Impact Factor ISSN: 1449-2288
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Material and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2016; 12(4):466-477. doi:10.7150/ijbs.12815 This issue Cite
Research Paper
Hyperglycemia Differentially Affects Maternal and Fetal DNA Integrity and DNA Damage Response
Jusciele B. Moreli1, Janine H. Santos2, Aline Rodrigues Lorenzon-Ojea3, Simone Corrêa-Silva1,3, Rodrigo S. Fortunato4, Clarissa Ribeiro Rocha5, Marilza V. Rudge1, Débora C. Damasceno1, Estela Bevilacqua3, Iracema M. Calderon1 ![]()
1. Graduate Program in Gynecology, Obstetrics and Mastology, Botucatu Medical School, São Paulo State University / UNESP, São Paulo, Brazil.
2. Laboratory of Molecular Carcinogenesis, National Institute of Environmental Health Sciences / NIEHS, North Carolina, USA.
3. Department of Cell and Developmental Biology, Institute of Biomedical Sciences, University of São Paulo / USP, São Paulo, Brazil.
4. Laboratory of Molecular Radiobiology, Carlos Chagas Filho Biophysics Institute, Federal University of Rio de Janeiro / UFRJ, Rio de Janeiro, Brazil.
5. DNA Repair Laboratory, Department of Microbiology, Institute of Biomedical Sciences, University of São Paulo / USP, São Paulo, Brazil.
Received 2015-5-28; Accepted 2016-1-13; Published 2016-2-12
Abstract

Objective: Investigate the DNA damage and its cellular response in blood samples from both mother and the umbilical cord of pregnancies complicated by hyperglycemia. Methods: A total of 144 subjects were divided into 4 groups: normoglycemia (ND; 46 cases), mild gestational hyperglycemia (MGH; 30 cases), gestational diabetes mellitus (GDM; 45 cases) and type-2 diabetes mellitus (DM2; 23 cases). Peripheral blood mononuclear cell (PBMC) isolation and/or leukocytes from whole maternal and umbilical cord blood were obtained from all groups at delivery. Nuclear and mitochondrial DNA damage were measured by gene-specific quantitative PCR, and the expression of mRNA and proteins involved in the base excision repair (BER) pathway were assessed by real-time qPCR and Western blot, respectively. Apoptosis was measured in vitro experiments by caspase 3/7 activity and ATP levels. Results: GDM and DM2 groups were characterized by an increase in oxidative stress biomarkers, an increase in nuclear and mitochondrial DNA damage, and decreased expression of mRNA (APE1, POLβ and FEN1) and proteins (hOGG1, APE1) involved in BER. The levels of hyperglycemia were associated with the in vitro apoptosis pathway. Blood levels of DNA damage in umbilical cord were similar among the groups. Newborns of diabetic mothers had increased expression of BER mRNA (APE1, POLβ and FEN1) and proteins (hOGG1, APE1, POLβ and FEN1). A diabetes-like environment was unable to induce apoptosis in the umbilical cord blood cells. Conclusions: Our data show relevant asymmetry between maternal and fetal blood cell susceptibility to DNA damage and apoptosis induction. Maternal cells seem to be more predisposed to changes in an adverse glucose environment. This may be due to differential ability in upregulating multiple genes involved in the activation of DNA repair response, especially the BER mechanism. However if this study shows a more effective adaptive response by the fetal organism, it also calls for further studies to determine the limit of this response that definitely changes the fate of a fetus under these conditions of cellular stress.
Keywords: Diabetes, Pregnancy, DNA damage, DNA repair.
Introduction
Diabetes mellitus (DM) is a metabolic disease characterized by hyperglycemia, resulting from a defect in insulin action and/or production [1]. In pregnancy, hyperglycemia poses both short-term and long-term risks to the health of women and their offspring [2]. Newborns of hyperglycemic mothers have an increased risk for malformations, macrossomia, hypoxia and perinatal death, these being associated with hypoglycemia, hyperbilirubinemia, hyperinsulinemia and hyperleptinemia [3-5]. During adult life, such metabolic alterations increase the risk of metabolic, cardiovascular and malignant diseases [2,5].
Oxidative stress is widely explored as a key factor in complications related to hyperglycemia, including alterations in embryonic and fetal development during pregnancy [6-8]. The fundamental explanation is that hyperglycemia leads to overproduction of mitochondrial reactive oxygen species (ROS), and thus induces protein oxidation, lipid peroxidation and DNA damage in mitochondrial (mtDNA) and nuclear DNA (nDNA) [6,9-11]. Different processes can remove oxidized lipids and proteins; however, DNA has to be repaired or, in the case of some types of mtDNA damage, the genome must be removed [10,11].
mtDNA is more vulnerable than nDNA to a variety of genotoxins, including ROS-mediated lesions [10-12]. One of the reasons that underlies this observation is that only base excision repair (BER) operates in mitochondria [13]. BER is the main mechanism for removal of ROS-induced lesions i.e., oxidized bases, AP sites and single-strand breaks. BER mechanism is initiated by DNA glycosylases (i.e., human 8-oxoguanine DNA glycosylase - hOGG1), which recognize and remove specifically damaged or inappropriate bases, forming apurinic/apyrimidinic (AP) sites. These are cleaved by an AP endonuclease, e.g. human AP endonuclease 1 (APE1), resulting in a single-strand break. This break can be processed by either short-patch or long-patch BER; in the former, APE1 interacts with DNA polymerase beta (POLβ), leading to single-nucleotide gap synthesis. In long-patch BER, APE1 similarly interacts with Flap endonuclease 1 (FEN1) for synthesis of 2-10 new nucleotides [14]. Repair capacity of DNA damage is critical for the maintenance of genome stability as, when not removed, DNA damage can result in apoptosis, an irreversible cell cycle checkpoint (cell senescence) or mutations [9,15]. Studies investigating the association between genome instability and hyperglycemia are scarce; data in human diabetic pregnancies are even more limited. For instance, it has been shown that patients with DM type 2 (DM2) have nuclear DNA damage, increased lymphocyte apoptosis, and decreased capacity to repair damage caused by hydrogen peroxide (H2O2) and doxorubicin, two well-known inducers of oxidative damage [16,17].A pilot study of healthy pregnant women on DNA damage and risk of GDM has been carried out but pregnant diabetic women were not included [18]. Data from animal models support the association between diabetes and DNA damage. Indeed, nuclear DNA damage in maternal and newborns leukocytes has been seen in streptozotocin-induced diabetes models in rats [19-21].
The hypothesis was tested that hyperglycemia-induced oxidative stress, decreases maternal and/or fetal DNA integrity, increasing cell death in peripheral blood cells during pregnancy. This may be associated either with the adverse outcomes seen in diabetic pregnancies or to factors that largely may determine newborn's health in adulthood. Our data indicate that while hyperglycemia does increase DNA damage and cell death in maternal cells; fetal cells are protected from these effects in the intrauterine environment.
