Impact Factor ISSN: 1449-2288
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. Regulation of mitochondrial...
3. Regulation of cellular...
4. Targeting mitochondrial Stat3...
Remarks
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2016; 12(5):532-544. doi:10.7150/ijbs.15153 This issue Cite
Review
Mitochondrial Stat3, the Need for Design Thinking
Rui Yang, Mercedes Rincon ![]()
Department of Medicine/Immunobiology Program, University of Vermont, Burlington, VT, USA, 05405
Received 2016-1-31; Accepted 2016-2-12; Published 2016-2-29
Abstract

Stat3 has been studied extensively as a transcription factor, however the finding that Stat3 also localizes to mitochondria has opened a new area to discover non-classical functions. Here we review the current knowledge of mitochondrial Stat3 as a regulator of the electron transport chain (ETC) and its impact on mitochondrial production of ATP and ROS. We also describe recent findings identifying Stat3 as a regulator of mitochondrial Ca2+ homeostasis through its effect on the ETC. It is becoming evident that these non-classical functions of Stat3 can have a major impact on cancer progression, cardiovascular diseases, and inflammatory diseases. Therefore, mitochondrial Stat3 functions challenge the current design of therapies that solely target Stat3 as a transcription factor and suggest the need for “design thinking,” which leads to the development of novel strategies, to intervene the Stat3 pathway.
Keywords: Stat3
1. Introduction
Signal Transducers and Activators of Transcription family 3 (Stat3) plays a crucial role in normal development, acute phase response, chronic inflammation, autoimmunity, metabolism and cancer progression [1]. Stat3 was first identified as an acute phase response factor (APRF) which selectively interacts with the promoter regions of acute phase genes in IL-6-stimulated hepatocytes [2-5]. Stat3 is activated by the a family of cytokines which share the same signal transducer gp130, including IL-6, IL-11, LIF (leukaemia inhibitory factor), OSM (oncostatin M), ciliary neurotrophic factor (CNTF), cardiotrophin-1 and cardiotrophin-like cytokines [6]. Several gp130 independent stimuli including epidermal growth factor (EGF), hepatocyte growth factor (HGF), IL-10, leptin also activate Stat3 [6]. Stat3 is abundantly expressed in most tissues. Stat3 is structurally similar to other Stat proteins, containing a conserved N-terminal domain involved in tetramerization and cooperativity, an Src homology 2 (SH2) domain for homodimerization, a DNA-binding domain and a C-terminal transactivating domain [7, 8]. There are two alternatively spliced Stat3 isoforms: the full-length Stat3α (a 770 amino acid protein) and truncated isoform Stat3β (containing amino acid 1-715 and 7 unique amino acid due to a frame-shift) [9-12]. While Stat3α and Stat3β share most features in cellular signaling and functions, Stat3α has some non-redundant features in regulating IL-6 signaling, including the upregulation of the negative feeback protein SOCS3 in response to IL-6 [9].
As a major stimulus of Stat3, engagement of IL-6 with IL-6 receptor α (IL-6Rα) recruits the ubiquitously expressed signal transducer gp130. Engagement of gp130 by IL-6 and other cytokines that signal through Stat3 leads to the activation of Jak1, Jak2 and Tyk2 [3, 13, 14]. Activation of Jak family kinases stimulates downstream transcription factor Stat3 [15, 16]. Stat3 is phosphorylated at two well-studied phosphorylation sites, Tyr705 and Ser727. Tyr705 phosphorylation in response to cytokine stimulation is mediated by Janus kinases, Jak1, Jak2 and Tyk2 with Jak1 being the preferential kinase [17]. Phosphorylation of Tyr705 leads to Stat3 homodimerization, nuclear translocation, DNA binding and downstream transcriptional activities (Fig. 1). The Ser727 phosphorylation is more complex because different activation signals lead to serine phosphorylation by different kinasese, including ERK1, ERK2, p38, JNK and MAP kinases [18]. Ser727 phosphorylation at the transcativating domain is considered a secondary event after Tyr705 phosphorylation (Fig. 1). This is required for the maximal transcriptional activity of Stat3 [19]. In addition, Stat3 has also been shown to form heterodimer with Stat1 in response to IL-6 stimulation [20].
Classical and non-classical pathways of Stat3. Classical pathway where Stat3 translocates to the nucleus and mediates gene transcription. Non-classical pathway where Stat3 is recruited to mitochondria and regulates functions alternative to transcription.
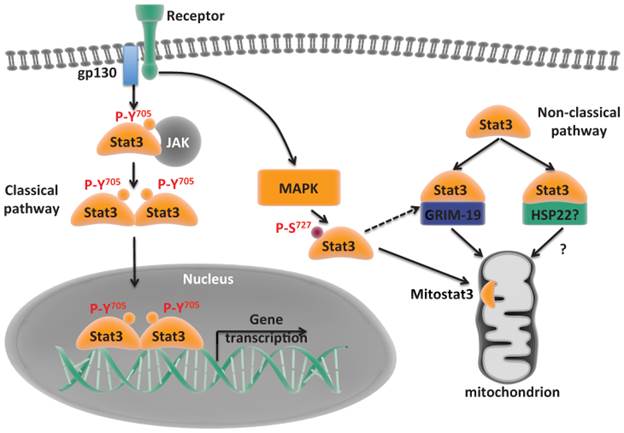
Although Stat3 has been studied as a transcription factor for decades, a small pool of Stat3 (mitoStat3) was found localized in mitochondria, where it functions as a positive regulator of mitochondrial electron transport chain (ETC) [21, 22]. A number of follow up studies have further confirmed the presence of Stat3 in mitochondria in different cells and tissues. Besides the pivotal role in cellular metabolism, the functions of mitoStat3 have been expanded to include cell death, development, cancer transformation, ischemia/reperfusion (I/R) heart injury, sperms motility, T cell immunity and others [21-30].
In light of the recent findings supporting new aspects of mitoStat3 in regulating mitochondrial metabolism and cellular function, and the rapidly growing interest in mitoStat3 from the therapeutical point of view, the goal of this review is to summarize these new discoveries and provide an overview of where the field is heading.
2. Regulation of mitochondrial function by mitochondrial Stat3
As a transcription factor, the role of Stat3 in the regulation of genes from acute phase response, chronic inflammation, and cancer development has been well characterized [1, 31]. Null disruption of Stat3 gene in mice leads to early embryonic degeneration and lethality [32]. Interestingly, however, many autosomal-dominant Hyper-IgE syndrome patients who bear a dominant negative mutation in Stat3 that disrupts its DNA binding transcriptional activity do not exhibit major developmental defects [33-35]. These findings posed the question whether some of the initial Stat3 functions might be independent of its activity as transcription factor. While it was generally believed that Stat3 was just a transcription factor, several studies observed that GRIM-19, a component of Complex I of ETC, binds to Stat3 directly [36, 37]. These studies paved the way for later revolutionary findings that some Stat3 localizes in mitochondria [21, 22]. Despite several studies reporting controversial results that GFP tagged Stat3 is excluded from mitochondria [38] and disproportioned stoichiometry analysis [39] between Stat3 and ETC complexes, growing evidences have confirmed the presence of mitoStat3 and its role in regulating cellular metabolism and various functions.
2.1 The role of mitoStat3 in the regulation of electron transport chain and mitochondrial ATP synthesis
MitoStat3 is associated with the inner mitochondrial membrane where the Complex I resides [40]. As with most of mitochondrial proteins encoded by nuclear genes, the mechanisms by which Stat3 is transported to the mitochondria remain unclear. Although the C-terminus of Stat3 has been shown to be required for mitochondrial transport no specific mitochondrial targeting signal has been identified. Since Stat3 associates with GRIM-19, a component of the ETC Complex I, most likely Stat3 transport to mitochondria is mediated by GRIM-19 through the mitochondrial importer Tom20 [26, 30, 36, 37, 40] (Fig. 1). In addition, heat shock protein 22 (Hsp22) has also been suggested to be an alternative shuttle for mitoStat3 [41] (Fig. 1). MitoStat3 is not required for mitochondrial maintenance or formation [22]. However, it enhances the activities of complex I and complex II of ETC [21]. In Stat3 deficient pro-B cells, the activities of Complex I and Complex II are reduced, but expression of a mitochondrial-targeted Stat3 (MLS-Stat3) rescues the ETC Complex I and II activities [21]. This shows mitoStat3 maintains ETC activities independent of nuclear activities. MitoStat3 has also been shown to enhance the activity of Complex V in cells undergoing oncogenic transformation [22]. Intriguingly, mitoStat3 also enhances ETC Complex IV activity in FGFR-R388 single nucleotide polymorphic variant cells [42]. The biochemical mechanisms by which mitoStat3 regulates ETC complex activities have been investigated by several groups. MitoStat3 directly interacts with Complex I and possibily Complex II of ETC [21], suggesting that mitoStat3 cooperatively enhances the enzyme activities of Complex I and II of ETC. Interestingly, both Tyr705 and Ser727 phosphorylation of Stat3 has been found in mitochondria [21, 23, 25, 28]. However, Stat3 Tyr705 phosphorylation does not seem to be required for the optimal activities of ETC in several cell types [21]. In contrast, Ser727 phosphorylation is critical for mitoStat3 to enhance ETC Complex I and Complex II activities [21, 22].
