal of andrology. 2010;12:639-57
15. Dehm SM, Tindall DJ. Alternatively spliced androgen receptor variants. Endocrine-related cancer. 2011;18:R183-96
16. Dehm SM, Tindall DJ. Androgen receptor structural and functional elements: role and regulation in prostate cancer. Mol Endocrinol. 2007;21:2855-63
17. Black BE, Paschal BM. Intranuclear organization and function of the androgen receptor. Trends in endocrinology and metabolism: TEM. 2004;15:411-7
18. van der Steen T, Tindall DJ, Huang H. Posttranslational modification of the androgen receptor in prostate cancer. International journal of molecular sciences. 2013;14:14833-59
19. Bevan CL, Hoare S, Claessens F, Heery DM, Parker MG. The AF1 and AF2 domains of the androgen receptor interact with distinct regions of SRC1. Molecular and cellular biology. 1999;19:8383-92
20. Alen P, Claessens F, Verhoeven G, Rombauts W, Peeters B. The androgen receptor amino-terminal domain plays a key role in p160 coactivator-stimulated gene transcription. Molecular and cellular biology. 1999;19:6085-97
21. He B, Kemppainen JA, Voegel JJ, Gronemeyer H, Wilson EM. Activation function 2 in the human androgen receptor ligand binding domain mediates interdomain communication with the NH(2)-terminal domain. The Journal of biological chemistry. 1999;274:37219-25
22. Li J, Fu J, Toumazou C, Yoon HG, Wong J. A role of the amino-terminal (N) and carboxyl-terminal (C) interaction in binding of androgen receptor to chromatin. Mol Endocrinol. 2006;20:776-85
23. Cleutjens KB, van der Korput HA, Ehren-van Eekelen CC, Sikes RA, Fasciana C, Chung LW. et al. A 6-kb promoter fragment mimics in transgenic mice the prostate-specific and androgen-regulated expression of the endogenous prostate-specific antigen gene in humans. Mol Endocrinol. 1997;11:1256-65
24. Cleutjens KB, van der Korput HA, van Eekelen CC, van Rooij HC, Faber PW, Trapman J. An androgen response element in a far upstream enhancer region is essential for high, androgen-regulated activity of the prostate-specific antigen promoter. Mol Endocrinol. 1997;11:148-61
25. Cleutjens KB, van Eekelen CC, van der Korput HA, Brinkmann AO, Trapman J. Two androgen response regions cooperate in steroid hormone regulated activity of the prostate-specific antigen promoter. The Journal of biological chemistry. 1996;271:6379-88
26. Balk SP, Ko YJ, Bubley GJ. Biology of prostate-specific antigen. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2003;21:383-91
27. Shaffer PL, Jivan A, Dollins DE, Claessens F, Gewirth DT. Structural basis of androgen receptor binding to selective androgen response elements. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:4758-63
28. Zhou ZX, Sar M, Simental JA, Lane MV, Wilson EM. A ligand-dependent bipartite nuclear targeting signal in the human androgen receptor. Requirement for the DNA-binding domain and modulation by NH2-terminal and carboxyl-terminal sequences. The Journal of biological chemistry. 1994;269:13115-23
29. Fu M, Wang C, Reutens AT, Wang J, Angeletti RH, Siconolfi-Baez L. et al. p300 and p300/cAMP-response element-binding protein-associated factor acetylate the androgen receptor at sites governing hormone-dependent transactivation. The Journal of biological chemistry. 2000;275:20853-60
30. Gaughan L, Logan IR, Cook S, Neal DE, Robson CN. Tip60 and histone deacetylase 1 regulate androgen receptor activity through changes to the acetylation status of the receptor. The Journal of biological chemistry. 2002;277:25904-13
31. Li Y, Lambert MH, Xu HE. Activation of nuclear receptors: a perspective from structural genomics. Structure. 2003;11:741-6
32. Saporita AJ, Zhang Q, Navai N, Dincer Z, Hahn J, Cai X. et al. Identification and characterization of a ligand-regulated nuclear export signal in androgen receptor. The Journal of biological chemistry. 2003;278:41998-2005
33. Xu K, Shimelis H, Linn DE, Jiang R, Yang X, Sun F. et al. Regulation of androgen receptor transcriptional activity and specificity by RNF6-induced ubiquitination. Cancer cell. 2009;15:270-82
34. Lin HK, Wang L, Hu YC, Altuwaijri S, Chang C. Phosphorylation-dependent ubiquitylation and degradation of androgen receptor by Akt require Mdm2 E3 ligase. The EMBO journal. 2002;21:4037-48