Material and Methods
Ethical statement
This study was conducted in the Diabetes and Pregnancy Service of the Botucatu Medical School/UNESP, Brazil, with the approval of the local Research Ethics Committee (protocol #507/2012). Written informed consent was obtained from all subjects, according to the principles of the Declaration of Helsinki.
Study design and subjects selection
One hundred forty-four pregnant women with hyperglycemic disorders were enrolled to this cross-sectional study. Pregnant women with DM2 (n = 23) were referred to our service with a confirmed diagnosis. The research subjects without pre-gestational diabetes underwent 75-g glucose tolerance test (75-gGTT), recommended by the American Diabetes Association [1], and the glucose profile (GP) test as recommended by Rudge [22], between 24th and 28th gestational weeks. According to the 75-gGTT and GP results, pregnant women were classified into the following study groups: non-diabetic (ND; normal 75-g GTT and GP; n = 46), mild gestational hyperglycemia (MGH; normal 75-g GTT and abnormal GP; n= 30) or gestational diabetes mellitus (GDM; abnormal 75-g GTT first reported during the pregnancy; n = 45). Supporting information can be assessed in Figure S1.
The inclusion criteria were as follows: (a) hyperglycemia defined at the minimum gestational age of 28-30 weeks for women with MGH and GDM, and 20 weeks for DM2 pregnant women; (b) prenatal and delivery care at the Service; (c) absence of clinically diagnosed infections and negative serology for HIV and syphilis, multiple pregnancies, DM1, fetal malformations, fetal death, alcohol or illicit drugs habits; (d) deliveries before the 36th week of gestation.
In MGH or GDM pregnant women, hyperglycemia was controlled by a combination of changes in lifestyle, individualized diet and exercise. Insulin was used when the glycemic goals were not achieved. Patients with DM2 followed the same protocol but received insulin therapy from the start of treatment to replace oral hypoglycemic agents. The goals of maternal glycemic control were fasting glucose ≤ 95 mg/dL, 1 h postmeal ≤ 140 mg/dL, and 2 h postmeal ≤ 120 mg/dL, resulting in a daily glycemic mean ≤ 120 mg/dL [1].
Sample collection
Maternal blood samples were collected at 36th week of pregnancy, prior to the beginning of labor. The umbilical cord blood was collected intra-delivery, shortly after clamping. The samples of maternal or umbilical cord blood were collected in Vacutainer tubes (Becton Dickinson, USA) treated with EDTA, serum or heparin tubes.
Characterization of the population
Characterization of the population of pregnant women included information on , age, body mass index (BMI) in pre-pregnancy and third trimester of pregnancy, weight gain during pregnancy, gestational age at delivery, hypertension (gestational or chronic hypertension), glycemic mean (GM) and glycated hemoglobin (HbA1c) levels as well as markers of oxidative stress (Table 1, Figure 1). GM was calculated from the arithmetic mean of plasma glucose levels measured in all glucose profiles during treatment. Plasma glucose levels were measured by the oxidase method (Glucose Analyzer II Beckman, Fullerton, CA, USA) and HbA1c levels by chromatography (high-performance liquid chromatography—D10™ Hemoglobin Testing System, Bio-Rad Laboratories, Hercules, CA, USA).
Maternal clinical background and perinatal results.
| ND | MGH | GDM | DM2 | p | |
|---|---|---|---|---|---|
| Mothers | |||||
| Age (years) | 26.1±7.9a0 | 28.4±6.4a0 | 30.9±4.9b0 | 33.2±7.1b0 | <0.0001 |
| BMI 1 (kg/m2) | 26.4±5.0a1 | 31.6±7.9b1 | 33.3±7.1b1 | 34.8±5.9b1 | < 0.0001 |
| BMI 2 (kg/m2) | 31.1±5.5a2 | 35.0±7.3a2 | 37.4±6.5b2 | 37.2±9.9b2 | 0.0003 |
| Weight gain (kg) | 12.3±5.1 | 9.9±6.1 | 10.3±8.0 | 10.2±6.5 | ns |
| GA (weeks) | 39.5±1.7a3 | 39.3±1.1a3 | 38.9±1.3a3 | 37.5±0.8b3 | <0.0001 |
| Hypertension | 03 (6.52) | 12 (40.00) | 07 (15.55) | 11 (47.82) | ns |
| GM (mg/dL) | 83.1±7.8a5 | 91.3±11.7b5 | 101.9±14.2c5 | 109.6±13.8d5 | < 0.0001 |
| HbA1c (%) | 5.3±0.4a6 | 5.5±0.5b6 | 5.6±0.5b6 | 6.5±1.1c6 | < 0.0001 |
| Newborns | |||||
| Glycemia (mg/dL) | 65.5±19.3 | 61.50±15.9 | 73.5±30.9 | 68.9±22.6 | ns |
| Insulin (μU/mL) | 5.3±5.4 | 9.3±13.7 | 8.9±9.1 | 8.0±5.5 | ns |
| Leptin (pg/mL) | 52.1±63.5a0 | 219.4±311.9b0 | 147.3±107.2b0 | 253.8±361.5b0 | 0.0162 |
| Weight (g) | 3198.8±421.2a1 | 3637.2±579.2b1 | 3317.2±559.7a1 | 3070.2±488.5a1 | <0.0001 |
| PI (g/cm3) | 0.028±0.002 | 0.028±0.003 | 0.029±0.003 | 0.030±0.003 | ns |
| SGA | 03 (11.1) | 03 (15.0) | 0.0 (0.0) | 0.0 (0.0) | <0.0001 |
| AGA | 22 (81.5) | 13 (65.0) | 20 (95.2) | 16 (84.2) | |
| LGA | 02 (7.4) | 04 (20.0) | 01 (4.8) | 03 (15.8) | |
| Ht (%) | 48.0±5.5a2 | 49.4±4.7b2 | 51.8±5.5b2 | 53.4±7.5c2 | 0.0026 |
| Hb (g/dL) | 15.9±2.2 | 16.1±1.5 | 16.6±1.8 | 17.0±2.3 | ns |
| pH | 7.26±0.12 | 7.28±0.10 | 7.27±0.09 | 7.30±0.07 | ns |
| Bilirubin (mg/dL) | 2.02±0.57 | 2.10±0.73 | 2.03±0.74 | 2.22±0.63 | ns |
| Apgar 1 < 7 | 05 (18.52) | 05 (25.00) | 03 (14.29) | 02 (10.53) | ns |
| Apgar 5 < 7 | 01 (3.70) | 02 (10.00) | 0 (0) | 0 (0) | ns |
Data presented as means ± standard deviation or n (%).BMI: body mass index; 1: Evaluated in pre-pregnancy; 2: Evaluated in the third trimester of pregnancy; GA: Gestational age at delivery; GM: Glycemic mean; HbA1c: Glycated hemoglobin; PI: ponderal index (weight/length3 x100); Newborn classification (weight/gestational age): small (SGA), adequate (AGA), and large (LGA) for gestational age; Ht: hematocrit; Hb: hemoglobin. Values followed by different letters and the same index significantly differ (p < 0.05). ns: not significant (p > 0.05).