Complex I largely contributes to establishing the mitochondrial membrane potential (MMP) across the inner membrane by pumping protons to the intermembrane space. The contribution of Stat3 to the maintenance of MMP appears to be dependent on cell types or metabolic requirements of cells. In H-ras transformed fibroblasts, Stat3 does not seem to be required for MMP [22]. In contrast, disruption of Stat3 reduces MMP in other conditions, such as astrocytes [43]. While naïve CD4 T lymphocytes prior activation have high MMP, we have shown that upon activation MMP decreases over time [23]. However, if IL-6 is present during the activation of CD4 T cells, MMP remains high [23]. The effect of IL-6 on preventing mitochondrial depolarization in these cells is Stat3-dependent [23]. Furthermore, we have shown that the effect of Stat3 on MMP is independent of its transcription-mediated activities [23]. Thus, the contribution of mitoStat3 to MMP is most likely determined by the metabolic state of the cells and specific requirements of mitochondrial respiration. Alternatively, since in addition to Complex I both Complex III and IV contribute to the generation of MMP, the role of mitoStat3 in MMP could be determined by the relative proportion of contribution by Complex I.
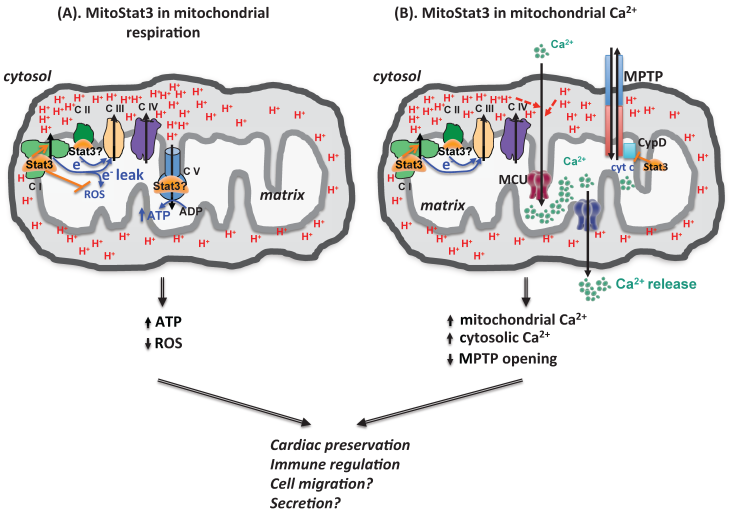
MMP is primarily used for ATP synthesis by Complex V of ETC, this process is defined as oxidative phosphorylation (OXPHOS) because it is coupled to oxygen consumption. The lack of mitoStat3 lowers the levels of ATP in cells undergoing oncogenic transformation [22]. Tissue specific disruption of Stat3 also reduces ATP production in astrocytes [43]. IL-6 promotes mitoStat3 translocation in pancreatic cancer cell lines panc02 and panc02.03, and leads to a significant increase in the levels of ATP [44]. Together, in most tissues and cells, mitoStat3 positively regulates ETC activities and this results in elevated ATP production (Fig. 2).
mitoStat3 as a regulator of mitochondria functions. (A) mitoStat3 promotes mitochondrial respiration (ATP synthesis) by increasing ETC activity and MMP. (B) mitoStat3 regulates mitochondrial Ca2+ and cytosolic Ca2+ through the regulation of ETC and MPTP.
Interestingly, we have recently reported that IL-6 also facilitates mitoStat3 translocation in activated mouse CD4 T cells. However, the increase in mitoStat3 does not result in any changes in ATP levels [23]. This suggests that the effect of mitoStat3 in the regulation of ATP production could be cell type and tissue type specific.
2.2. Exploring the role of mitoStat3 in calcium homeostasis
While a major function of the mitochondrial membrane potential is to drive the synthesis of ATP by OXPHOS, it is also used to regulate Ca2+ uptake into the mitochondria [45] (Fig. 2). Mitochondria were the one of the first intracellular organelles to be associated with Ca2+ storage. In recent years, mitochondria are emerging as a critical organelle in buffering cytosolic Ca2+. The uptake of Ca2+ into the mitochondrial matrix through ion impermeable inner mitochondrial membrane is mediated by the recently identified mitochondrial calcium uniporter (MCU), using the mitochondrial membrane potential gradient [46-48]. Mitochondrial Ca2+ is then released to the cytosol by the mitochondrial Na+/Ca+ exchanger (mNCX) and mitochondrial H+/Ca2+ exchanger (mHCX) [45, 49, 50] (Fig. 2).
The uptake and release of Ca2+ by mitochondria contributes to the cytosolic Ca2+ homeostasis. Although mitoStat3 is known to regulate ETC activity and contribute to establishment of MMP, no studies had revealed its role in mitochondrial Ca2+ homeostasis until recently. We have recently shown that IL-6 sustains mitochondrial membrane polarization during the activation of CD4 cells, and this effect is mediated by Stat3 independently of its transcriptional activity [23]. Surprisingly, despite its effect on mitochondrial membrane potential, we found that IL-6 does not affect ATP synthesis, oxygen consumption rate or glycolytic rate [23]. However, we discovered that the sustained mitochondrial polarization triggered by IL-6 is essential to promote mitochondrial Ca2+ influx through MCU, and requires Stat3 but not its transcriptional activity [23]. Thus, in addition of its role in modulating mitochondrial respiration, mitoStat3 can also regulate mitochondrial Ca2+ through its effect on the ETC and mitochondrial membrane potential (Fig. 2). While endoplasmic reticulum (ER) is the best characterized intracellular Ca2+ storage that regulates cytosolic Ca2+, a number of studies have now proposed that mitochondria could be even more essential since it serves as a vehicle to deliver Ca2+ from ER to the cytosol [45, 51-53] (Fig. 2). We have shown that CD4 cells activated with IL-6 through mitoStat3 maintain higher cytosolic Ca2+ levels using mitochondrial Ca2+ as a source [23]. In addition, mitoStat3 can also regulate cytosolic Ca2+ through mitochondrial permeability transition pore (MPTP), a Ca2+-dependent nonselective pore that is opened to release Ca2+ and cytochrome C from mitochondria matrix to cytosol, leading to cell death (Fig. 2). Therefore, in addition to the regulation of mitochondrial ATP production, mitoStat3 also plays a role in Ca2+ homeostasis.
2.3. MitoStat3 in the regulation of ROS
In mitochondria, ETC Complex I, II and III shuttle electrons for Complex IV (cytochrome c oxidase) to complete reduction of O2 to H2O. However, electron leakage also occurs when electrons pass down the respiratory chain exit prior to the reduction of O2 to H2O at Complex IV, forming reactive oxygen species (ROS). Accumulation of ROS has been linked to multiple pathological conditions, including neurodegenerative diseases, ischemia/reperfusion injuries, diabetes, cancer and aging [54, 55]. Although mitoStat3 enhances ETC activity, the role of mitoStat3 in the regulation of mitochondrial ROS is less clear. TNFα induces Ser727 phosphorylation of Stat3 in L929 cells. This leads to increased Stat3-GRIM-19 interaction, which results in increased mitoStat3 translocation [26, 56]. Increased ROS production by TNFα could be mediated by mitoStat3 [26, 56, 57]. Similarly, increased ROS production by nerve growth factor (NGF) in PC12 cells has also been associated with increased Stat3 mitochondrial localization [29]. In contrast to these studies suggesting that mitoStat3 promotes ROS production, several other studies suggest that mitoStat3 prevents ROS production. Stat3 deficient astrocytes produce high levels of ROS, leading to increased cell death [43]. The levels of ROS are also higher in several mouse bone marrow subtype cells of Stat3 deficient mice relative to wild-type [58]. Upon induction of cardiac ischemia, expression of transcriptional inactive mitochondria-targeted Stat3 in heart in a transgenic mouse model (MLS-STAT3E mice) significantly reduces the production of ROS, preserving heart tissues during ischemia/reperfusion injuries [27]. Similarly, expression of MLS-Stat3 also reduces ROS production in the murine breast cancer 4T1 cells [25]. We have also shown that IL-6 increases MMP by mitoStat3 in mouse CD4 cells, but this effect is associated with a reduced ROS production [23]. Therefore, although not yet clear, there is growing evidence to suggest that while mitoStat3 can promote ETC activity in mitochondria, it can also suppress the production of mitochondrial ROS (Fig. 2).
The mechanisms by which mitoStat3 could minimize the production of mitochondrial ROS while increasing electron transport at the ETC also remain unclear. However, a recent metabolomics study has revealed that mitoStat3 is required for the functions of gamma-glutamyl cycle, and the formation of glutathione, which can balance the normal redox stage [59]. Thus, while electron leaks may still occur, mitoStat3 could neutralize the ROS through this mechanism.