35. He B, Bai S, Hnat AT, Kalman RI, Minges JT, Patterson C. et al. An androgen receptor NH2-terminal conserved motif interacts with the COOH terminus of the Hsp70-interacting protein (CHIP). The Journal of biological chemistry. 2004;279:30643-53
36. Heemers HV, Tindall DJ. Androgen receptor (AR) coregulators: a diversity of functions converging on and regulating the AR transcriptional complex. Endocrine reviews. 2007;28:778-808
37. Cano LQ, Lavery DN, Bevan CL. Mini-review: Foldosome regulation of androgen receptor action in prostate cancer. Molecular and cellular endocrinology. 2013;369:52-62
38. Fu M, Rao M, Wang C, Sakamaki T, Wang J, Di Vizio D. et al. Acetylation of androgen receptor enhances coactivator binding and promotes prostate cancer cell growth. Molecular and cellular biology. 2003;23:8563-75
39. Massie CE, Lynch A, Ramos-Montoya A, Boren J, Stark R, Fazli L. et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. The EMBO journal. 2011;30:2719-33
40. Gomez L, Kovac JR, Lamb DJ. CYP17A1 inhibitors in castration-resistant prostate cancer. Steroids. 2015;95:80-7
41. Byar DP. Proceedings: The Veterans Administration Cooperative Urological Research Group's studies of cancer of the prostate. Cancer. 1973;32:1126-30
42. Sharifi N, Gulley JL, Dahut WL. Androgen deprivation therapy for prostate cancer. JAMA. 2005;294:238-44
43. Heemers HV. Targeting androgen receptor action for prostate cancer treatment: does the post-receptor level provide novel opportunities? International journal of biological sciences. 2014;10:576-87
44. Matsuo H, Baba Y, Nair RM, Arimura A, Schally AV. Structure of the porcine LH- and FSH-releasing hormone. I. The proposed amino acid sequence. Biochemical and biophysical research communications. 1971;43:1334-9
45. Teutonico D, Montanari S, Ponchel G. Leuprolide acetate: pharmaceutical use and delivery potentials. Expert opinion on drug delivery. 2012;9:343-54
46. Tolis G, Ackman D, Stellos A, Mehta A, Labrie F, Fazekas AT. et al. Tumor growth inhibition in patients with prostatic carcinoma treated with luteinizing hormone-releasing hormone agonists. Proceedings of the National Academy of Sciences of the United States of America. 1982;79:1658-62
47. Roach M 3rd, Izaguirre A. Goserelin acetate in combination with radiotherapy for prostate cancer. Expert opinion on pharmacotherapy. 2007;8:257-64
48. Ploussard G, Mongiat-Artus P. Triptorelin in the management of prostate cancer. Future oncology. 2013;9:93-102
49. Deeks ED. Histrelin: in advanced prostate cancer. Drugs. 2010;70:623-30
50. Seidenfeld J, Samson DJ, Hasselblad V, Aronson N, Albertsen PC, Bennett CL. et al. Single-therapy androgen suppression in men with advanced prostate cancer: a systematic review and meta-analysis. Annals of internal medicine. 2000;132:566-77
51. Gommersall LM, Hayne D, Shergill IS, Arya M, Wallace DM. Luteinising hormone releasing hormone analogues in the treatment of prostate cancer. Expert opinion on pharmacotherapy. 2002;3:1685-92
52. Van Poppel H, Klotz L. Gonadotropin-releasing hormone: an update review of the antagonists versus agonists. International journal of urology: official journal of the Japanese Urological Association. 2012;19:594-601
53. Theobald P, Porter J, Rivier C, Corrigan A, Hook W, Siraganian R. et al. Novel gonadotropin-releasing hormone antagonists: peptides incorporating modified N omega-cyanoguanidino moieties. Journal of medicinal chemistry. 1991;34:2395-402
54. Rivier JE, Jiang G, Porter J, Hoeger CA, Craig AG, Corrigan A. et al. Gonadotropin-releasing hormone antagonists: novel members of the azaline B family. Journal of medicinal chemistry. 1995;38:2649-62
55. Moul JW. Utility of LHRH antagonists for advanced prostate cancer. The Canadian journal of urology. 2014;21:22-7
56. Klotz L, Boccon-Gibod L, Shore ND, Andreou C, Persson BE, Cantor P. et al. The efficacy and safety of degarelix: a 12-month, comparative, randomized, open-label, parallel-group phase III study in patients with prostate cancer. BJU international. 2008;102:1531-8
57. Yin L, Hu Q. CYP17 inhibitors-abiraterone, C17,20-lyase inhibitors and multi-targeting agents. Nature reviews Urology. 2014;11:32-42
