ISSN: 1449-2288International Journal of Biological Sciences
- Current issue
- Volume 20; 2024
- Volume 19; 2023
- Volume 18; 2022
- Volume 17; 2021
- Volume 16; 2020
- Archive
- Advance articles
- Cover images
- Index & coverage
- Cover suggestion
- Special issues
Introduction
Materials and Methods
Results
Discussion
Conclusions
Abbreviations
Acknowledgements
Conflicts of interest
References

Int J Biol Sci 2016; 12(5):580-593. doi:10.7150/ijbs.14578 This issue Cite
Research Paper
Methacryloxylethyl Cetyl Ammonium Chloride Induces DNA Damage and Apoptosis in Human Dental Pulp Cells via Generation of Oxidative Stress
Yang Jiao1,#, Sai Ma1,#, Yirong Wang2,#, Jing Li3, Lequn Shan4, Jinlong Sun1, Jihua Chen1, ![]()
1. State Key Laboratory of Military Stomatology & National Clinical Research Center for Oral Diseases & Shaanxi Key Laboratory of Oral Diseases, Department of Prosthodontics, School of Stomatology, The Fourth Military Medical University, Xi'an, PR China
2. State Key Laboratory of Military Stomatology & National Clinical Research Center for Oral Diseases & Shaanxi Key Laboratory of Oral Diseases, Department of Operative Dentistry and Endodontics, School of Stomatology, The Fourth Military Medical University, Xi'an, PR China
3. Department of Orthopaedic Oncology, Xijing Hospital, the Fourth Military Medical University, Xi'an, PR China
4. Department of Orthopaedic Surgery, Tangdu hospital, the Fourth Military Medical University, Xi'an, PR China
# These authors have contributed equally to this work.
Abstract

The polymerizable antibacterial monomer methacryloxylethyl cetyl ammonium chloride (DMAE-CB) has provided an effective strategy to combat dental caries. However, the application of such material raises the question about the biological safety and the question remains open. The mechanism of this toxic action, however, is not yet clearly understood. The present study aims at providing novel insight into the possible causal link between cellular oxidative stress and DNA damage, as well as apoptosis in human dental pulp cells exposed to DMAE-CB. The enhanced formation of reactive oxygen species and depletion of glutathione, as well as differential changes in activities of superoxide dismutase, glutathione peroxidase, and catalase in DMAE-CB-treated cells indicated oxidative stress. By using substances that can alter GSH synthesis, we found that GSH was the key component in the regulation of cell response towards oxidative stress induced by DMAE-CB. The increase in oxidative stress-sensitive 8-Oxo-2'-deoxyguanosine (8-OHdG) content, formation of γ-H2AX and cell cycle G1 phase arrest indicated that DNA damage occurred as a result of the interaction between DNA base and ROS beyond the capacities of antioxidant mechanisms in cells exposed to DMAE-CB. Such oxidative DNA damage thus triggers the activation of ataxia telangiectasia-mutated (ATM) signaling, the intrinsic apoptotic pathway, and destruction of mitochondrial morphology and function.
Keywords: methacryloxylethyl cetyl ammonium chloride, oxidative stress, DNA damage, apoptosis, mitochondria.
Introduction
Dental caries is one of the most prevalent bacteria-related infectious diseases worldwide [1]. Data analysis from American Dental Association indicates that nearly 200 million dental restorations have been placed in the U.S. in one year [2]. Dental caries shorten the service life dental restorations and more than 50% of dental restorations need replacement [3]. The annual cost for dental restorations in the U.S. has been reported to be nearly $50 billion in 2005 [4]. Thus, dental caries brings a heavy economic burden for individuals and society. The cariogenic bacterial biofilms tend to accumulate on the restorations and surrounding tooth tissues, and they generate acids to cause the demineralization of enamel and dentin, which is responsible for dental caries [5]. In order to reduce secondary caries and prolong the service life, great efforts have been made to develop dental restorative materials with antibacterial properties. Accordingly, novel polymers containing quaternary ammonium monomers, such as methacryloxylethyl cetyl ammonium chloride (DMAE-CB) have been developed [6, 7] (Fig. 1A). These antibacterial monomers can be blended with other resin monomers, and subsequently copolymerized within the polymer network to develop antibacterial dental materials, which has been considered as an effective strategy to combat dental caries [8-10].