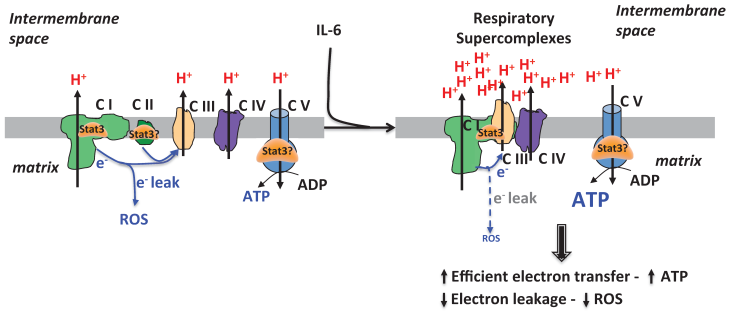
Alternatively, it is also possible that mitoStat3 can directly reduce the production of electron leak from ETC. Historically, respiratory ETC complexes were thought to be randomly distributed as independent entities [60]. However, this view has been challenged based on both functional and structural analyses that support a model where the individual ETC complexes interact to form stable supramolecular structures, called respiratory chain supercomplexes (RCS) or respirasome [61-63] (Fig. 3). RCS containing Complexe I and III, or Complexe I, III and IV have been the most studied [64]. The objective of RCS is to facilitate electron transfer within the individual complexes in RCS and to minimize the generation of ROS. Our recent study has revealed the presence of Stat3 in RCS in CD4 cells activated in the presence of IL-6 [23]. Increased levels of mitoStat3 containing RCS correlates with decreased mitochondrial ROS levels in these cells [23] (Fig. 3). Similar to this study, Stat3 also plays a protective role against ROS production in heart, astrocytes and sperms as reviewed above. Because RCS minimize the formation of ROS while facilitating normal ETC functions, the presence of mitoStat3 in RCS could be a mechanism by which mitoStat3 can promote ETC activity while still prevents ROS formation (Fig. 3). Although more studies are needed to address whether Stat3 regulates the formation of respiratory supercomplexes, the formation of RCS elicited by mitoStat3 could be another mechanism for mitoStat3 to mediate a very efficient mitochondrial respiration in other tissues, such as heart and cancer cells.
mitoStat3 as a regulator of respiratory supercomplexes. Stat3 can be recruited to respiratory supercomplexes to promote ETC activity and ATP synthesis without increasing ROS production.
3. Regulation of cellular function by mitochondrial Stat3
As such an important positive regulator in mitochondrial ETC, as described above, mitoStat3 results in various major physiological consequences in following areas, cardioprotection, cancer progression and immunological regulation.
3.1. Cardioprotective role of mitochondrial Stat3
Reperfusion of an ischaemic heart (I/R) may exacerbate the damage caused by ischaemia itself, as reflected in increased necrosis and release of cellular enzymes, accompanying by dysfunction in mitochondria [65]. Upon reperfusion, rapid accumulation of mitochondrial Ca2+ is accompanied by increased mitochondrial membrane potential, extensive production of ROS and adenine nucleotide depletion. These lead to opening of MPTP which enables free passage across the mitochondrial membranes of molecules of less than 1.5 kDa [66, 67] to release protons, accumulated Ca2+ and pro-apoptotic proteins like cytochrome c to initiate downstream apoptosis [66, 68, 69]. The presence of Stat3 in mitochondria was first discovered in heart [21], specifically in the mitochondrial of subsarcolemmal and interfibrillar cardiomyocytes [30]. Importantly, mitoStat3 provides cardioprotection during heart I/R injury [27, 28, 30, 70]. For instance, expression of a transcriptional inactive mitochondria-targeted Stat3 with a mutation in DNA-binding domain in a transgenic mouse model (MLS-STAT3E mice) leads to better cardiac tissue preservation during heart I/R injury [27, 70]. It is believed that the cardioprotection elicited by mitoStat3 is mediated by delaying MPTP opening [27, 28, 30, 70, 71]. Several studies have proposed that this is an indirect effect of mitoStat3 on ETC, primarily through the suppression of ROS [27, 28, 30, 70, 71]. Alternatively, it has also been suggested that mitoStat3 delays MPTP opening during heart I/R injury through direct interaction with cyclophilin D, one of the MPTP components [30]. Therefore, mitoStat3 provides cardioprotective effects, although additional studies will be needed to dissect the specific mechanisms.
3.2. MitoStat3 promotes cancer progression
Unlike most other cells, cancer cells produce most of their ATP through glycolysis, even under aerobic conditions, instead of mitochondrial OXPHOS, known as the Warburg effect [72]. However, mitochondria still play a role in ROS production and Ca2+ homeostasis in cancer cells [73]. The nuclear role of Stat3 in cancer progression has been studied extensively [31], but the findings of Stat3 being present in mitochondria opened a new area in cancer research in terms of how mitoStat3 might regulate cancer physiology.
Malignant transformation of mouse embryonic fibroblasts using RAS requires the presence of mitoStat3, and blocking mitoStat3 halts tumor growth [22, 74]. Similarly, mitoStat3 is also critical to the oncogenesis in precancerous Barrett's epithelial cells, especially during Ras-dependent transformation [75]. This is dependent on the Ser727 phosphorylation of mitoStat3. MitoStat3 contributes to the Ras-dependent oncogenic transformation by augmenting ETC complexes activities [22]. MitoStat3 is also required for optimal gamma-glutathione cycle activities. This restrains ROS levels in cells undergoing oncogenic transformation. In the absence of mitoStat3, significant cell death accompanied by decreased reducing GSH and increased mitochondrial ROS is observed [59]. Importantly, the actions of mitoStat3 in controlling respiration and Ras transformation are mediated by the phosphorylation of Ser727. In addition, expression of MLS-Stat3 in Stat3 null background promotes breast cancer cell growth, and Ser727 phosphorylation is required for the effects [25]. Moreover, in pancreatic cells, IL-6 induces mitoStat3 translocation to mitochondria. This leads to an enhanced ETC complex I activity and ATP production, suggesting that mitoStat3 could accelerate early neoplastic lesions through enhanced bioenergetics [44, 76, 77]. Interestingly, mitoStat3 has also been shown to induce autophagy in pancreatic cancer cells, which is a common strategy for cancer cell growth, survival and therapeutic resistance. This suggests another mechanism for mitoStat3 to promote carcinogenesis [44]. FGFR4-R388 variant cells rely on mitoStat3 to enhance cytochrome c oxidase (complex IV) activities. This leads to pituitary tumorigenesis [42]. MitoStat3 has also been proposed to contribute to the tumorigenesis in keratinocytes through the regulation of mitochondrial gene expression by binding to mitochondrial DNA [78], however this effect on mitochondrial gene expression has not been corroborated by any other study. Therefore, mitoStat3 holds critical functions in promoting tumorigenesis and cancer progression, but the mechanism remains to be clarified.
A number of studies have also linked Stat3 to chemoresistance and metastasis in cancer [79-85]. However, no study has reported a direct effect of mitoStat3 in these important aspects of cancer biology.
3.3. Perspective of mitoStat3 in Immune Regulation
As a transcription factor, Stat3 has a pleiotropic function in the immune response. Stat3 is a key regulator in T cell differentiation. Naïve CD4 cells can differentiate into T helper 1 (Th1), Th2, Th17, regulatory T helper cells (Treg) and follicular T helper cells (Tfh). The balance of these T helper subsets is critical for maintainance of host immune response and preservation of immune tolerance [86-88]. Stat3 is required for Th17 cell differentiation, which is primarily driven by IL-6, and also mediates autoimmune diseases [89-95]. Stat3 is also required for the differentiation of Tfh cells which produce IL-21 and help B cell antibody production [96-98]. Stat3 binds to regulatory regions of genes including Il17 and Il21 to regulate important immune gene expression [99]. Patients with Stat3 dominant-negative mutation develop Job's Syndrome or Hyper IgE syndrome (HIES) in which patients have defective Th17 responses and defective IL-6 response with increased IgE levels [33-35, 100]. Patients with gain-of-function Stat3 mutations develop early-onset autoimmune disorders [92, 93, 101].
Although the function of Stat3 as a transcription factor in immune response has been well studied, how mitoStat3 may contribute to some aspects previously assigned to Stat3 as a transcription factor had remained unknown until recently. Our study was the first identifying Stat3 in mitochondria in CD4 cells activated with IL-6 [23]. IL-6 promotes the production of IL-4 and IL-21 by CD4 cells [102-104]. Stat3, as a transcription factor, has been shown to induce early expression IL-21 by IL-6 [102]. Instead, we have shown that Stat3 also contributes to sustaining late production of IL-21 and IL-4 independently of its transcription activity [23]. Increased mitochondrial Ca2+ mediated by Stat3 contributes to a sustained NFAT activities and this further sustains the expression of IL-4 and IL-21 by IL-6 [23]. This correlates with the finding that Tfh cells have higher Ca2+ to help expressing IL-4 and IL-21 in vivo [105]. Moreover, although mitochondria play a key role in the metabolism of memory CD8 cells, and Stat3 is critical in the development and maintenance of memory T cells [106-108], no studies have reported a role for mitoStat3 in this process. Investigating how mitoStat3 contributes to other T cell differentiation and effector function could be a promising field in future immune research. Mitochondria are also essential in leukocyte chemotaxis due to their dynamic characteristics and their ability to relocate using coupled microtubules to the adhesion zone where they can provide both ATP and Ca2+ needed for migration [109-111]. Since mitoStat3 promotes the increase in both mitochondrial ATP and Ca2+, it could also contribute to migration of immune cells in inflammatory response, as well as migration of cancer cells in metastasis (Fig. 2).
Other than T cells, Stat3 regulates the functions of other immune cells including macrophages, dendritic cells, mast cells. Interestingly, a recent study has shown that mitoStat3 directly contributes to mast cell degranulation in response to stimuli [112]. Regulation of mitochondrial activities by mitoStat3 could also be involved in the cytokine secretion.