58. Geller J. Rationale for blockade of adrenal as well as testicular androgens in the treatment of advanced prostate cancer. Seminars in oncology. 1985;12:28-35
59. Locke JA, Guns ES, Lubik AA, Adomat HH, Hendy SC, Wood CA. et al. Androgen levels increase by intratumoral de novo steroidogenesis during progression of castration-resistant prostate cancer. Cancer Res. 2008;68:6407-15
60. Montgomery RB, Mostaghel EA, Vessella R, Hess DL, Kalhorn TF, Higano CS. et al. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res. 2008;68:4447-54
61. Titus MA, Schell MJ, Lih FB, Tomer KB, Mohler JL. Testosterone and dihydrotestosterone tissue levels in recurrent prostate cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2005;11:4653-7
62. Lam JS, Leppert JT, Vemulapalli SN, Shvarts O, Belldegrun AS. Secondary hormonal therapy for advanced prostate cancer. The Journal of urology. 2006;175:27-34
63. Loose DS, Kan PB, Hirst MA, Marcus RA, Feldman D. Ketoconazole blocks adrenal steroidogenesis by inhibiting cytochrome P450-dependent enzymes. The Journal of clinical investigation. 1983;71:1495-9
64. Trump DL, Havlin KH, Messing EM, Cummings KB, Lange PH, Jordan VC. High-dose ketoconazole in advanced hormone-refractory prostate cancer: endocrinologic and clinical effects. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 1989;7:1093-8
65. Small EJ, Baron AD, Fippin L, Apodaca D. Ketoconazole retains activity in advanced prostate cancer patients with progression despite flutamide withdrawal. The Journal of urology. 1997;157:1204-7
66. Potter GA, Barrie SE, Jarman M, Rowlands MG. Novel steroidal inhibitors of human cytochrome P45017 alpha (17 alpha-hydroxylase-C17,20-lyase): potential agents for the treatment of prostatic cancer. Journal of medicinal chemistry. 1995;38:2463-71
67. Jarman M, Barrie SE, Llera JM. The 16,17-double bond is needed for irreversible inhibition of human cytochrome p45017alpha by abiraterone (17-(3-pyridyl)androsta-5, 16-dien-3beta-ol) and related steroidal inhibitors. Journal of medicinal chemistry. 1998;41:5375-81
68. O'Donnell A, Judson I, Dowsett M, Raynaud F, Dearnaley D, Mason M. et al. Hormonal impact of the 17alpha-hydroxylase/C(17,20)-lyase inhibitor abiraterone acetate (CB7630) in patients with prostate cancer. British journal of cancer. 2004;90:2317-25
69. Attard G, Reid AH, Yap TA, Raynaud F, Dowsett M, Settatree S. et al. Phase I clinical trial of a selective inhibitor of CYP17, abiraterone acetate, confirms that castration-resistant prostate cancer commonly remains hormone driven. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2008;26:4563-71
70. de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L. et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364:1995-2005
71. Ryan CJ, Smith MR, de Bono JS, Molina A, Logothetis CJ, de Souza P. et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med. 2013;368:138-48
72. Sartor O. Combination therapy: Abiraterone prolongs survival in metastatic prostate cancer. Nature reviews Clinical oncology. 2011;8:515-6
73. Kaku T, Hitaka T, Ojida A, Matsunaga N, Adachi M, Tanaka T. et al. Discovery of orteronel (TAK-700), a naphthylmethylimidazole derivative, as a highly selective 17,20-lyase inhibitor with potential utility in the treatment of prostate cancer. Bioorganic & medicinal chemistry. 2011;19:6383-99
74. Yamaoka M, Hara T, Hitaka T, Kaku T, Takeuchi T, Takahashi J. et al. Orteronel (TAK-700), a novel non-steroidal 17,20-lyase inhibitor: effects on steroid synthesis in human and monkey adrenal cells and serum steroid levels in cynomolgus monkeys. The Journal of steroid biochemistry and molecular biology. 2012;129:115-28
75. Fizazi K, Jones R, Oudard S, Efstathiou E, Saad F, de Wit R. et al. Phase III, randomized, double-blind, multicenter trial comparing orteronel (TAK-700) plus prednisone with placebo plus prednisone in patients with metastatic castration-resistant prostate cancer that has progressed during or after docetaxel-based therapy: ELM-PC 5. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2015;33:723-31