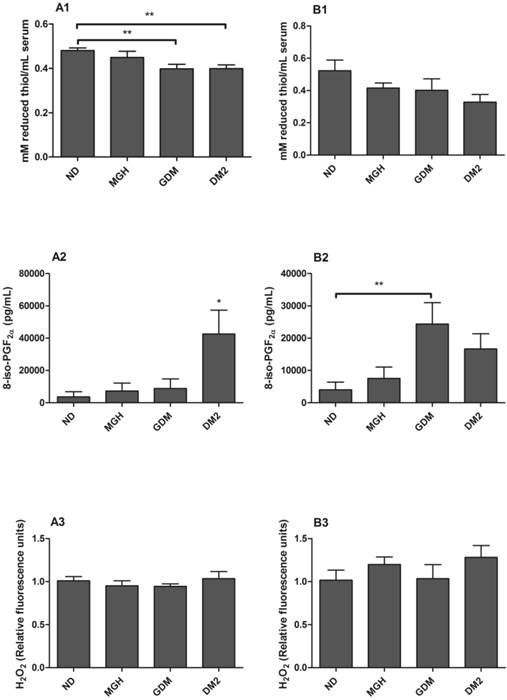
Evaluation of oxidative stress markers in serum from mothers [A] and umbilical cord blood [B]. A1 and B1: Total reduced thiols determined by the DTNB method. A2 and B2: 8-iso-PGF2α evaluated by EIA method. A3 and B3: H2O2 scavenging capacity after incubation with 20 μM hydrogen peroxide for 30 minutes at 37°C. Hydrogen peroxide not degraded by the samples was determined by Amplex Red/horseradish peroxidase fluorescence assay. Values as mean ± SEM, *p < 0.05 vs others groups; ** p < 0.05 vs appointed study group. n = 15/group.
The perinatal results evaluated included glucose, insulin and leptin levels (predictor of newborns weight), hematocrit, hemoglobin, pH and total bilirubin in umbilical cord blood. The corporal weight, ponderal index (weight/length3 X 100), newborns classification (weight/gestational age) in small (SGA), adequate (AGA) or large (LGA) for the gestational age, and 1st and 5th min Apgar scores were assessed at delivery. Hematological parameters (i.e., hematocrit and hemoglobin) were determined in total blood samples with a Sysmex KX-21N (Roche, Switzerland). After examination of the hematological parameters, total blood aliquots were centrifuged (4°C for 15 minutes at 1.000×g) for plasma collection and further analyses. Bilirubin concentrations were measured colorimetrically using BuBc slides (VITROS Chemistry products, Johnson & Johnson), and insulin levels were measured by a chemiluminescent immunoassay using microparticles (ARCHITECT insulin, Abbott Laboratories, São Paulo, SP, Brazil) and leptin by ELISA kit (R&D system, MN, USA) (Table 1).
Evaluation of oxidative stress
Total protein reduced thiols- indirect marker of protein oxidation
Total reduced thiols were determined in a spectrophotometer (Spectra Max PLUS 384, Molecular Devices) using 5,5-dithionitrobenzoic acid (DTNB- Sigma Aldrich, St Louis, MO, USA). Thiol residues react with DTNB, cleaving the disulfide bond to give 2-nitro-5-thiobenzoate (NTB2), which ionizes to the NTB22 di-anion in water at neutral and alkaline pH. NTB22 was quantified in a spectrophotometer at an absorbance of 412 nm, and was expressed as mM reduced thiols/mL serum.
8-Isoprostaglandin F2𝛼 levels (8-iso-PGF2α) - lipid peroxidation marker
8-iso-PGF2α was detected using a commercially available Direct 8-iso-PGF2α EIA kit (Enzo Life Sciences, Farmingdale, USA).
H2O2 scavenging capacity
H2O2 scavenging capacity of serum samples was measured using Amplex Red/horseradish peroxidase fluorescence assay (Invitrogen, Paisley, UK). Serum samples were incubated with 20 μM hydrogen peroxide for 30 minutes at 37ºC, after which they were incubated with Amplex Red (50 mM) and horseradish peroxidase type II (0.1 U/mL) to quantify the remaining hydrogen peroxide in each sample. Fluorescence readings were determined in GloMax®-Multi+ Microplate Multimode Reader (Promega Corp. Madison, Wisc., USA) with Ex/Em at 530/590 nm.
DNA damage analysis
Gene-specific quantitative PCR (QPCR) was used to assay nDNA and mtDNA damage [10,11]. Briefly, total genomic DNA was isolated using QIAGEN Genomic Tip and Genomic DNA Buffer Set Kit (QIAGEN, Valencia, CA). Purified genomic DNA was quantified fluorimetrically using Pico Green dsDNA reagent (Molecular Probes, Life Technologies, USA). Lambda (λ)/HindDIII DNA (Gibco Invitrogen, Paisley, UK) was used to generate a standard curve and adjust the final DNA concentration to 3 ng/μL. The “hot start” PCR used the Gene Amp XL PCR Kit (Applied Biosystems, Foster City, CA, USA) with 15 ng DNA, 1X buffer, 100 ng/μL BSA, 200 μM dNTPs, 20 pmol of each primer (Table S1), 1.3 mM Mg++ and water to 45μL. The reaction was brought to 75ºC before adding 1U/reaction enzyme (0.5μL of polymerase in 4.5 μL water). We quantitatively amplified an 8.9-kb and 221-bp fragment of the mitochondrial genome and 13.5-Kb of nuclear genome. Amplification of hyperglycemic samples (MGH, GDM and DM2 groups) was compared to non-diabetic samples (ND group) and relative amplifications were calculated. These measurements were used to estimate the frequency of lesions in the DNA based on a Poisson distribution.
Isolation of Peripheral Blood Mononuclear Cells (PBMC)
Blood samples were collected in tubes containing heparin before being diluted in phosphate-buffered saline (PBS), layered onto Ficoll-Pacque (Pharmacia Biotech, Uppsala, Sweden), and centrifuged for 40 minutes at 1.000×g. The intermediate phase with the PBMC was collected and washed twice in Dulbecco's modified Eagle's medium (DMEM) with low glucose (12320-032-Gibco Invitrogen, Paisley, UK) supplemented with 10% fetal bovine serum. The 2x106 cell aliquots were frozen in FBS with 10% of dimethyl sulfoxide (Sigma Aldrich, St Louis, MO, USA) and stored in liquid nitrogen. The samples were thawed in the same medium. To verify the viability of PBMC after isolation and thawing, 1% trypan blue (Gibco, Invitrogen, Paisley, UK) solution was added at a 1:1 ratio. The number of dead PBMC in a sample of 100 cells was counted. Only samples with viability > 95% were used in the experiments.
RNA extraction and cDNA synthesis
RNA extraction and cDNA synthesis was carried out in PBMC using power SYBR® Green Cells-to-CT™ Kit (Ambion, Carlsbad, CA, USA).