4. Targeting mitochondrial Stat3 for treatment of cancer
As reviewed above, mitoStat3 is crucial in cancer progression and immunological effector function. Targeting nuclear functions of Stat3 has been studied extensively in treating cancer and autoimmune diseases experimentally [113-115]. As a successful experimental target, Stat3 is currently targeted by cancer therapies in several clinical trials, including cyclic Stat3 decoy in head and neck cancer, AZD9150 anti-sense oligonucleotide in malignant ascites, OPB-51602 in advanced solid tumors and WP1066 in central nervous system melanoma and recurrent glioblastoma multiforme. Although many drugs were developed to target nuclear function of Stat3, the potential therapeutic values of targeting mitoStat3 have not been carefully investigated. In addition, several recent studies imply the significance of targeting mitoStat3. Importantly, several known Stat3 inhibitors have been demonstrated to have promising mitochondrial effects, suggesting that Stat3 inhibitors might have inhibitory effects on mitoStat3. The findings of these inhibitors, the potential therapeutic values of targeting mitoStat3 and perspectives of targeting mitoStat3 as a new R&D strategy are summarized below.
Aberrant activation of Stat3 occurs commonly in human tumors. It leads to elevated anti-apoptotic mechanism and cell cycle regulating proteins. Many cancers express constitutively active Stat3 for better resistance to cell death [31]. Importantly, mitoStat3 holds critical functions in tumorigenesis and cancer progression. MitoStat3 is required for metabolic switch during cancer cell oncogenic transformation and tumorigenesis [22, 42, 74]. MitoStat3 promotes ETC complexes activities in cancer cells. This leads to increased ATP production [44, 76, 77]. MitoStat3 suppresses ROS production during canceriogenesis [59]. A number of pharmacological Stat3 inhibitors have been developed by directly targeting Stat3. Most of these Stat3 inhibitors target either Stat3 dimerization or Stat3 DNA binding activities [114]. Interestingly, some of these inhibitors have shown promising effects in inhibiting mitoStat3. Moreover, specific mitoStat3 inhibitors have been discovered recently. These Stat3 inhibitors are selectively described below.
Several DNA-based Stat3 inhibitors have been already tested in cancer treatment in clinical trials in US. AZD9150 or ISIS481464 is a synthetic bicyclic nucleic acid-containing antisense oligonucleotide that is complementary to the mRNA for Stat3. AZD9150 down-regulates Stat3 expression. AZD9150 was first found effective in inhibiting growth in human lymphoma cell lines and tumor xenografts and primary human tumor explants [116]. AZD9150 is currently tested clinically in patients with diffused large B cell lymphoma or advanced/metastatic hepatocellular carcinoma. Cyclic stat3 decoy is another DNA-based Stat3 inhibitor. Cyclic Stat3 decoy contains double-stranded oligonucleotides that correspond to the Stat3 response element. This effectively blocks Stat3 DNA binding activities [117]. Cyclic Stat3 decoy has demonstrated selective binding for Stat3 protein and inhibited the growth of head and neck squamous cell carcinoma cells in vitro and in xenograft models [117, 118]. Cyclic Stat3 decoy is now being tested clinically in patients with head and neck squamous cell carcinoma. Although targeting Stat3 using DNA-based inhibitors has shown promising preclinical anti-tumor effects, whether they could interfere with mitoStat3 function is still unknown.
Peptide-based Stat3 inhibitors have been shown very effective in experimental cancer treatment. Peptide derivatives block Stat3 dimerization and downstream Stat3 functions by mimicking Stat3 SH2 domain. PY*LKTK (Y* represents phosphorylated tyrosine residue) peptide is one of the Stat3 SH2 domain binding peptides. Tagging a membrane translocation sequence to the PY*LKTK peptide suppresses cancer transformation and cancer growth [119, 120]. ISS 610, another peptide derivative inhibitor, is more potent in disrupting Stat3 dimerization [121]. ISS 610 induces cancer cell growth inhibition and apoptosis. S3I-M2001, an oxazole-based peptide inhibitor, also inhibits cancer growth by blocking Stat3 dimerization [122]. Although peptidomimetics of Stat3 have been effective in inhibiting cancer progression in experimental models, none of them have been tested in clinical trials so far in US and the effect of peptidomimetics on mitoStat3 has not been investigated.
The majority of Stat3 inhibitors are small molecule inhibitors. Stat3 protein consists of six structural domains, including N-terminal, coiled-coil, DNA binding domain, Src homology 2 (SH2) domain for dimerization, transactivation domain and C-terminal end. Most Stat3 small molecule inhibitors were designed to target SH2 and DNA binding domains. Intriguingly, some small molecule Stat3 inhibitors have been suggested to inhibit mitoStat3. However, the molecular mechanisms and demonstrations of how these inhibitors interact with mitoStat3 need further investigations. Stattic, a common Stat3 inhibitor which first developed to target Stat3 phosphorylation and dimerization, is effective in experimental cancer therapies. Cells treated with Stattic have reduced Tyr phosphorylation and decreased Stat3 homodimerization. Stattic induces cancer cell apoptosis and inhibits cancer cell growth and progression experimentally [123-126]. More interestingly, Stattic is implied to target mitochondrial function by inhibiting mitoStat3 in several tissues other than cancers (Table 1). In isolated mitochondria from heart, Stattic increases ROS generation and facilitates MPTP opening [71]. This suggests that Stattic has direct effects on mitoStat3. Stattic causes exacerbated myocardial I/R cellular injuries due to increased MPTP opening during myocardial I/R injuries [28, 30, 71] (Table 1). Similar to cells lacking mitoStat3, Stattic reduces mitochondrial ETC activities, mitochondrial membrane potential, ATP production and increases ROS production [24, 71]. Since Stattic is well known to inhibit cancer cell growth and promote apoptosis, the relative contribution of its effects on mitoStat3 could be substantial to Stattic's anti-tumor functions. However, more efforts are needed to distinguish its effects on mitoStat3 from nuclear Stat3. In addition, FLLL32, a new small molecule inhibitor, selectively inhibits Stat3 Tyr705 phosphorylation [127, 128]. FLLL32 has promising anti-tumor effects by facilitating cancer cell apoptosis [127, 128]. Similar to experimental models lacking mitoStat3, FLLL32 significantly depolarizes mitochondrial membrane potential in melanoma cells [127] (Table 1). However, the specific molecular interactions need further investigations. Recently, the first mitoStat3 inhibitor has been demonstrated recently. MDC-1112 or Phospho-valproic acid was first found to inhibit pancreatic cancer cell growth in xenograft model (Table 1). Antibody pathway screening reveals that MDC-1112 targets the phosphorylation of Stat3. Besides inhibiting IL-6-stimulated Stat3 phosphorylation and nuclear DNA binding, surprisingly, MDC-1112 also inhibits the mitochondrial accumulation of mitoStat3. This leads to depolarized mitochondrial membrane potential and increased ROS production, and subsequent apoptotic cell death [129], which can be reversed by overexpression of transcriptional inactive Stat3 (Stat3Y705F) or mitochondrial localized Stat3 (MLS-Stat3). This indicates that MDC-1112 inhibits pancreatic cancer growth by potentially inhibiting mitoStat3 activity. Further investigations into the functions of MDC-1112, as a novel inhibitor, may lead to new understanding of mitoStat3 activities as well as novel cancer therapies.