76. Lu K. The Negative Is Not So Negative: What Do We Learn From Trials With Orteronel?. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2015
77. Rafferty SW, Eisner JR, Moore WR, Schotzinger RJ, Hoekstra WJ. Highly-selective 4-(1,2,3-triazole)-based P450c17a 17,20-lyase inhibitors. Bioorganic & medicinal chemistry letters. 2014;24:2444-7
78. Toren PJ, Kim S, Pham S, Mangalji A, Adomat H, Guns ES. et al. Anticancer activity of a novel selective CYP17A1 inhibitor in preclinical models of castrate-resistant prostate cancer. Molecular cancer therapeutics. 2015;14:59-69
79. Yin L, Hu Q, Hartmann RW. Recent progress in pharmaceutical therapies for castration-resistant prostate cancer. International journal of molecular sciences. 2013;14:13958-78
80. Njar VC, Brodie AM. Discovery and development of Galeterone (TOK-001 or VN/124-1) for the treatment of all stages of prostate cancer. Journal of medicinal chemistry. 2015;58:2077-87
81. Handratta VD, Vasaitis TS, Njar VC, Gediya LK, Kataria R, Chopra P. et al. Novel C-17-heteroaryl steroidal CYP17 inhibitors/antiandrogens: synthesis, in vitro biological activity, pharmacokinetics, and antitumor activity in the LAPC4 human prostate cancer xenograft model. Journal of medicinal chemistry. 2005;48:2972-84
82. Yu Z, Cai C, Gao S, Simon NI, Shen HC, Balk SP. Galeterone prevents androgen receptor binding to chromatin and enhances degradation of mutant androgen receptor. Clinical cancer research: an official journal of the American Association for Cancer Research. 2014;20:4075-85
83. Kwegyir-Afful AK, Senthilmurugan R, Purushottamachar P, Ramamurthy VP, Njar VC. Galeterone and VNPT55 induce proteasomal degradation of AR/AR-V7, induce significant apoptosis via cytochrome c release and suppress growth of castration resistant prostate cancer xenografts in vivo. Oncotarget. 2015
84. Aggarwal S, Thareja S, Verma A, Bhardwaj TR, Kumar M. An overview on 5alpha-reductase inhibitors. Steroids. 2010;75:109-53
85. Gormley GJ, Stoner E, Bruskewitz RC, Imperato-McGinley J, Walsh PC, McConnell JD. et al. The effect of finasteride in men with benign prostatic hyperplasia. The Finasteride Study Group. N Engl J Med. 1992;327:1185-91
86. Roehrborn CG, Boyle P, Nickel JC, Hoefner K, Andriole G, Aria A. et al. Efficacy and safety of a dual inhibitor of 5-alpha-reductase types 1 and 2 (dutasteride) in men with benign prostatic hyperplasia. Urology. 2002;60:434-41
87. Tiwari A, Krishna NS, Nanda K, Chugh A. Benign prostatic hyperplasia: an insight into current investigational medical therapies. Expert opinion on investigational drugs. 2005;14:1359-72
88. Iehle C, Radvanyi F, Gil Diez de Medina S, Ouafik LH, Gerard H, Chopin D. et al. Differences in steroid 5alpha-reductase iso-enzymes expression between normal and pathological human prostate tissue. The Journal of steroid biochemistry and molecular biology. 1999;68:189-95
89. Lacy JM, Kyprianou N. A tale of two trials: The impact of 5alpha-reductase inhibition on prostate cancer (Review). Oncology letters. 2014;8:1391-6
90. Thompson IM, Goodman PJ, Tangen CM, Lucia MS, Miller GJ, Ford LG. et al. The influence of finasteride on the development of prostate cancer. N Engl J Med. 2003;349:215-24
91. Andriole GL, Bostwick DG, Brawley OW, Gomella LG, Marberger M, Montorsi F. et al. Effect of dutasteride on the risk of prostate cancer. N Engl J Med. 2010;362:1192-202
92. FDA Drug Safety Communication. 5-alpha reductase inhibitors (5-ARIs) may increase the risk of a more serious form of prostate cancer. http://www.fda.gov/Drugs/DrugSafety/ucm258314.htm
93. Nickel JC. Comparison of clinical trials with finasteride and dutasteride. Reviews in urology. 2004;6(Suppl 9):S31-9
94. Azoulay L, Eberg M, Benayoun S, Pollak M. 5alpha-Reductase Inhibitors and the Risk of Cancer-Related Mortality in Men With Prostate Cancer. JAMA oncology. 2015;1:314-20
95. Denmeade SR, Jakobsen CM, Janssen S, Khan SR, Garrett ES, Lilja H. et al. Prostate-specific antigen-activated thapsigargin prodrug as targeted therapy for prostate cancer. Journal of the National Cancer Institute. 2003;95:990-1000
96. Fleshner NE, Lucia MS, Egerdie B, Aaron L, Eure G, Nandy I. et al. Dutasteride in localised prostate cancer management: the REDEEM randomised, double-blind, placebo-controlled trial. Lancet. 2012;379:1103-11