Real-time qPCR
Real-time qPCR was used to measure the expression of mRNA involved in different steps of BER. This technique was performed with Power SYBR® Green PCR Master Mix in an Applied Biosystems Step One Plus System (both from Applied Biosystems, Foster City, CA). Real-time qPCR was carried out using specific primers for hOGG1, APE1, FEN1, POLβ and GAPDH (Table S2).The 2^ΔΔCT method of analysis was used with the GAPDH gene for normalization.
Western blot
Western blot was used to quantify the expression of proteins involved in BER. The same number of PBMC from maternal and umbilical cord blood was lysed in ice-cold RIPA buffer, supplemented with a cocktail of protease inhibitors (Sigma Chemical Co, St Louis, MO). Proteins were separated electrophoretically using 15% SDS-PAGE before being transferred to a 0.45 µm to nitrocellulose membrane (Millipore, Bedford, MA, USA). Transfer was confirmed by staining the membranes with a 10% Ponceau S solution (Sigma Aldrich, St Louis, MO, USA). The blotted membranes were blocked with TBS-T-milk 3% for 1 h and washed 3 times with TBS buffer before being incubated at 4°C with anti-hOGG1, anti-APE1 and anti- FEN1 (all of them at 1:1000; Novus Biologicals, Littleton, CO, USA) or anti-POLβ (at 1:500, Santa Cruz, CA) in TBS-T-milk 3% overnight and washed 3 times with TBS buffer. The membranes were exposed to horseradish peroxidase-conjugated antibody (1:1000, Jackson Immuno Research, USA) in TBS-T-milk 3% for 1 h and washed 3 times with TBS buffer. Immunoreactive bands (peroxidase activity) were detected by the Enhanced chemiluminescence method (ECL). The quantitative analysis of all DNA repair protein expression levels was measured by densitometry using Image J software (v. 1.43 u, NIH, Bethesda, MD, USA). β-actin was used as the loading control.
PBMC cultures
PBMC were suspended in DMEM low glucose with 2% FBS, and 1% gentamicin and plated in 96-well culture dishes. Three cultures groups contained D-glucose (Sigma Aldrich, St Louis, MO) at the following concentrations: i) 5 mmol/L (90 mg/dL, control sample - physiological concentration of glucose - equivalent of normoglycemia in the case of patients); ii) 17.5 mmol/L (315 mg/dL, moderated concentration of glucose) and, iii) 30 mmol/L (540 mg/dL high glucose concentration - equivalent of severe hyperglycemia in patients). Glucose concentrations in culture medium followed values found in patients and the literature [9,23]. Cells were incubated in 5% CO2 incubator at 37°C for 48 h. Caspase 3/7 activity (Caspase-Glo® 3/7 Assay, Promega Corp. Madison, WI) and ATP levels (Cell Titer-Glo® Luminescent Cell Viability Assay, Promega Corp. Madison, WI) were determined by luminescence in GloMax® - Multi+ Microplate Multimode Reader (Promega Corp. Madison, Wisc., USA). Staurosporine 1 mM (Abcam, Cambridge, UK) was used as a positive control of caspase and a negative control of ATP analysis.
Statistical Analysis
Analysis of variance and Tukey's multiple comparison tests were used for quantitative variables with normal distribution. For those with an abnormal distribution, the generalized linear model with gamma distribution and the log-link function and the LS Means tests were used for multiple comparisons. Analyses used SAS software, version 9.1 and Prism, taking statistical significance as p < 0.05.
Results
Maternal clinical background and perinatal outcomes evaluation
GDM and DM2 patients were older (p < 0.0001), with the highest BMI in the third trimester of pregnancy (p = 0.0003). The diabetic and MGH groups had the highest pre-gestational BMI (p < 0.0001). The gestational age at delivery was lower in the DM2 group (p < 0.0001).The GM got progressively higher with the severity of the clinical condition of the mother (p < 0.0001); the HbA1c levels in the third trimester (p < 0.0001) confirmed the GM levels. Nevertheless, the values of GM did not exceed the glycemic goals recommended by the American Diabetes Association [1] (Table 1).
Newborns from MGH, GDM and DM2 groups had the highest leptin (p = 0.0162) and hematocrit levels (p = 0.0026). The newborn weight (p < 0.0001) and percentage of large for gestational age percentage (p < 0.0001) were increased only in MGH groups (Table 1).
Oxidative stress
Serum levels of reduced thiols (for protein oxidation evaluation) were significantly decreased in mothers with GDM (p = 0.0043) and DM2 (p = 0.027) compared to ND group (Figure 1-A1). No statistically significant difference was found between groups in umbilical cord serum (Figure 1-B1).
Maternal 8-iso-PGF2α serum levels were higher in the DM2 group (p = 0.0061) compared to others (Figure 1-A2), whereas umbilical cord serum had a significant increase in the GDM group (p = 0.0217) when compared to ND group (Figure 1-B2).
No significant differences in H2O2 scavenging capacity were detected either in maternal (Figure 1-A3) or in newborns serum (Figure 1-B3).
Nuclear and mitochondrial DNA damage
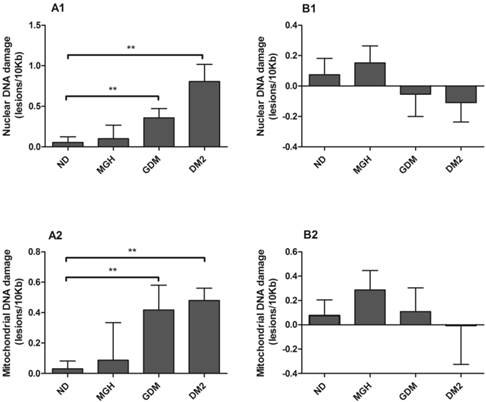
nDNA and mtDNA damage (Figure 2-A1; Figure 2-A2, respectively) in maternal leukocytes were higher in both GDM (p = 0.02) and DM2 groups (p = 0.0007 in nDNA and p = 0.002 in mtDNA) compared to ND group. In contrast, no significant differences were found in umbilical cord blood leukocytes (Figure 2-B1 and Figure 2-B2). A summary is given in Table 2.
Nuclear [1] and mitochondrial [2] DNA damage from maternal [A] and umbilical cord blood [B]. DNA damage determined by Gene-specific quantitative PCR (QPCR). Values as mean ± SEM, ** p < 0.05 vs appointed study group. n = 15/group.