However, the application of such materials raises concerns regarding their potential harm to the ecosystem and human health. According to in vitro studies with multiple target cells, DMAE-CB can interfere with various cellular functions, and induce apoptosis via the intrinsic mitochondrial apoptotic pathway [11-13]. Although not yet clearly understood, current experimental evidence strongly suggests that the mechanism behind these specific cell responses is the generation of oxidative stress [14, 15]. It has been firmly established that DMAE-CB, like other acrylic and methacrylic monomers, after its intake by cells, causes a depletion of the intracellular antioxidant glutathione (GSH) and over-production of reactive oxygen species (ROS) [16-18]. There exists a highly sophisticated anti-oxidative system consisting of non-enzymatic and enzymatic elements to maintain a balanced intracellular redox homeostasis. GSH is the key component of the anti-oxidative defense system, and the activities of many enzymatic elements depend on the availability of GSH [19]. Based on the understanding of the involvement of oxidative stress in dental monomer-related cytotoxicity, antioxidants, such as N-acetyl cysteine (NAC), have been proved effective to protect cells from DMAE-CB-induced cell damage [12].
Genotoxicity or mutagenicity has been reported in various types of cells exposed to methacrylic-based monomers such as 2-hydroxy ethyl methacrylic (HEMA) and triethylene glycol dimethacrylic (TEGDMA) [20-27]. The overproduced ROS beyond the capacities of anti-oxidative mechanisms can react with cellular macromolecules, such as lipids, proteins, and DNA [17, 28]. The interactions of ROS with DNA may result in DNA lesions and block progression of replication, which causes the formation of DNA double-strand breaks (DSBs) in the chromosome and triggers related signal transduction pathways that establish cell-cycle arrest and the induction of programmed cell death [20]. Based on the facts that DMAE-CB induced oxidative stress similar to conventional monomers, we hypothesized that DNA damage and subsequent cell cycle arrest as well as apoptosis might be induced after DAME-CB treatment as a result of ROS over-production [29].
To test our hypothesis, first, we investigated the levels of ROS and GSH and activities of anti-oxidative enzymes after DMAE-CB treatment. Then, DMAE-CB-induced DNA damage was examined by checking the formation of DSBs, the disturbed cell cycle, and the altered expression of several related genes. Finally, since DMAE-CB is known to induce intrinsic mitochondrial apoptosis [11-13], we further investigated whether DMAE-CB impacted the morphologies and functions of mitochondria. To investigate these mechanisms, human dental pulp cells (hDPCs) from primary cultures were used as model cells, which previously have been demonstrated to be sensitive to DMAE-CB-induced cytotoxicity [12]. The role of oxidative stress in the current investigation was examined by employing substances modifying GSH synthesis [30].
Materials and Methods
Cell cultures
Human dental pulp cells (hDPCs) were obtained from dental pulps of clinically healthy teeth from 18-25 year old patients who had their noncarious third molars extracted. The procedure was reviewed and approved by the Ethics Committee of the Fourth Military Medical University. After removal of the dental pulp tissues from the tooth, hDPCs were isolated and expanded as described in previous studies [31]. Experiments for this study were performed with hDPCs between passages two to five.
Biomarkers of oxidative stress
Intracellular ROS analysis
The cells (1×105 cells/well) were cultivated in 6-well plates at 37 °C for 24 h. Next, cell cultures were subsequently preincubated with either 50 μM buthionine sulfoximine (BSO) or 5 mM 2-oxo-4-thiazolidine-carboxylic acid (OTC) for 20 h prior to exposure to DMAE-CB as described before [30]. Then, the cells were treated with DMAE-CB (0-0.01-0.05 mM) in the presence or absence of BSO or OTC at 37 °C for 6 h. Subsequently, a commercial kit (Beyotime Biotechnology, China) was used to measure the level of intracellular ROS according to the manufacturer's recommendations. For each sample, 10,000 cells were analyzed.
Determination of glutathione (GSH) content
The cells were cultivated in 6-well plates at 37 °C for 24 h. Next, cell cultures were subsequently preincubated with either 50 μM BSO or 5 mM OTC for 20 h prior to exposure to DMAE-CB. Then, the cells were treated with DMAE-CB (0-0.01-0.05 mM) in the presence or absence of BSO or OTC at 37 °C for 24 h. Subsequently, intracellular levels of GSH were measured using a commercially available kit (Nanjing Jiancheng Bioengineering Institute, China) according to the manufacturer's recommendations.