97. Schroder F, Bangma C, Angulo JC, Alcaraz A, Colombel M, McNicholas T. et al. Dutasteride treatment over 2 years delays prostate-specific antigen progression in patients with biochemical failure after radical therapy for prostate cancer: results from the randomised, placebo-controlled Avodart After Radical Therapy for Prostate Cancer Study (ARTS). European urology. 2013;63:779-87
98. Chu FM, Sartor O, Gomella L, Rudo T, Somerville MC, Hereghty B. et al. A randomised, double-blind study comparing the addition of bicalutamide with or without dutasteride to GnRH analogue therapy in men with non-metastatic castrate-resistant prostate cancer. European journal of cancer. 2015;51:1555-69
99. Mohler JL. Concept and viability of androgen annihilation for advanced prostate cancer. Cancer. 2014;120:2628-37
100. Schroder FH. Cyproterone acetate-mechanism of action and clinical effectiveness in prostate cancer treatment. Cancer. 1993;72:3810-5
101. Jacobi GH, Altwein JE, Kurth KH, Basting R, Hohenfellner R. Treatment of advanced prostatic cancer with parenteral cyproterone acetate: a phase III randomised trial. British journal of urology. 1980;52:208-15
102. Pavone-Macaluso M, de Voogt HJ, Viggiano G, Barasolo E, Lardennois B, de Pauw M. et al. Comparison of diethylstilbestrol, cyproterone acetate and medroxyprogesterone acetate in the treatment of advanced prostatic cancer: final analysis of a randomized phase III trial of the European Organization for Research on Treatment of Cancer Urological Group. The Journal of urology. 1986;136:624-31
103. Venner P. Megestrol acetate in the treatment of metastatic carcinoma of the prostate. Oncology. 1992;49(Suppl 2):22-7
104. Neri R, Florance K, Koziol P, Van Cleave S. A biological profile of a nonsteroidal antiandrogen, SCH 13521 (4'-nitro-3'trifluoromethylisobutyranilide). Endocrinology. 1972;91:427-37
105. Labrie F. Mechanism of action and pure antiandrogenic properties of flutamide. Cancer. 1993;72:3816-27
106. Lefebvre FA, Seguin C, Belanger A, Caron S, Sairam MR, Raynaud JP. et al. Combined long-term treatment with an LHRH agonist and a pure antiandrogen blocks androgenic influence in the rat. Prostate. 1982;3:569-78
107. Crawford ED, Eisenberger MA, McLeod DG, Spaulding JT, Benson R, Dorr FA. et al. A controlled trial of leuprolide with and without flutamide in prostatic carcinoma. N Engl J Med. 1989;321:419-24
108. Schellhammer P, Sharifi R, Block N, Soloway M, Venner P, Patterson AL. et al. Maximal androgen blockade for patients with metastatic prostate cancer: outcome of a controlled trial of bicalutamide versus flutamide, each in combination with luteinizing hormone-releasing hormone analogue therapy. Casodex Combination Study Group. Urology. 1996;47:54-60 discussion 80-4
109. Tyagi RK, Lavrovsky Y, Ahn SC, Song CS, Chatterjee B, Roy AK. Dynamics of intracellular movement and nucleocytoplasmic recycling of the ligand-activated androgen receptor in living cells. Molecular endocrinology (Baltimore, Md). 2000;14:1162-74
110. Farla P, Hersmus R, Trapman J, Houtsmuller AB. Antiandrogens prevent stable DNA-binding of the androgen receptor. J Cell Sci. 2005;118:4187-98
111. Kang Z, Janne OA, Palvimo JJ. Coregulator recruitment and histone modifications in transcriptional regulation by the androgen receptor. Mol Endocrinol. 2004;18:2633-48
112. Hodgson MC, Astapova I, Cheng S, Lee LJ, Verhoeven MC, Choi E. et al. The androgen receptor recruits nuclear receptor CoRepressor (N-CoR) in the presence of mifepristone via its N and C termini revealing a novel molecular mechanism for androgen receptor antagonists. The Journal of biological chemistry. 2005;280:6511-9
113. Baek SH, Ohgi KA, Nelson CA, Welsbie D, Chen C, Sawyers CL. et al. Ligand-specific allosteric regulation of coactivator functions of androgen receptor in prostate cancer cells. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:3100-5
114. Bambury RM, Scher HI. Enzalutamide: Development from bench to bedside. Urologic oncology. 2015;33:280-8
115. Culig Z. Targeting the androgen receptor in prostate cancer. Expert opinion on pharmacotherapy. 2014;15:1427-37