Promising anti-tumor Stat3 inhibitors that target mitochondrial functions
| Inhibitor | Target Site | Function | Mitochondrial Effects |
|---|---|---|---|
| AG490 | JAK2 kinases, Stat3 tyr phosphorylation [138, 139] | ↑ malignant cells apoptosis, ↓ malignant cell growth, ↓ malignant cell invasion [140-143] | ↓ mitochondrial membrane potential in astrocytes [43] ↑ ROS production in astrocytes [43] ↓ cardioprotective preservation of mitochondrial function by inhibition of mitoStat3 in myocardial I/R injury [28] ↑ MPTP opening [149, 150] ↓ neuroprotective effects by leptin-mediated mitochondrial stabilization [144] ↓ cardioprotective effects by melatonin-mediated mitochondrial preservation in myocardial I/R injury [151] ↓ cardioprotective effects by the reduced MPTP opening elicited by leptin and atorvastatin in myocardial I/R injury [149, 150] |
| Stattic | Stat3 phosphorylation, SH2 domain [71] | ↑ malignant cells apoptosis, ↓ malignant cell growth, ↑ tumor chemo- and radiosensitivity [123-125] | ↓ mitochondrial ADP-stimulated oxygen consumption in cardiomyocytes [30] ↓ ATP production in human sperms and mast cells [24, 112] ↓ oxygen consumption rates and ↓ ETC complex II and complex III activities in mast cells [112] ↓ mitochondrial membrane potential in human sperms [24] ↑ ROS production in human sperms [24] ↓ cardioprotective preservation of mitochondrial function by mitoStat3 activation in myocardial I/R injury [28] ↑ ROS production in cardiomyocytes, and subsequent ↑ MPTP opening and ↓ ATP production [71] ↑ cellular injuries due to ↑ MPTP opening by mitoStat3 inhibition in myocardial I/R injury [30] ↓ mast cell degranulation through the inhibition of mitochondrial function [112] |
| Cucurbitacin-I/JSI-124 | Stat3 tyr phosphorylation [130] | ↑ malignant cells apoptosis, ↓ malignant cell growth [130-136] | ↓ mitochondrial membrane potential [137] ↑ ROS production and subsequent ↑ autophagy [136] ↑ mitochondrial apoptotic cell death in cancer cells [131-133] |
| MDC-1112/phospho-valproic acid | Stat3 phosphorylation, mitochondrial Stat3 accumulation [129] | ↑ malignant cells apoptosis, ↓ malignant cell growth [129] | ↓ mitoStat3 mitochondrial accumulation [129] ↓ mitochondrial membrane potential [129] ↑ mitochondrial apoptotic cell death [129] |
| FLLL32 | Stat3 tyr phosphorylation [127, 128] | ↑ malignant cells apoptosis [127, 128] | ↓ mitochondrial membrane potential [127] ↑ mitochondrial apoptotic cell death in cancer cells [127, 128] |
A number of small molecules targeting upstream JAKs have also been proposed as potential modulators of Stat3. JSI-124 or cucurbitacin I inhibits Stat3 phosphorylation by suppressing levels of Janus kinases and also activities of Janus kinases [130]. JSI-124 inhibits growth, invasion and induces apoptotic cell death in multiple cancer cells, including leukemia, lymphoma, lung cancer, breast cancer, glioma, glioblastoma, endometrial cancer and cervical cancers [130-136]. Interestingly, JSI-124 also inhibits mitochondria-related functions (Table 1). JSI-124 leads to a significant reduction in mitochondrial membrane potential in SW480 colon cancer cells [137]. In addition, JSI-124 induces mitochondrial ROS production in a Stat3 dependent manner, and subsequently, downstream activation of autophagy [136]. Although JSI-124 contributes to multiple aspects of mitochondrial function, whether the effects of JSI-124 are due to direct modulation of mitoStat3 needs further investigations. Another small molecule inhibitor AG490 selectively inhibits Janus kinases, especially Jak2 to suppress Stat3 phosphorylation [138, 139]. AG490 induces cancer cell apoptosis and inhibits malignant cell growth and invasion in multiple cancer cells [140-143]. Intriguingly, recent studies have revealed that AG490 also elicits many functions by targeting mitochondria in various cell and tissue models (Table 1). AG490 has been implied to inhibit mitoStat3 pathway directly and abolish the cardioprotective effects elicited by mitoStat3 in myocardial I/R injury by postconditioning treatment [28]. AG490 induces apoptosis in astrocytes by inhibiting mitochondrial membrane potential and increasing ROS generation [43], and blocks Stat3-mediated lectin neuroprotection by destabilizing mitochondrial function [144]. Thus, some of the anti-tumor effects of AG490 might be in part due to its effects on mitoStat3. Thus, these JAKs inhibitors have shown promising effects in inhibiting mitoStat3, however, the specificity of these inhibitors for Stat3 relative to other downstream factors needs more careful investigations.
In summary, in many of conventional Stat3 inhibitors, some of them have shown significant direct effects on mitoStat3. Others have shown evidences suggesting their inhibitory effects on mitochondria. New screening systems have also discovered novel mitoStat3 inhibitors. Although the direct mitoStat3 inhibition effect of many of these inhibitors needs to be validated, further investigations into this topic may shed light into new targets in anti-tumor therapies.
Remarks
In summary, in addition to the well-characterized role of Stat3 as a transcription factor, we have described here the molecular mechanisms and functions of mitochondrial Stat3 as well as promising ways to target mitoStat3 as a strategy to modulate biological functions. Different from several other important review papers [145-148], this review summarizes several recent findings related to mitoStat3, including calcium homeostasis, respiratory supercomplexes, regulation of immune system and the inhibitors that might modulate mitoStat3 activities. The role of mitoStat3 in the regulation of supercomplexes and calcium should be also confirmed in other tissues. As mentioned above, additional studies exploring the mechanisms by which mitoStat3 could regulate supercomplexes formation and mitochondrial calcium homeostasis are urgently needed to understand a broader aspects of mitoStat3. Moreover, testing existing Stat3 inhibitors with their inhibitory effects on mitoStat3, as well as developing novel mitoStat3 specific inhibitors are of great therapeutic and research values.
Acknowledgements
Some image components were obtained from http://www.somersault1824.com image library. We thank Dr. Edward Lesnefsky (Medical College of Virginia School of Medicine, Virginia Commonwealth University) for his insightful reading of the review and his feedback. We also thank Devin P. Champagne and Tina M. Thornton for carefully proof-reading the manuscript. This work was supported by National Institutes of Health Grant R56AI094027 (to M.R.) and P20GM103496 (to R.Y. and M.R.). R.Y. was supported through the American Association of Immunologists Careers in Immunology Fellowship Program.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Levy DE, Lee CK. What does Stat3 do? J Clin Invest. 2002;109:1143-8
2. Akira S, Nishio Y, Inoue M, Wang XJ, Wei S, Matsusaka T. et al. Molecular cloning of APRF, a novel IFN-stimulated gene factor 3 p91-related transcription factor involved in the gp130-mediated signaling pathway. Cell. 1994;77:63-71
3. Lutticken C, Wegenka UM, Yuan J, Buschmann J, Schindler C, Ziemiecki A. et al. Association of transcription factor APRF and protein kinase Jak1 with the interleukin-6 signal transducer gp130. Science. 1994;263:89-92
4. Zhong Z, Wen Z, Darnell JE Jr. Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science. 1994;264:95-8
5. Raz R, Durbin JE, Levy DE. Acute phase response factor and additional members of the interferon-stimulated gene factor 3 family integrate diverse signals from cytokines, interferons, and growth factors. The Journal of biological chemistry. 1994;269:24391-5
6. Heinrich PC, Behrmann I, Haan S, Hermanns HM, Muller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. The Biochemical journal. 2003;374:1-20
7. Xu X, Sun YL, Hoey T. Cooperative DNA binding and sequence-selective recognition conferred by the STAT amino-terminal domain. Science. 1996;273:794-7
8. Becker S, Groner B, Muller CW. Three-dimensional structure of the Stat3beta homodimer bound to DNA. Nature. 1998;394:145-51
9. Maritano D, Sugrue ML, Tininini S, Dewilde S, Strobl B, Fu X. et al. The STAT3 isoforms alpha and beta have unique and specific functions. Nature immunology. 2004;5:401-9
10. Yoo JY, Huso DL, Nathans D, Desiderio S. Specific ablation of Stat3beta distorts the pattern of Stat3-responsive gene expression and impairs recovery from endotoxic shock. Cell. 2002;108:331-44
11. Caldenhoven E, van Dijk TB, Solari R, Armstrong J, Raaijmakers JA, Lammers JW. et al. STAT3beta, a splice variant of transcription factor STAT3, is a dominant negative regulator of transcription. The Journal of biological chemistry. 1996;271:13221-7
12. Schaefer TS, Sanders LK, Nathans D. Cooperative transcriptional activity of Jun and Stat3 beta, a short form of Stat3. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:9097-101
13. Stahl N, Boulton TG, Farruggella T, Ip NY, Davis S, Witthuhn BA. et al. Association and activation of Jak-Tyk kinases by CNTF-LIF-OSM-IL-6 beta receptor components. Science. 1994;263:92-5