Summary results of DNA damage and repair.
| ND | MGH | GDM | DM2 | |||||
|---|---|---|---|---|---|---|---|---|
| DNA Damage | ||||||||
| nDNA | mtDNA | nDNA | mtDNA | nDNA | mtDNA | nDNA | mtDNA | |
| Mothers | ↑* | ↑* | ↑* | ↑* | ||||
| Newborns | ns | ns | ns | ns | ns | ns | ns | ns |
| DNA Repair | ||||||||
| mRNA | Protein | mRNA | Protein | mRNA | Protein | mRNA | Protein | |
| Mothers | ||||||||
| hOGG1 | nd | nd | nd | ↓§ | nd | |||
| APE1 | ↓* | ↓ | ↓* | |||||
| FEN1 | ns | ns | ↓*# | ns | ↓# | ns | ||
| POLβ | ↓* | ↓* | ↓* | |||||
| Newborns | ||||||||
| hOGG1 | nd | nd | nd | nd | ↑*# | |||
| APE1 | ↑ | ↑ | ||||||
| FEN1 | ↑* | ↑*# | ↑*# | |||||
| POLβ | ↑* | ↑ | ||||||
↑ increase of expression or ↓ decrease of expression compared to all groups; *compared to ND group; # compared to MGH group; § compared to DM2 group. nd: not detected; ns: not significant.
mRNA expression of APE1, FEN1 and POLβ
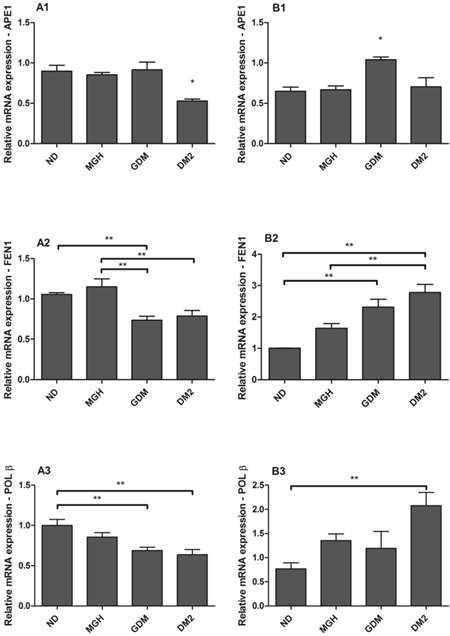
hOGG1 mRNA was not found in maternal or in newborns PBMC, differently from the assays using a choriocarcinoma cell line (BeWo, assay control, data not shown). APE1, FEN1 and POLβ were expressed in all groups but with distinct differences among the maternal and newborns groups (Figure 3). Pregnant women with GDM and DM2 had reduced expression of FEN1 (p = 0.0006; Figure 3-A2) and POLβ (p = 0.0044; Figure 3-A3) compared to the ND group. Expression of APE1 was lower in DM2 (p = 0.0008; Figure 3-A1) compared to other groups. In newborns, PBMC of GDM had higher expression of APE1 (p = 0.0032; Figure 3-B1) compared to other groups and FEN1 (p = 0.0016; Figure 3-B2) compared to ND group. Newborns of the DM2 group also had increased expression of FEN1 (Figure 3-B2) and POLβ (p = 0.0036) (Figure 3-B3) when compared to ND group. A summary is given in Table 2.
mRNA expression of APE1 [1], FEN1 [2] and POLβ [3] in PBMC isolated from maternal [A] and umbilical cord blood [B]. Each reaction was run in triplicate. All expression was normalized to GAPDH. hOGG1 expression was not detected by the techniques used. Values as mean ± SEM; *p < 0.05 vs others groups; ** p < 0.05 vs appointed study group. n = 15/group.
Protein expression levels of hOGG1, APE1, FEN1 and POLβ
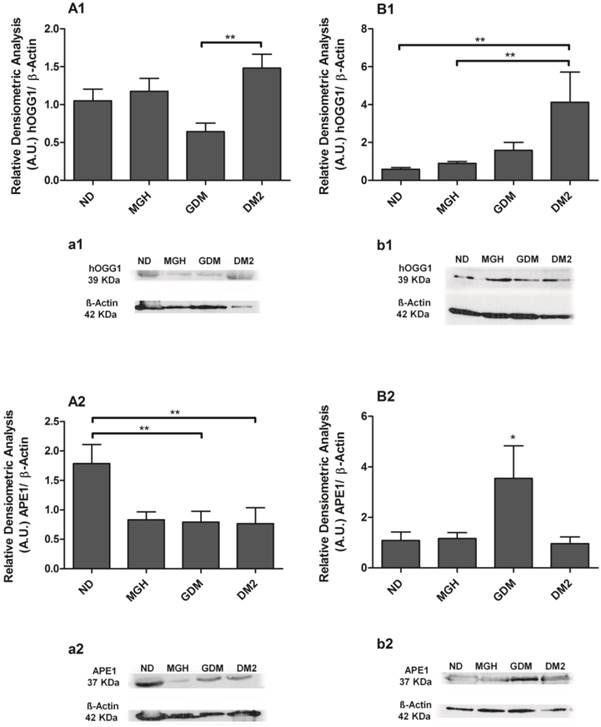
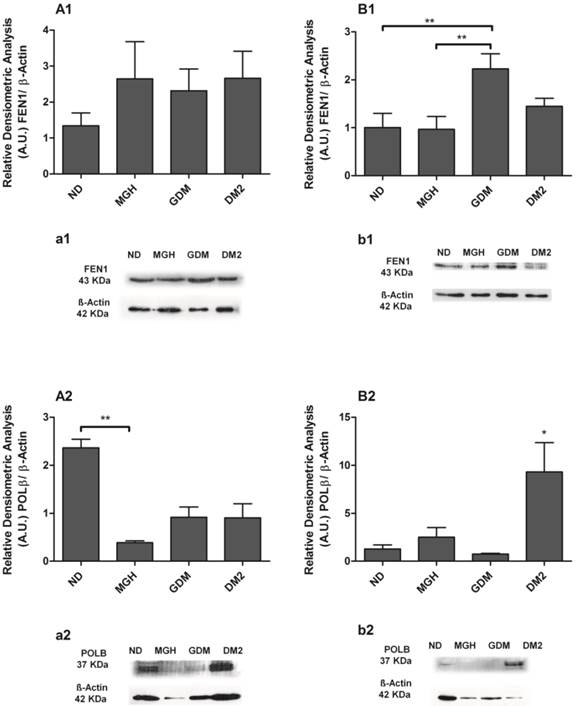
BER proteins were detected in all groups, and the results confirmed the different responses observed at the mRNA level between mothers and newborns of the diabetic groups (Figures 4 and 5). Pregnant women with GDM had lower hOGG1 protein expression (p = 0.0105; Figures 4A1, a1) compared to DM2 group, and lower APE1 expression (p = 0.0145; Figure 4-A2; a2) when compared to ND group. Pregnant women with DM2 had decreased APE1 expression (p = 0.0145; Figure 4-A2; a2) compared to ND group. We detected a significant reduction of only POLβ (p = 0.0363; Figure 5-A2; a2) in the MGH group when compared to ND group. In newborns, umbilical cord blood PBMC of the GDM group had increased APE1 protein expression (p = 0.0164; Figure 4-B2; b2) compared to other groups and an increase in FEN1 (p = 0.0099; Figure 5-B1; b1) compared to ND and MGH groups. Newborns of the DM2 group also had increased hOGG1protein expression (p = 0.0039; Figure 4-B1; b1) compared to ND and MGH groups and in POLβ (p = 0.0077; Figure 5-B2; b2) compared to remaining groups. These results are summarized in Table 2.