116. Karantanos T, Corn PG, Thompson TC. Prostate cancer progression after androgen deprivation therapy: mechanisms of castrate resistance and novel therapeutic approaches. Oncogene. 2013;32:5501-11
117. Van Dort ME, Robins DM, Wayburn B. Design, synthesis, and pharmacological characterization of 4-[4, 4-dimethyl-3-(4-hydroxybutyl)-5-oxo-2-thioxo-1-imidazolidinyl]- 2-iodobenzonitrile as a high-affinity nonsteroidal androgen receptor ligand. Journal of medicinal chemistry. 2000;43:3344-7
118. Tran C. Development of a Second-Generation Antiandrogen for Treatment of Advanced Prostate Cancer. Science. 2006
119. Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R. et al. Molecular determinants of resistance to antiandrogen therapy. Nature medicine. 2004;10:33-9
120. Scher HI, Beer TM, Higano CS, Anand A, Taplin ME, Efstathiou E. et al. Antitumour activity of MDV3100 in castration-resistant prostate cancer: a phase 1-2 study. Lancet. 2010;375:1437-46
121. Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K. et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012;367:1187-97
122. Beer TM, Armstrong AJ, Rathkopf DE, Loriot Y, Sternberg CN, Higano CS. et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N Engl J Med. 2014;371:424-33
123. Loriot Y, Miller K, Sternberg CN, Fizazi K, De Bono JS, Chowdhury S. et al. Effect of enzalutamide on health-related quality of life, pain, and skeletal-related events in asymptomatic and minimally symptomatic, chemotherapy-naive patients with metastatic castration-resistant prostate cancer (PREVAIL): results from a randomised, phase 3 trial. The Lancet Oncology. 2015;16:509-21
124. Joseph JD, Lu N, Qian J, Sensintaffar J, Shao G, Brigham D. et al. A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer Discov. 2013;3:1020-9
125. Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC. et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med. 2014;371:1028-38
126. Nadiminty N, Tummala R, Liu C, Yang J, Lou W, Evans CP. et al. NF-kappaB2/p52 induces resistance to enzalutamide in prostate cancer: role of androgen receptor and its variants. Molecular cancer therapeutics. 2013;12:1629-37
127. Korpal M, Korn JM, Gao X, Rakiec DP, Ruddy DA, Doshi S. et al. An F876L mutation in androgen receptor confers genetic and phenotypic resistance to MDV3100 (enzalutamide). Cancer Discov. 2013;3:1030-43
128. Clegg NJ, Wongvipat J, Joseph JD, Tran C, Ouk S, Dilhas A. et al. ARN-509: a novel antiandrogen for prostate cancer treatment. Cancer Res. 2012;72:1494-503
129. Rathkopf DE, Morris MJ, Fox JJ, Danila DC, Slovin SF, Hager JH. et al. Phase I study of ARN-509, a novel antiandrogen, in the treatment of castration-resistant prostate cancer. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2013;31:3525-30
130. Moilanen AM, Riikonen R, Oksala R, Ravanti L, Aho E, Wohlfahrt G. et al. Discovery of ODM-201, a new-generation androgen receptor inhibitor targeting resistance mechanisms to androgen signaling-directed prostate cancer therapies. Scientific reports. 2015;5:12007
131. Fizazi K, Massard C, Bono P, Jones R, Kataja V, James N. et al. Activity and safety of ODM-201 in patients with progressive metastatic castration-resistant prostate cancer (ARADES): an open-label phase 1 dose-escalation and randomised phase 2 dose expansion trial. The Lancet Oncology. 2014;15:975-85
132. Liu C, Lou W, Zhu Y, Nadiminty N, Schwartz CT, Evans CP. et al. Niclosamide inhibits androgen receptor variants expression and overcomes enzalutamide resistance in castration-resistant prostate cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2014;20:3198-210
133. Lai KP, Huang CK, Chang YJ, Chung CY, Yamashita S, Li L. et al. New therapeutic approach to suppress castration-resistant prostate cancer using ASC-J9 via targeting androgen receptor in selective prostate cells. Am J Pathol. 2013;182:460-73
134. Yamashita S, Lai KP, Chuang KL, Xu D, Miyamoto H, Tochigi T. et al. ASC-J9 suppresses castration-resistant prostate cancer growth through degradation of full-length and splice variant androgen receptors. Neoplasia. 2012;14:74-83