14. Narazaki M, Witthuhn BA, Yoshida K, Silvennoinen O, Yasukawa K, Ihle JN. et al. Activation of JAK2 kinase mediated by the interleukin 6 signal transducer gp130. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:2285-9
15. Heinrich PC, Behrmann I, Muller-Newen G, Schaper F, Graeve L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. The Biochemical journal. 1998;334( Pt 2):297-314
16. Boulanger MJ, Chow DC, Brevnova EE, Garcia KC. Hexameric structure and assembly of the interleukin-6/IL-6 alpha-receptor/gp130 complex. Science. 2003;300:2101-4
17. Guschin D, Rogers N, Briscoe J, Witthuhn B, Watling D, Horn F. et al. A major role for the protein tyrosine kinase JAK1 in the JAK/STAT signal transduction pathway in response to interleukin-6. The EMBO journal. 1995;14:1421-9
18. Decker T, Kovarik P. Serine phosphorylation of STATs. Oncogene. 2000;19:2628-37
19. Wen Z, Zhong Z, Darnell JE Jr. Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell. 1995;82:241-50
20. Zhong Z, Wen Z, Darnell JE Jr. Stat3 and Stat4: members of the family of signal transducers and activators of transcription. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:4806-10
21. Wegrzyn J, Potla R, Chwae YJ, Sepuri NB, Zhang Q, Koeck T. et al. Function of mitochondrial Stat3 in cellular respiration. Science. 2009;323:793-7
22. Gough DJ, Corlett A, Schlessinger K, Wegrzyn J, Larner AC, Levy DE. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science. 2009;324:1713-6
23. Yang R, Lirussi D, Thornton TM, Jelley-Gibbs DM, Diehl SA, Case LK. et al. Mitochondrial Ca(2)(+) and membrane potential, an alternative pathway for Interleukin 6 to regulate CD4 cell effector function. eLife. 2015:4
24. Lachance C, Goupil S, Leclerc P. Stattic V, a STAT3 inhibitor, affects human spermatozoa through regulation of mitochondrial activity. Journal of cellular physiology. 2013;228:704-13
25. Zhang Q, Raje V, Yakovlev VA, Yacoub A, Szczepanek K, Meier J. et al. Mitochondrial localized Stat3 promotes breast cancer growth via phosphorylation of serine 727. The Journal of biological chemistry. 2013;288:31280-8
26. Shulga N, Pastorino JG. GRIM-19-mediated translocation of STAT3 to mitochondria is necessary for TNF-induced necroptosis. Journal of cell science. 2012;125:2995-3003
27. Szczepanek K, Chen Q, Derecka M, Salloum FN, Zhang Q, Szelag M. et al. Mitochondrial-targeted Signal transducer and activator of transcription 3 (STAT3) protects against ischemia-induced changes in the electron transport chain and the generation of reactive oxygen species. The Journal of biological chemistry. 2011;286:29610-20
28. Heusch G, Musiolik J, Gedik N, Skyschally A. Mitochondrial STAT3 activation and cardioprotection by ischemic postconditioning in pigs with regional myocardial ischemia/reperfusion. Circulation research. 2011;109:1302-8
29. Zhou L, Too HP. Mitochondrial localized STAT3 is involved in NGF induced neurite outgrowth. PloS one. 2011;6:e21680
30. Boengler K, Hilfiker-Kleiner D, Heusch G, Schulz R. Inhibition of permeability transition pore opening by mitochondrial STAT3 and its role in myocardial ischemia/reperfusion. Basic research in cardiology. 2010;105:771-85
31. Yu H, Lee H, Herrmann A, Buettner R, Jove R. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nature reviews Cancer. 2014;14:736-46
32. Takeda K, Noguchi K, Shi W, Tanaka T, Matsumoto M, Yoshida N. et al. Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:3801-4
33. Minegishi Y, Saito M, Nagasawa M, Takada H, Hara T, Tsuchiya S. et al. Molecular explanation for the contradiction between systemic Th17 defect and localized bacterial infection in hyper-IgE syndrome. The Journal of experimental medicine. 2009;206:1291-301
34. Milner JD, Brenchley JM, Laurence A, Freeman AF, Hill BJ, Elias KM. et al. Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature. 2008;452:773-6
35. de Beaucoudrey L, Puel A, Filipe-Santos O, Cobat A, Ghandil P, Chrabieh M. et al. Mutations in STAT3 and IL12RB1 impair the development of human IL-17-producing T cells. The Journal of experimental medicine. 2008;205:1543-50
36. Lufei C, Ma J, Huang G, Zhang T, Novotny-Diermayr V, Ong CT. et al. GRIM-19, a death-regulatory gene product, suppresses Stat3 activity via functional interaction. The EMBO journal. 2003;22:1325-35
37. Zhang J, Yang J, Roy SK, Tininini S, Hu J, Bromberg JF. et al. The cell death regulator GRIM-19 is an inhibitor of signal transducer and activator of transcription 3. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:9342-7
38. Cimica V, Chen HC, Iyer JK, Reich NC. Dynamics of the STAT3 transcription factor: nuclear import dependent on Ran and importin-beta1. PloS one. 2011;6:e20188
39. Phillips D, Reilley MJ, Aponte AM, Wang G, Boja E, Gucek M. et al. Stoichiometry of STAT3 and mitochondrial proteins: Implications for the regulation of oxidative phosphorylation by protein-protein interactions. The Journal of biological chemistry. 2010;285:23532-6
40. Tammineni P, Anugula C, Mohammed F, Anjaneyulu M, Larner AC, Sepuri NB. The import of the transcription factor STAT3 into mitochondria depends on GRIM-19, a component of the electron transport chain. The Journal of biological chemistry. 2013;288:4723-32
41. Qiu H, Lizano P, Laure L, Sui X, Rashed E, Park JY. et al. H11 kinase/heat shock protein 22 deletion impairs both nuclear and mitochondrial functions of STAT3 and accelerates the transition into heart failure on cardiac overload. Circulation. 2011;124:406-15
42. Tateno T, Asa SL, Zheng L, Mayr T, Ullrich A, Ezzat S. The FGFR4-G388R polymorphism promotes mitochondrial STAT3 serine phosphorylation to facilitate pituitary growth hormone cell tumorigenesis. PLoS genetics. 2011;7:e1002400
43. Sarafian TA, Montes C, Imura T, Qi J, Coppola G, Geschwind DH. et al. Disruption of astrocyte STAT3 signaling decreases mitochondrial function and increases oxidative stress in vitro. PloS one. 2010;5:e9532
44. Kang R, Loux T, Tang D, Schapiro NE, Vernon P, Livesey KM. et al. The expression of the receptor for advanced glycation endproducts (RAGE) is permissive for early pancreatic neoplasia. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:7031-6
45. Rizzuto R, De Stefani D, Raffaello A, Mammucari C. Mitochondria as sensors and regulators of calcium signalling. Nature reviews Molecular cell biology. 2012;13:566-78
46. Shanmughapriya S, Rajan S, Hoffman NE, Zhang X, Guo S, Kolesar JE. et al. Ca2+ signals regulate mitochondrial metabolism by stimulating CREB-mediated expression of the mitochondrial Ca2+ uniporter gene MCU. Science signaling. 2015;8:ra23
47. Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y. et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011;476:341-5
48. De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature. 2011;476:336-40
49. Palty R, Silverman WF, Hershfinkel M, Caporale T, Sensi SL, Parnis J. et al. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:436-41
50. Crompton M, Kunzi M, Carafoli E. The calcium-induced and sodium-induced effluxes of calcium from heart mitochondria. Evidence for a sodium-calcium carrier. European journal of biochemistry / FEBS. 1977;79:549-58
51. Tang Y, Zucker RS. Mitochondrial involvement in post-tetanic potentiation of synaptic transmission. Neuron. 1997;18:483-91
52. Billups B, Forsythe ID. Presynaptic mitochondrial calcium sequestration influences transmission at mammalian central synapses. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2002;22:5840-7
53. Medler K, Gleason EL. Mitochondrial Ca(2+) buffering regulates synaptic transmission between retinal amacrine cells. Journal of neurophysiology. 2002;87:1426-39
54. Sena LA, Chandel NS. Physiological roles of mitochondrial reactive oxygen species. Molecular cell. 2012;48:158-67
55. Murphy MP. How mitochondria produce reactive oxygen species. The Biochemical journal. 2009;417:1-13
56. Shulga N, Pastorino JG. Mitoneet mediates TNFalpha-induced necroptosis promoted by exposure to fructose and ethanol. Journal of cell science. 2014;127:896-907
57. Zouein FA, Duhe RJ, Arany I, Shirey K, Hosler JP, Liu H. et al. Loss of STAT3 in mouse embryonic fibroblasts reveals its Janus-like actions on mitochondrial function and cell viability. Cytokine. 2014;66:7-16
58. Mantel C, Messina-Graham S, Moh A, Cooper S, Hangoc G, Fu XY. et al. Mouse hematopoietic cell-targeted STAT3 deletion: stem/progenitor cell defects, mitochondrial dysfunction, ROS overproduction, and a rapid aging-like phenotype. Blood. 2012;120:2589-99
59. Garama DJ, Harris TJ, White CL, Rossello FJ, Abdul-Hay M, Gough DJ. et al. A synthetic lethal interaction between glutathione synthesis and mitochondrial reactive oxygen species provides a tumor specific vulnerability dependent on STAT3. Molecular and cellular biology. 2015
60. Hackenbrock CR, Chazotte B, Gupte SS. The random collision model and a critical assessment of diffusion and collision in mitochondrial electron transport. Journal of bioenergetics and biomembranes. 1986;18:331-68
61. Acin-Perez R, Fernandez-Silva P, Peleato ML, Perez-Martos A, Enriquez JA. Respiratory active mitochondrial supercomplexes. Molecular cell. 2008;32:529-39
62. Eubel H, Heinemeyer J, Braun HP. Identification and characterization of respirasomes in potato mitochondria. Plant physiology. 2004;134:1450-9