Protein expression of hOGG1 [1] and APE1 [2] in PBMC isolated from maternal [A,a] and umbilical cord blood [B,b]. The relative band intensities from western blot experiments were normalized to β-actin and analyzed with Image J software [A1, A2, B1, B2]. Values as mean ± SEM; *p < 0.05 vs others groups; ** p < 0.05 vs appointed study group. n = 08/group.
Protein expression of FEN1 [1] and POLβ [2] in PBMC isolated from maternal [A,a] and umbilical coord blood [B,b]. The relative band intensities from western blot experiments were normalized to β-actin and analyzed with Image J software [A1, A2, B1, B2]. Values as mean ± SEM; *p < 0.05 vs others groups; ** p < 0.05 vs appointed study group. n = 08/group.
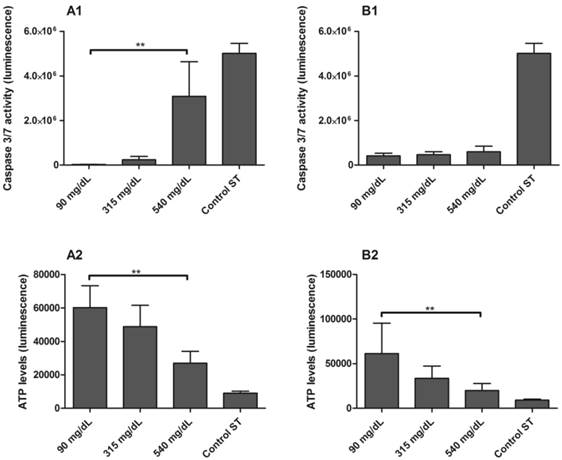
Apoptosis and ATP production analysis
The activity of effector caspases-3 and caspase-7 were higher in PBMC isolated from ND mothers and exposed to 540 mg/dL glucose in comparison to 90 mg/dL glucose concentrations (p < 0.001; Figure 6-A1). To confirm these results, ATP levels were measured in the same samples, and were found to be significantly reduced in 540 mg/dL glucose concentration (p = 0.0480; Figure 6-A2). Activation of caspase-3 and caspase-7 did not occur in PBMC from newborns (Figure 6-B1), although the ATP levels were reduced in 540 mg/dL glucose concentration (p = 0.0439; Figure 6-B2).
Caspase 3/7 activity [1] and ATP levels [2] in PBMC isolated from maternal [A] and umbilical cord blood [B] of non-diabetic group. PBMC were exposed to 5 mmol/L (90 mg/dL), 17.5 mmol/L (315 mg/dL) and 30 mmol/L (540 mg/dL) glucose for 48h. Control ST: staurosporine 1 mM. Values as mean ± SEM, ** p < 0.05 vs appointed study group. n = 06/group.
Discussion
Our results demonstrate that hyperglycemia differentially affects maternal and fetal DNA integrity and DNA damage response in peripheral blood cells. The detrimental effects of hyperglycemia were particularly found in cells from GDM and DM2 pregnant groups. Maternal blood serum showed increased oxidative stress markers and both nuclear and mitochondrial DNA damage in leukocytes. In addition, same genes related to the BER pathway were decreased both at the mRNA and protein levels. Higher glucose levels were also associated with increased apoptosis in vitro assays. Conversely, the absence of DNA damage and apoptosis in fetal samples were associated with higher expression of BER genes.
It is widely accepted that hyperglycemia leads to oxidative stress [7,24-26]. Increased biomarkers of oxidative damage and abnormalities in the antioxidant defenses have been reported in diabetic patients [7,24-26]. Plasma and urinary concentrations of 8-iso-PGF2α are associated with DM2 in non-pregnant women [27,28], and with GDM in pregnant mothers [29]. In this and previous studies [30,31] we found that patients from the DM2 group have increased hyperglycemia, are overweight and obese, are insulin resistant and show increased levels of cytokines production, all of which may be related to oxidative stress. Indeed, we found lower levels of thiols in the reduced form in GDM and DM2, and higher levels of 8-iso-PGF2α in DM2 group indicating the presence of oxidized proteins and lipid peroxidation, respectively, reinforcing the association between hyperglycemia and oxidative stress. Such findings are consistent with studies in rats that demonstrated higher levels of lipid peroxidation as gauged by malondialdehyde levels in maternal blood of streptozotocin-induced diabetic pregnant rats [7].
Although our results have showed increased oxidative stress, we could not differentiate at this stage whether this results from increased ROS production or decreased antioxidant defenses. The data on mtDNA damage supports the possibility of increased mitochondrial ROS production, which is further reinforced by the results obtained with Amplex red that showed no differences in H2O2 scavenging capacity in the serum from mothers, irrespective of the glycemia levels. However, given that most antioxidants present in serum do not react directly with H2O2, at this time we may not exclude the possibility that antioxidant defenses are also affected. Data from literature suggests that GDM is associated with down regulation of antioxidant status in blood samples [32,33], which supports this possibility.
Blood cells from GDM and DM2 showed significant increase in nDNA and mtDNA damage and decrease of BER enzymes expression. mtDNA damage causes mitochondrial dysfunction, promoting and maintaining increased ROS production, which could leak out of the mitochondria, eventually affecting the nuclear genome [10,11]. DNA damage induction can impair genome integrity, which in turn, can increase individual susceptibility to several diseases, including cancers [34]. Epidemiological evidence suggests a connection between diabetes and a risk of neoplastic diseases (in liver, pancreas, endometrium, colon and rectum, breast and bladder) [35]. Sella and collaborators [36] showed a clear correlation between GDM and Non-Hodgkin's lymphoma, Hodgkin's lymphoma and acute myeloid leukemia in a cohort in Israel. These studies did not explain the underlying mechanisms but clearly show an association with hyperinsulinemia, hyperglycemia, and inflammation [35-37]. DNA damage in diabetic pregnant women seen here might be part of this mechanism; however, it was not addressed in this study. In this context, a further cohort study in our population might explore this association.
Our previous work showed higher levels of oxidative DNA damage in both streptozotocin-induced diabetic pregnant rats and newborns when glucose levels were ≥ 300mg/dL [19-21], which is opposite to the results shown here. This discrepancy may be explained by the fact that in the present study maternal glucose was maintained at levels lower than those in the diabetic rats. Although limited in number of mothers and newborns evaluated, and the difference of age in mothers with GDM and DM2, our results highlight that mechanisms to protect the fetus from DNA-damage may depend on glycemic levels, reinforcing the importance of adequate maternal glycemic control in pregnancies complicated by diabetes.