135. Loddick SA, Ross SJ, Thomason AG, Robinson DM, Walker GE, Dunkley TP. et al. AZD3514: a small molecule that modulates androgen receptor signaling and function in vitro and in vivo. Molecular cancer therapeutics. 2013;12:1715-27
136. Omlin A, Jones RJ, van der Noll R, Satoh T, Niwakawa M, Smith SA. et al. AZD3514, an oral selective androgen receptor down-regulator in patients with castration-resistant prostate cancer - results of two parallel first-in-human phase I studies. Invest New Drugs. 2015;33:679-90
137. Zhang Y, Castaneda S, Dumble M, Wang M, Mileski M, Qu Z. et al. Reduced expression of the androgen receptor by third generation of antisense shows antitumor activity in models of prostate cancer. Molecular cancer therapeutics. 2011;10:2309-19
138. Bianchini D, Omlin A, Pezaro C, Lorente D, Ferraldeschi R, Mukherji D. et al. First-in-human Phase I study of EZN-4176, a locked nucleic acid antisense oligonucleotide to exon 4 of the androgen receptor mRNA in patients with castration-resistant prostate cancer. British journal of cancer. 2013;109:2579-86
139. De Maeseneer DJ, Van Praet C, Lumen N, Rottey S. Battling resistance mechanisms in antihormonal prostate cancer treatment: Novel agents and combinations. Urologic oncology. 2015
140. Oh WK, Galsky MD, Stadler WM, Srinivas S, Chu F, Bubley G. et al. Multicenter phase II trial of the heat shock protein 90 inhibitor, retaspimycin hydrochloride (IPI-504), in patients with castration-resistant prostate cancer. Urology. 2011;78:626-30
141. Heath EI, Hillman DW, Vaishampayan U, Sheng S, Sarkar F, Harper F. et al. A phase II trial of 17-allylamino-17-demethoxygeldanamycin in patients with hormone-refractory metastatic prostate cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2008;14:7940-6
142. Rocchi P, Jugpal P, So A, Sinneman S, Ettinger S, Fazli L. et al. Small interference RNA targeting heat-shock protein 27 inhibits the growth of prostatic cell lines and induces apoptosis via caspase-3 activation in vitro. BJU international. 2006;98:1082-9
143. Wang Y, Lonard DM, Yu Y, Chow DC, Palzkill TG, O'Malley BW. Small molecule inhibition of the steroid receptor coactivators, SRC-3 and SRC-1. Mol Endocrinol. 2011;25:2041-53
144. Meng Y, Tang W, Dai Y, Wu X, Liu M, Ji Q. et al. Natural BH3 mimetic (-)-gossypol chemosensitizes human prostate cancer via Bcl-xL inhibition accompanied by increase of Puma and Noxa. Molecular cancer therapeutics. 2008;7:2192-202
145. Liu G, Kelly WK, Wilding G, Leopold L, Brill K, Somer B. An open-label, multicenter, phase I/II study of single-agent AT-101 in men with castrate-resistant prostate cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2009;15:3172-6
146. Sonpavde G, Matveev V, Burke JM, Caton JR, Fleming MT, Hutson TE. et al. Randomized phase II trial of docetaxel plus prednisone in combination with placebo or AT-101, an oral small molecule Bcl-2 family antagonist, as first-line therapy for metastatic castration-resistant prostate cancer. Annals of oncology: official journal of the European Society for Medical Oncology / ESMO. 2012;23:1803-8
147. Andersen RJ, Mawji NR, Wang J, Wang G, Haile S, Myung JK. et al. Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor. Cancer cell. 2010;17:535-46
148. Sadar MD. Small molecule inhibitors targeting the "achilles' heel" of androgen receptor activity. Cancer Res. 2011;71:1208-13
149. Myung JK, Banuelos CA, Fernandez JG, Mawji NR, Wang J, Tien AH. et al. An androgen receptor N-terminal domain antagonist for treating prostate cancer. The Journal of clinical investigation. 2013;123:2948-60
150. Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF. et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363:411-22
151. Small EJ, Schellhammer PF, Higano CS, Redfern CH, Nemunaitis JJ, Valone FH. et al. Placebo-controlled phase III trial of immunologic therapy with sipuleucel-T (APC8015) in patients with metastatic, asymptomatic hormone refractory prostate cancer. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2006;24:3089-94
152. Higano CS, Schellhammer PF, Small EJ, Burch PA, Nemunaitis J, Yuh L. et al. Integrated data from 2 randomized, double-blind, placebo-controlled, phase 3 trials of active cellular immunotherapy with sipuleucel-T in advanced prostate cancer. Cancer. 2009;115:3670-9