63. Eubel H, Heinemeyer J, Sunderhaus S, Braun HP. Respiratory chain supercomplexes in plant mitochondria. Plant physiology and biochemistry: PPB / Societe francaise de physiologie vegetale. 2004;42:937-42
64. Winge DR. Sealing the mitochondrial respirasome. Molecular and cellular biology. 2012;32:2647-52
65. Honda HM, Korge P, Weiss JN. Mitochondria and ischemia/reperfusion injury. Annals of the New York Academy of Sciences. 2005;1047:248-58
66. Halestrap AP. What is the mitochondrial permeability transition pore? Journal of molecular and cellular cardiology. 2009;46:821-31
67. Halestrap AP, Clarke SJ, Javadov SA. Mitochondrial permeability transition pore opening during myocardial reperfusion-a target for cardioprotection. Cardiovascular research. 2004;61:372-85
68. Doran E, Halestrap AP. Cytochrome c release from isolated rat liver mitochondria can occur independently of outer-membrane rupture: possible role of contact sites. The Biochemical journal. 2000;348(Pt 2):343-50
69. Petronilli V, Nicolli A, Costantini P, Colonna R, Bernardi P. Regulation of the permeability transition pore, a voltage-dependent mitochondrial channel inhibited by cyclosporin A. Biochimica et biophysica acta. 1994;1187:255-9
70. Szczepanek K, Xu A, Hu Y, Thompson J, He J, Larner AC. et al. Cardioprotective function of mitochondrial-targeted and transcriptionally inactive STAT3 against ischemia and reperfusion injury. Basic research in cardiology. 2015;110:53
71. Boengler K, Ungefug E, Heusch G, Schulz R. The STAT3 inhibitor stattic impairs cardiomyocyte mitochondrial function through increased reactive oxygen species formation. Current pharmaceutical design. 2013;19:6890-5
72. Warburg O, Wind F, Negelein E. The Metabolism of Tumors in the Body. The Journal of general physiology. 1927;8:519-30
73. Gogvadze V, Orrenius S, Zhivotovsky B. Mitochondria in cancer cells: what is so special about them? Trends in cell biology. 2008;18:165-73
74. Gough DJ, Koetz L, Levy DE. The MEK-ERK pathway is necessary for serine phosphorylation of mitochondrial STAT3 and Ras-mediated transformation. PloS one. 2013;8:e83395
75. Yu C, Huo X, Agoston AT, Zhang X, Theiss AL, Cheng E. et al. Mitochondrial STAT3 contributes to transformation of Barrett's epithelial cells that express oncogenic Ras in a p53-independent fashion. American journal of physiology Gastrointestinal and liver physiology. 2015;309:G146-61
76. Kang R, Tang D, Lotze MT, Zeh HJ 3rd. AGER/RAGE-mediated autophagy promotes pancreatic tumorigenesis and bioenergetics through the IL6-pSTAT3 pathway. Autophagy. 2012;8:989-91
77. Kang R, Tang D. Autophagy in pancreatic cancer pathogenesis and treatment. American journal of cancer research. 2012;2:383-96
78. Macias E, Rao D, Carbajal S, Kiguchi K, DiGiovanni J. Stat3 binds to mtDNA and regulates mitochondrial gene expression in keratinocytes. The Journal of investigative dermatology. 2014;134:1971-80
79. Du L, Subauste MC, DeSevo C, Zhao Z, Baker M, Borkowski R. et al. miR-337-3p and its targets STAT3 and RAP1A modulate taxane sensitivity in non-small cell lung cancers. PloS one. 2012;7:e39167
80. Han Z, Feng J, Hong Z, Chen L, Li W, Liao S. et al. Silencing of the STAT3 signaling pathway reverses the inherent and induced chemoresistance of human ovarian cancer cells. Biochemical and biophysical research communications. 2013;435:188-94
81. Chen RJ, Ho YS, Guo HR, Wang YJ. Long-term nicotine exposure-induced chemoresistance is mediated by activation of Stat3 and downregulation of ERK1/2 via nAChR and beta-adrenoceptors in human bladder cancer cells. Toxicological sciences: an official journal of the Society of Toxicology. 2010;115:118-30
82. Bhardwaj A, Sethi G, Vadhan-Raj S, Bueso-Ramos C, Takada Y, Gaur U. et al. Resveratrol inhibits proliferation, induces apoptosis, and overcomes chemoresistance through down-regulation of STAT3 and nuclear factor-kappaB-regulated antiapoptotic and cell survival gene products in human multiple myeloma cells. Blood. 2007;109:2293-302
83. Real PJ, Sierra A, De Juan A, Segovia JC, Lopez-Vega JM, Fernandez-Luna JL. Resistance to chemotherapy via Stat3-dependent overexpression of Bcl-2 in metastatic breast cancer cells. Oncogene. 2002;21:7611-8
84. Wen K, Fu Z, Wu X, Feng J, Chen W, Qian J. Oct-4 is required for an antiapoptotic behavior of chemoresistant colorectal cancer cells enriched for cancer stem cells: effects associated with STAT3/Survivin. Cancer letters. 2013;333:56-65
85. Guo L, Chen C, Shi M, Wang F, Chen X, Diao D. et al. Stat3-coordinated Lin-28-let-7-HMGA2 and miR-200-ZEB1 circuits initiate and maintain oncostatin M-driven epithelial-mesenchymal transition. Oncogene. 2013;32:5272-82
86. Yang XO, Nurieva R, Martinez GJ, Kang HS, Chung Y, Pappu BP. et al. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity. 2008;29:44-56
87. Stockinger B, Veldhoen M. Differentiation and function of Th17 T cells. Current opinion in immunology. 2007;19:281-6
88. Weaver CT, Hatton RD, Mangan PR, Harrington LE. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annual review of immunology. 2007;25:821-52
89. Nishihara M, Ogura H, Ueda N, Tsuruoka M, Kitabayashi C, Tsuji F. et al. IL-6-gp130-STAT3 in T cells directs the development of IL-17+ Th with a minimum effect on that of Treg in the steady state. International immunology. 2007;19:695-702
90. Bettelli E, Korn T, Oukka M, Kuchroo VK. Induction and effector functions of T(H)17 cells. Nature. 2008;453:1051-7
91. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M. et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235-8
92. Flanagan SE, Haapaniemi E, Russell MA, Caswell R, Lango Allen H, De Franco E. et al. Activating germline mutations in STAT3 cause early-onset multi-organ autoimmune disease. Nature genetics. 2014;46:812-4
93. Milner JD, Vogel TP, Forbes L, Ma CA, Stray-Pedersen A, Niemela JE. et al. Early-onset lymphoproliferation and autoimmunity caused by germline STAT3 gain-of-function mutations. Blood. 2015;125:591-9
94. Lee YK, Turner H, Maynard CL, Oliver JR, Chen D, Elson CO. et al. Late developmental plasticity in the T helper 17 lineage. Immunity. 2009;30:92-107
95. Weaver CT, Elson CO, Fouser LA, Kolls JK. The Th17 pathway and inflammatory diseases of the intestines, lungs, and skin. Annual review of pathology. 2013;8:477-512
96. Ma CS, Avery DT, Chan A, Batten M, Bustamante J, Boisson-Dupuis S. et al. Functional STAT3 deficiency compromises the generation of human T follicular helper cells. Blood. 2012;119:3997-4008
97. Crotty S. Follicular helper CD4 T cells (TFH). Annual review of immunology. 2011;29:621-63
98. Nurieva RI, Chung Y, Hwang D, Yang XO, Kang HS, Ma L. et al. Generation of T follicular helper cells is mediated by interleukin-21 but independent of T helper 1, 2, or 17 cell lineages. Immunity. 2008;29:138-49
99. Durant L, Watford WT, Ramos HL, Laurence A, Vahedi G, Wei L. et al. Diverse targets of the transcription factor STAT3 contribute to T cell pathogenicity and homeostasis. Immunity. 2010;32:605-15
100. Steward-Tharp SM, Laurence A, Kanno Y, Kotlyar A, Villarino AV, Sciume G. et al. A mouse model of HIES reveals pro- and anti-inflammatory functions of STAT3. Blood. 2014;123:2978-87
101. Walker S, Wang C, Walradt T, Hong BS, Tanner JR, Levinsohn JL. et al. Identification of a gain-of-function STAT3 mutation (p.Y640F) in lymphocytic variant hypereosinophilic syndrome. Blood. 2015
102. Dienz O, Eaton SM, Bond JP, Neveu W, Moquin D, Noubade R. et al. The induction of antibody production by IL-6 is indirectly mediated by IL-21 produced by CD4+ T cells. The Journal of experimental medicine. 2009;206:69-78
103. Diehl S, Chow CW, Weiss L, Palmetshofer A, Twardzik T, Rounds L. et al. Induction of NFATc2 expression by interleukin 6 promotes T helper type 2 differentiation. The Journal of experimental medicine. 2002;196:39-49
104. Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T. et al. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nature immunology. 2007;8:967-74
105. Shulman Z, Gitlin AD, Weinstein JS, Lainez B, Esplugues E, Flavell RA. et al. Dynamic signaling by T follicular helper cells during germinal center B cell selection. Science. 2014;345:1058-62
106. van der Windt GJ, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E. et al. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity. 2012;36:68-78
107. Cui W, Liu Y, Weinstein JS, Craft J, Kaech SM. An interleukin-21-interleukin-10-STAT3 pathway is critical for functional maturation of memory CD8+ T cells. Immunity. 2011;35:792-805
108. Siegel AM, Heimall J, Freeman AF, Hsu AP, Brittain E, Brenchley JM. et al. A critical role for STAT3 transcription factor signaling in the development and maintenance of human T cell memory. Immunity. 2011;35:806-18
109. Campello S, Lacalle RA, Bettella M, Manes S, Scorrano L, Viola A. Orchestration of lymphocyte chemotaxis by mitochondrial dynamics. The Journal of experimental medicine. 2006;203:2879-86
110. Morlino G, Barreiro O, Baixauli F, Robles-Valero J, Gonzalez-Granado JM, Villa-Bellosta R. et al. Miro-1 links mitochondria and microtubule Dynein motors to control lymphocyte migration and polarity. Molecular and cellular biology. 2014;34:1412-26