Maternal glucose readily crosses the placenta and it has always been associated with adverse perinatal outcomes [38-40]. In our study, newborns of hyperglycemic mothers were characterized by elevated levels of leptin, increased body weight, higher hematocrit levels and high rate of large for gestational age babies. The most intriguing result, contrary to our initial hypothesis, is the differential response to DNA insults under hyperglycemia in the mother and the fetus's blood. nDNA and mtDNA damage seen in blood cells from GDM and DM2 mothers were not found in the blood cells of their newborns. These results were corroborated by previous study using the comet assay technique [41] and may be due to differential ability in upregulating multiple genes involved in the activation of DNA repair response, especially the BER mechanism.
Regardless of the fact that we found some differences between mRNA and protein levels in PBMC from mothers, these pregnant women's cells expressed low levels of BER mRNA (APE1, FEN1, POLβ) and proteins (hOGG1, APE1) and the newborns had both levels increased. The period of hyperglycemia exposure in utero (longer time in DM2 than GDM) may modulate the BER pathway (short or long path BER) in newborns. This may justify our observation that DM2 group showed more hOGG1 and POLβ expression and GDM group showed more APE1 and FEN1 expression. However, the reason why they exhibited different alteration of BER enzymes needs to be refined to better understand this scenario. Analysis of apoptosis in blood cells in vitro follows the same pattern. No apoptosis was observed in high glucose concentrations in the newborn blood cells, in opposition to the results found in the maternal cells.
The umbilical vein is a crucial compartment in maternal-fetal interface. It receives oxygen and nutrients from the maternal blood through the placenta and that will contribute to the development of fetal peripheral blood profile. It is also a possible site of blood cell susceptibility due to the action of other elements that may cross the placental barrier, such as glucose [42]. Small numbers of maternal cells also traffic across the placenta, reaching fetal capillaries and then, umbilical vein [42]. However, despite these changes in umbilical vein contents at placental sites, the response of its blood cells to the diabetogenic maternal environment had its own characteristics. Taken together, our results from umbilical vein blood indicate that the fetal genome is robustly protected from a detrimental uterine environment, which include increase in DNA repair capacity. Of note, newborns from GDM mothers showed increased lipid peroxidation but it was not associated with DNA damage.
Some mechanisms have been proposed to explain these results. The umbilical cord contains at least three populations of stem cells, each with unique features and properties [43]. These stem cells possess highly efficient DNA repair pathways that become less efficient after differentiation. They also have anaerobic metabolism, which reduces mitochondrial number and oxidative stress [43,44]. The higher telomerase activity [45] and the increased mitochondrial/nuclei ratio of human translocation telomerase reverse transcriptase (hTERT) [29] are also considered as possible protective factors for DNA umbilical blood cells in hyperglycemic pregnancies. Thus, collective the previously published and our current data suggest that umbilical cord blood cells have several mechanisms to protect the DNA against genomic insults associated with hyperglycemic and oxidative stress conditions.
In conclusion, our results showed that hyperglycemia affects maternal and fetal DNA integrity and DNA damage response differently. Blood cells from the maternal organism seem to be more susceptible to DNA damage and apoptosis induction compared to fetal blood cells. These findings may be related to the different expression of genes involved in DNA repair mechanisms, especially in the BER mechanism. If this study shows a more effective adaptive response by the fetal organism, it also calls for further studies to determine the limit of this response that can change the fate of a fetus. This fate generated under conditions which may engage intrinsic responses to cellular stress and, therefore have an effect on the susceptibility for several diseases.
Supplementary Material
Figure S1 and tables S1-S2.
Acknowledgements
The authors would like to thank Fundação de Amparo à Pesquisa do Estado de São Paulo - FAPESP (grant number 2011/18240-2; 2011/13562-1 and 2012/23296-0) for financial support and for the fellowships of JB Moreli. Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq). Dr. Carlos Frederico Martins Menck for scientific support and Dr. José Eduardo Corrente for statistics support.
Competing Interests
The authors have declared that no competing interest exists.
References
1. American Diabetes Association (ADA). Standards of medical care in Diabetes - 2014. Diabetes Care. 2014;37:S14-S80
2. Ornoy A. Prenatal origin of obesity and their complications: Gestational diabetes, maternal overweight and the paradoxical effects of fetal growth restriction and macrosomia. Reprod Toxicol. 2011;32:205-12
3. Calderon IMP, Kerche LTRL, Damasceno DC, Rudge MVC. Diabetes and pregnancy: an update of the problem. ARBS. 2007;9:1-11
4. Yang J, Cummings EA, O'connell C, Jangaard K. Fetal and neonatal outcomes of diabetic pregnancies. Obstet Gynecol. 2006;10:644-50
5. Lehnen H, Zechner U, Haaf T. Epigenetics of gestational diabetes mellitus and offspring health: the time for action is in early stages of life. Mol Hum Reprod. 2013;19:415-22
6. Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813-20
7. Spada AP, Damasceno DC, Sinzato YK, Campos KE, Faria PA, Dallaqua B. et al. Oxidative stress in maternal blood and placenta from mild diabetic rats. Reprod Sci. 2014;21:973-77
8. Damasceno DC, Netto AO, Iessi IL, Gallego FQ, Corvino SB, Dallaqua B. et al. Streptozotocin-induced diabetes models: pathophysiological mechanisms and fetal outcomes. Biomed Res Int. doi: 10.1155/2014/819065
9. Arya AK, Pokharia D, Tripathi K. Relationship between oxidative stress and apoptotic markers in lymphocytes of diabetic patients with chronic non healing wound. Diabetes Res Clin Pract. 2011;94:377-84
10. Kovalenko OA, Santos JH. Analysis of oxidative damage by gene-specific quantitative PCR. Curr Protoc Hum Genet. 2009 Chapter 19:Unit 19.1
11. Santos JH, Meyer JN, Mandavilli BS, Van Houten B. Quantitative PCR-based measurement of nuclear and mitochondrial DNA damage and repair in mammalian cells. Methods Mol Biol. 2006;314:183-99
12. Yakes FM, Van Houten B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc Natl Acad Sci U S A. 1997;94:514-9