153. Rhee H, Gunter JH, Heathcote P, Ho K, Stricker P, Corcoran NM. et al. Adverse effects of androgen-deprivation therapy in prostate cancer and their management. BJU international. 2015;115(Suppl 5):3-13
154. Edelman S, Butler J, Hershatter BW, Khan MK. The effects of androgen deprivation therapy on cardiac function and heart failure: implications for management of prostate cancer. Clinical genitourinary cancer. 2014;12:399-407
155. Cheung AS, Zajac JD, Grossmann M. Muscle and bone effects of androgen deprivation therapy: current and emerging therapies. Endocrine-related cancer. 2014;21:R371-94
Author Biography
 Dr. Shahriar Koochekpour is a professor of cancer genetics, urology, and oncology at Roswell Park Cancer Institute, Buffalo, New York. He has authored and coauthored over 70 publications including Clinical Cancer Research, Cancer Research, Molecular Cancer, Molecular and Cellular Biology. He holds two US and International Patents and established two African American prostate cancer cell lines. Dr. Koochekpour's current basic and translational research interests include: (a) Development of therapeutic strategies targeted at AR and its spliced variants in prostate cancer; (b) Discovery of biomarkers of prostate cancer aggressiveness and progression; (c) Investigating inter-racial disparity of prostate cancer; and (d) Mechanistic understanding of castrate-recurrent progression of prostate cancer.
Dr. Shahriar Koochekpour is a professor of cancer genetics, urology, and oncology at Roswell Park Cancer Institute, Buffalo, New York. He has authored and coauthored over 70 publications including Clinical Cancer Research, Cancer Research, Molecular Cancer, Molecular and Cellular Biology. He holds two US and International Patents and established two African American prostate cancer cell lines. Dr. Koochekpour's current basic and translational research interests include: (a) Development of therapeutic strategies targeted at AR and its spliced variants in prostate cancer; (b) Discovery of biomarkers of prostate cancer aggressiveness and progression; (c) Investigating inter-racial disparity of prostate cancer; and (d) Mechanistic understanding of castrate-recurrent progression of prostate cancer.
 Dr. Kristine M Wadosky earned her Ph. D. degree in Pathology in 2014 at the University of North Carolina at Chapel Hill, Chapel Hill, NC, USA. In her dissertation work, she studied nuclear receptor signaling and protein degradation pathways in cardiac biology and heart failure. Her pre-doctoral work was funded by a fellowship awarded to her by the American Heart Association. Dr. Wadosky is continuing her training as a postdoctoral fellow in Prof. Shahriar Koochekpour's research team at Roswell Park Cancer Institute in Buffalo, NY, USA. Her current research aims to understand the mechanisms underlying recurrence of advanced prostate cancer.
Dr. Kristine M Wadosky earned her Ph. D. degree in Pathology in 2014 at the University of North Carolina at Chapel Hill, Chapel Hill, NC, USA. In her dissertation work, she studied nuclear receptor signaling and protein degradation pathways in cardiac biology and heart failure. Her pre-doctoral work was funded by a fellowship awarded to her by the American Heart Association. Dr. Wadosky is continuing her training as a postdoctoral fellow in Prof. Shahriar Koochekpour's research team at Roswell Park Cancer Institute in Buffalo, NY, USA. Her current research aims to understand the mechanisms underlying recurrence of advanced prostate cancer.
![]() Corresponding author: Dr. Shahriar Koochekpour, Departments of Cancer Genetics and Urology, Center for Genetics and Pharmacology, Roswell Park Cancer Institute, Elm and Carlton Streets, Buffalo, NY, 14263, USA, Telephone: 716-845-3345; Fax: 716-845-1698; Email: Shahriar.Koochekpourorg.
Corresponding author: Dr. Shahriar Koochekpour, Departments of Cancer Genetics and Urology, Center for Genetics and Pharmacology, Roswell Park Cancer Institute, Elm and Carlton Streets, Buffalo, NY, 14263, USA, Telephone: 716-845-3345; Fax: 716-845-1698; Email: Shahriar.Koochekpourorg.