111. Sanchez-Madrid F, Serrador JM. Mitochondrial redistribution: adding new players to the chemotaxis game. Trends in immunology. 2007;28:193-6
112. Erlich TH, Yagil Z, Kay G, Peretz A, Migalovich-Sheikhet H, Tshori S. et al. Mitochondrial STAT3 plays a major role in IgE-antigen-mediated mast cell exocytosis. The Journal of allergy and clinical immunology. 2014;134:460-9
113. Yu CR, Lee YS, Mahdi RM, Surendran N, Egwuagu CE. Therapeutic targeting of STAT3 (signal transducers and activators of transcription 3) pathway inhibits experimental autoimmune uveitis. PloS one. 2012;7:e29742
114. Yue P, Turkson J. Targeting STAT3 in cancer: how successful are we? Expert opinion on investigational drugs. 2009;18:45-56
115. Jing N, Tweardy DJ. Targeting Stat3 in cancer therapy. Anti-cancer drugs. 2005;16:601-7
116. Hong D, Kurzrock R, Kim Y, Woessner R, Younes A, Nemunaitis J. et al. AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Science translational medicine. 2015;7:314ra185
117. Leong PL, Andrews GA, Johnson DE, Dyer KF, Xi S, Mai JC. et al. Targeted inhibition of Stat3 with a decoy oligonucleotide abrogates head and neck cancer cell growth. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:4138-43
118. Sen M, Thomas SM, Kim S, Yeh JI, Ferris RL, Johnson JT. et al. First-in-human trial of a STAT3 decoy oligonucleotide in head and neck tumors: implications for cancer therapy. Cancer discovery. 2012;2:694-705
119. Turkson J, Ryan D, Kim JS, Zhang Y, Chen Z, Haura E. et al. Phosphotyrosyl peptides block Stat3-mediated DNA binding activity, gene regulation, and cell transformation. The Journal of biological chemistry. 2001;276:45443-55
120. Vultur A, Cao J, Arulanandam R, Turkson J, Jove R, Greer P. et al. Cell-to-cell adhesion modulates Stat3 activity in normal and breast carcinoma cells. Oncogene. 2004;23:2600-16
121. Turkson J, Kim JS, Zhang S, Yuan J, Huang M, Glenn M. et al. Novel peptidomimetic inhibitors of signal transducer and activator of transcription 3 dimerization and biological activity. Molecular cancer therapeutics. 2004;3:261-9
122. Zhang X, Yue P, Page BD, Li T, Zhao W, Namanja AT. et al. Orally bioavailable small-molecule inhibitor of transcription factor Stat3 regresses human breast and lung cancer xenografts. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:9623-8
123. Zhang Q, Zhang C, He J, Guo Q, Hu D, Yang X. et al. STAT3 inhibitor stattic enhances radiosensitivity in esophageal squamous cell carcinoma. Tumour biology: the journal of the International Society for Oncodevelopmental Biology and Medicine. 2015;36:2135-42
124. Pan Y, Zhou F, Zhang R, Claret FX. Stat3 inhibitor Stattic exhibits potent antitumor activity and induces chemo- and radio-sensitivity in nasopharyngeal carcinoma. PloS one. 2013;8:e54565
125. Adachi M, Cui C, Dodge CT, Bhayani MK, Lai SY. Targeting STAT3 inhibits growth and enhances radiosensitivity in head and neck squamous cell carcinoma. Oral oncology. 2012;48:1220-6
126. Schust J, Sperl B, Hollis A, Mayer TU, Berg T. Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chemistry & biology. 2006;13:1235-42
127. Bill MA, Fuchs JR, Li C, Yui J, Bakan C, Benson DM Jr. et al. The small molecule curcumin analog FLLL32 induces apoptosis in melanoma cells via STAT3 inhibition and retains the cellular response to cytokines with anti-tumor activity. Molecular cancer. 2010;9:165
128. Lin L, Hutzen B, Zuo M, Ball S, Deangelis S, Foust E. et al. Novel STAT3 phosphorylation inhibitors exhibit potent growth-suppressive activity in pancreatic and breast cancer cells. Cancer research. 2010;70:2445-54
129. Mackenzie GG, Huang L, Alston N, Ouyang N, Vrankova K, Mattheolabakis G. et al. Targeting mitochondrial STAT3 with the novel phospho-valproic acid (MDC-1112) inhibits pancreatic cancer growth in mice. PloS one. 2013;8:e61532
130. Blaskovich MA, Sun J, Cantor A, Turkson J, Jove R, Sebti SM. Discovery of JSI-124 (cucurbitacin I), a selective Janus kinase/signal transducer and activator of transcription 3 signaling pathway inhibitor with potent antitumor activity against human and murine cancer cells in mice. Cancer research. 2003;63:1270-9
131. Ishdorj G, Johnston JB, Gibson SB. Inhibition of constitutive activation of STAT3 by curcurbitacin-I (JSI-124) sensitized human B-leukemia cells to apoptosis. Molecular cancer therapeutics. 2010;9:3302-14
132. Premkumar DR, Jane EP, Agostino NR, Scialabba JL, Pollack IF. Dasatinib synergizes with JSI-124 to inhibit growth and migration and induce apoptosis of malignant human glioma cells. Journal of carcinogenesis. 2010:9
133. Su Y, Li G, Zhang X, Gu J, Zhang C, Tian Z. et al. JSI-124 inhibits glioblastoma multiforme cell proliferation through G(2)/M cell cycle arrest and apoptosis augment. Cancer biology & therapy. 2008;7:1243-9
134. Chen CL, Hsieh FC, Lieblein JC, Brown J, Chan C, Wallace JA. et al. Stat3 activation in human endometrial and cervical cancers. British journal of cancer. 2007;96:591-9
135. Shi X, Franko B, Frantz C, Amin HM, Lai R. JSI-124 (cucurbitacin I) inhibits Janus kinase-3/signal transducer and activator of transcription-3 signalling, downregulates nucleophosmin-anaplastic lymphoma kinase (ALK), and induces apoptosis in ALK-positive anaplastic large cell lymphoma cells. British journal of haematology. 2006;135:26-32
136. Zhang T, Li Y, Park KA, Byun HS, Won M, Jeon J. et al. Cucurbitacin induces autophagy through mitochondrial ROS production which counteracts to limit caspase-dependent apoptosis. Autophagy. 2012;8:559-76
137. Kim HJ, Park JH, Kim JK. Cucurbitacin-I, a natural cell-permeable triterpenoid isolated from Cucurbitaceae, exerts potent anticancer effect in colon cancer. Chemico-biological interactions. 2014;219:1-8
138. Wang LH, Kirken RA, Erwin RA, Yu CR, Farrar WL. JAK3, STAT, and MAPK signaling pathways as novel molecular targets for the tyrphostin AG-490 regulation of IL-2-mediated T cell response. Journal of immunology. 1999;162:3897-904
139. Levitzki A, Gazit A. Tyrosine kinase inhibition: an approach to drug development. Science. 1995;267:1782-8
140. Du W, Hong J, Wang YC, Zhang YJ, Wang P, Su WY. et al. Inhibition of JAK2/STAT3 signalling induces colorectal cancer cell apoptosis via mitochondrial pathway. Journal of cellular and molecular medicine. 2012;16:1878-88
141. Huang C, Cao J, Huang KJ, Zhang F, Jiang T, Zhu L. et al. Inhibition of STAT3 activity with AG490 decreases the invasion of human pancreatic cancer cells in vitro. Cancer science. 2006;97:1417-23
142. Miyamoto N, Sugita K, Goi K, Inukai T, Lijima K, Tezuka T. et al. The JAK2 inhibitor AG490 predominantly abrogates the growth of human B-precursor leukemic cells with 11q23 translocation or Philadelphia chromosome. Leukemia. 2001;15:1758-68
143. De Vos J, Jourdan M, Tarte K, Jasmin C, Klein B. JAK2 tyrosine kinase inhibitor tyrphostin AG490 downregulates the mitogen-activated protein kinase (MAPK) and signal transducer and activator of transcription (STAT) pathways and induces apoptosis in myeloma cells. British journal of haematology. 2000;109:823-8
144. Guo Z, Jiang H, Xu X, Duan W, Mattson MP. Leptin-mediated cell survival signaling in hippocampal neurons mediated by JAK STAT3 and mitochondrial stabilization. The Journal of biological chemistry. 2008;283:1754-63
145. Meier JA, Larner AC. Toward a new STATe: the role of STATs in mitochondrial function. Seminars in immunology. 2014;26:20-8
146. Szczepanek K, Lesnefsky EJ, Larner AC. Multi-tasking: nuclear transcription factors with novel roles in the mitochondria. Trends in cell biology. 2012;22:429-37
147. Szczepanek K, Chen Q, Larner AC, Lesnefsky EJ. Cytoprotection by the modulation of mitochondrial electron transport chain: the emerging role of mitochondrial STAT3. Mitochondrion. 2012;12:180-9
148. Zouein FA, Altara R, Chen Q, Lesnefsky EJ, Kurdi M, Booz GW. Pivotal Importance of STAT3 in Protecting the Heart from Acute and Chronic Stress: New Advancement and Unresolved Issues. Front Cardiovasc Med. 2015;2:36
149. Smith CC, Dixon RA, Wynne AM, Theodorou L, Ong SG, Subrayan S. et al. Leptin-induced cardioprotection involves JAK/STAT signaling that may be linked to the mitochondrial permeability transition pore. American journal of physiology Heart and circulatory physiology. 2010;299:H1265-70
150. Lemoine S, Zhu L, Legallois D, Massetti M, Manrique A, Hanouz JL. Atorvastatin-induced cardioprotection of human myocardium is mediated by the inhibition of mitochondrial permeability transition pore opening via tumor necrosis factor-alpha and Janus kinase/signal transducers and activators of transcription pathway. Anesthesiology. 2013;118:1373-84
151. Yang Y, Duan W, Jin Z, Yi W, Yan J, Zhang S. et al. JAK2/STAT3 activation by melatonin attenuates the mitochondrial oxidative damage induced by myocardial ischemia/reperfusion injury. Journal of pineal research. 2013;55:275-86
Author contact
![]() Corresponding author: University of Vermont College of Medicine, Given Medical Building Room C331, 89 Beaumont Ave., Burlington, VT 05405-0068, USA. Phone: 802-656-0937; email: mrinconedu
Corresponding author: University of Vermont College of Medicine, Given Medical Building Room C331, 89 Beaumont Ave., Burlington, VT 05405-0068, USA. Phone: 802-656-0937; email: mrinconedu