13. Mandavilli BS, Santos JH, Van Houten B. Mitochondrial DNA repair and aging. Mutat Res. 2002;509:127-51
14. Mitra S, Boldogh I, Izumi T, Hazra TK. Complexities of the DNA base excision repair pathway for repair of oxidative DNA damage. Environ Mol Mutagen. 2001;38:180-90
15. Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071-8
16. Blasiak J, Arabski M, Krupa R, Wozniak K, Zadrozny M, Kasznicki J. et al. DNA damage and repair in type 2 diabetes mellitus. Mutat Res. 2004;554:297-304
17. Tatsch E, Bochi GV, Piva SJ, De Carvalho JA, Kober H, Torbitz VD. et al. Association between DNA strand breakage and oxidative, inflammatory and endothelial biomarkers in type 2 diabetes. Mutat Res. 2012;732:16-20
18. Qiu C, Hevner K, Abetew D, Enquobahrie DA, Williams MA. Oxidative DNA damage in early pregnancy and risk of gestational diabetes mellitus: A pilot study. Clin Biochem. 2011;44:804-8
19. Lima PH, Sinzato YK, de Souza Mda S, Braz MG, Rudge MV, Damasceno DC. Evaluation of level of DNA damage in blood leukocytes of non-diabetic and diabetic rat exposed to cigarette smoke. Mutat Res. 2007;628:117-22
20. Lima PH, Damasceno DC, Sinzato YK, de Souza Mda S, Salvadori DM, Calderon I de M. et al. Levels of DNA damage in blood leukocyte samples from non-diabetic and diabetic female rats and their fetuses exposed to air or cigarette smoke. Mutat Res. 2008;653:44-9
21. Lima PH, Sinzato YK, Gelaleti RB, Calderon IM, Rudge MV, Damasceno DC. Genotoxicity evaluation in severe or mild diabetic pregnancy in laboratory animals. Exp Clin Endocrinol Diabetes. 2012;120:303-7
22. Rudge MVC, Peraçoli JC, Berezowski AT, Calderon IMP, Brasil MAM. The oral glucose tolerance test is a poor predictor of hyperglycemia during the pregnancy. Braz J Med Biol Res. 1990;23:1079-89
23. Oleszczak B, Szablewski L, Pliszka M. The effect of hyperglycemia and hypoglycemia on glucose transport and expression of glucose transporters in human lymphocytes B and T: an in vitro study. Diabetes Res Clin Pract. 2012;96:170-8
24. Zhu C, Yang H, Geng Q, Ma Q, Long Y, Zhou C. et al. Association of oxidative stress biomarkers with gestational diabetes mellitus in pregnant women: a case-control study. PLoS One. 2015;10:e0126490
25. West IC. Radicals and oxidative stress in diabetes. Diabet Med. 2000;17:171-80
26. Tangvarasittichai S. Oxidative stress, insulin resistance, dyslipidemia and type 2 diabetes mellitus. World J Diabetes. 2015;6:456-80
27. Davì G, Ciabattoni G, Consoli A, Mezzetti A, Falco A, Santarone S. et al. In vivo formation of 8-iso-prostaglandin f2alpha and platelet activation in diabetes mellitus: effects of improved metabolic control and vitamin E supplementation. Circulation. 1999;99:224-9
28. Devaraj S, Hirany SV, Burk RF, Jialal I. Divergence between LDL oxidative susceptibility and urinary F(2)-isoprostanes as measures of oxidative stress in type 2 diabetes. Clin Chem. 2001;47:1974-9
29. Li P, Tong Y, Yang H, Zhou S, Xiong F, Huo T. et al. Mitochondrial translocation of human telomerase reverse transcriptase in cord blood mononuclear cells of newborns with gestational diabetes mellitus mothers. Diabetes Res Clin Pract. 2014;103:310-8
30. Moreli JB, Morceli G, De Luca AK, Magalhães CG, Costa RA, Damasceno DC. et al. Influence of maternal hyperglycemia on IL-10 and TNF-α production: the relationship with perinatal outcomes. J Clin Immunol. 2012;32:604-10
31. Moreli JB, Corrêa-Silva S, Damasceno DC, Sinzato YK, Lorenzon-Ojea AR, Borbely AU. et al. Dynamics changes in the TNF-alpha/IL-10 ratio in hyperglycemic-associated pregnancies. Diabetes Res Clin Pract. 2015;107:362-69
32. Grissa O, Atègbo JM, Yessoufou A, Tabka Z, Miled A, Jerbi M. et al. Antioxidant status and circulating lipids are altered in human gestational diabetes and macrosomia. Transl Res. 2007;150:164-71
33. Karacay O, Sepici-Dincel A, Karcaaltincaba D, Sahin D, Yalvaç S, Akyol M. et al. A quantitative evaluation of total antioxidant status and oxidative stress markers in preeclampsia and gestational diabetic patients in 24-36 weeks of gestation. Diabetes Res Clin Pract. 2010;89:231-8
34. Matyszewski A, Czarnecka AM, Solarek W, Korzeń P, Safir IJ, Kukwa W. et al. Molecular basis of carcinogenesis in diabetic patients. Int J Oncol. 2015;46:1435-43
35. Zelenko Z, Gallagher EJ. Diabetes and cancer. Endocrinol Metab Clin North Am. 2014;43:167-85
36. Sella T, Chodick G, Barchana M, Heymann AD, Porath A, Kokia E. et al. Gestational diabetes and risk of incident primary cancer: a large historical cohort study in Israel. Cancer Causes Control. 2011;22:1513-20
37. Moreli JB, Santos JH, Rocha CR, Damasceno DC, Morceli G, Rudge MV. et al. DNA damage and its cellular response in mother and fetus exposed to hyperglycemic environment. Biomed Res Int. 2014;2014:676758. doi: 10.1155/2014/676758
38. Pedersen J, Bojsen-Moller B, Paulsen H. Blood sugar in newborn infants of diabetic mothers. Acta Endocrinol (Copenh). 1954;15:33-52
39. Jansson T, Powell TL. Role of the placenta in fetal programming: underlying mechanisms and potential interventional approaches. Clin Sci (Lond). 2007;113:1-13
40. Fraser A, Lawlor DA. Long-term health outcomes in offspring born to women with diabetes in pregnancy. Curr Diab Rep. 2014;14:489
41. Gelaleti RB, Damasceno DC, Santos DP, Calderon IM, Rudge MV. Increased DNA damage is related to maternal blood glucose levels in the offspring of women with diabetes and mild gestational hyperglycemia. Reprod Sci. 2015. 1933 719115602766
42. Cunningham FG, Leveno KJ, Bloom SL. et al. Williams Obstetrics. USA: Mc Graw-Hill Companies. 2010
43. Ali H, Mulla FA. Defining umbilical cord blood stem cells. Stem Cell Discovery. 2012;2:15-23
44. Rocha CR, Lerner LK, Okamoto OK, Marchetto MC, Menck CF. The role of DNA repair in the pluripotency and differentiation of human stem cells. Mutat Res. 2013;752:25-35
45. Cross JA, Temple RC, Hughes JC, Dozio NC, Brennan C, Stanley K. et al. Cord blood telomere length, telomerase activity and inflammatory markers in pregnancies in women with diabetes or gestational diabetes. Diabet Med. 2010;27:1264-70
Author contact
![]() Corresponding author: Iracema Calderon, Department of Obstetrics and Gynecology, Botucatu Medical School, UNESP-São Paulo State University, Distrito de Rubião Jr. s/n, CEP 18618-000, Botucatu, São Paulo, Brazil. Fone: +55 (14) 38801383. E-mails: calderonunesp.br and juscielemorelicom.
Corresponding author: Iracema Calderon, Department of Obstetrics and Gynecology, Botucatu Medical School, UNESP-São Paulo State University, Distrito de Rubião Jr. s/n, CEP 18618-000, Botucatu, São Paulo, Brazil. Fone: +55 (14) 38801383. E-mails: calderonunesp.br and juscielemorelicom.