Antioxidant enzyme activity assays
Briefly, the cells were seeded into a 6-well plate for 24 h at 37°C. Next, cells were preincubated with 50 μM BSO or 5 mM OTC for 20 h, and then exposed to 0.01 and 0.05 mM DMAE-CB in the absence or presence of BSO or OTC for 24 h as described above. The activities of glutathione peroxidase (GPx1/2), superoxide dismutase (SOD), and catalase (CAT) were all determined by using commercial assay kits (Beyotime Biotechnology, China) as described by our previous study [32].
Determination of apoptosis
DMAE-CB-induced cell apoptosis was assessed with an Annexin V-FITC apoptosis detection kit (Beyotime Biotechnology, China). The cells were cultivated in 6-well plates at 37 °C for 24 h. Next, the cells were treated with DMAE-CB (0-0.01-0.05 mM) in the presence or absence of BSO or OTC at 37 °C for 24 h as described above. Subsequently, the cells were washed with PBS, gently resuspended in Annexin V binding buffer, and incubated with Annexin V-FITC/propidium iodide (PI). Flow cytometry analysis was performed (FAC Scan, Becton Dickenson, USA). For each sample, 10,000 cells were analyzed.
Analyses of DNA damage
Measurement of 8-Oxo-2'-deoxyguanosine (8-OHdG)
The cells (1×104 cells/well) were cultivated in 96-well plates at 37 °C for 24 h. Next, the cells were treated with DMAE-CB (0-0.01-0.05 mM) in the presence or absence of BSO or OTC at 37 °C for 24 h as described above. Subsequently, DNA samples were extracted as described before [33]. The measurements of 8-OHdG were carried out using the OxiSelect Oxidative DNA Damage ELISA Kit (Cell Biolabs, San Diego, USA) according to the manufacturer's protocol.
Quantification of DNA damage
DMAE-CB-induced DNA damage in hDPCs was assessed and quantified using a commercial DNA damage assay kit (Epigentek, Farmingdale, NY, USA) according to the manufacturer's recommendations. The assay enables the measurements of the phosphorylation of histone H2AX to form γ-H2AX, which is a chromatin modification during DNA damage.
γ-H2AX immunofluorescence
DNA-DSBs formation was examined using the OxiSelect DNA Double-Strand Break Staining Kit (Cell Biolabs, San Diego, United States). The cells (5×105 cells/well) were cultivated on glass-bottomed cell culture dishes (Nest Biotechnology, China) at 37 °C for 24 h. Next, cell cultures were subsequently preincubated with either 50 μM BSO or 5 mM OTC for 20 h prior to exposure to DMAE-CB. Then, the cells were treated with 0.01 mM DMAE-CB in the presence or absence of BSO or OTC at 37 °C for 24 h. For immunofluorescent staining, the cells were fixed with 3.7% paraformaldehyde in PBS for 10 min at room temperature, washed with cold PBS (4ºC) once, and incubated with ice-cold 90% methanol for 10 minutes at 4ºC. Then the cells were incubated with blocking buffer for 30 minutes at room temperature on an orbital shaker and washed with cold PBS once. Thereafter, cells were incubated with anti-phospho-Histone (γ-H2AX) Antibody Solution, and incubated for 1 hour. After washed 5 times with Wash Buffer (PBST), the cells were incubated with secondary antibody, Cy3 Conjugate Solution for 1 hour at room temperature. Cells were further incubated with 10 mg/ml Hoechest 33342 (Sigma) for 5 min. Specimens were analyzed by laser scanning confocal microscopy (Keyence Co., Osaka, Japan).
Cell cycle analysis
The cells were cultivated in 6-well plates at 37 °C for 24 h. Next, the cells were treated with DMAE-CB (0-0.01-0.05 mM) in the presence or absence of BSO or OTC at 37 °C for 24 h as described before. Cells were then harvested and treated with mixed solution of RNase and PI using a cell cycle and apoptosis analysis kit (Beyotime Biotechnology, China) according to the manufacturer's recommendations. For each sample 10,000 cells were analyzed.
Quantitative reverse transcription-polymerase chain reaction (RT-PCR)
The cells were cultivated in 6-well plates at 37 °C for 24 h. Next, the cells were treated with 0.01 mM DMAE-CB in the presence or absence of BSO or OTC at 37 °C for 24 h as described above. Quantitative RT-PCR was used to examine the expression of apoptotic and cell-cycle-related gene markers, including B-cell lymphoma-2 (BCL-2), Bcl2-associated X (BAX), CASPASE-3, Ataxia telangiectasia mutated (ATM), P21 and tumor protein p53 (TP53). Primer sequences for the aforementioned genes are listed in Table 1. Total RNA was extracted with Trizol Reagent (Invitrogen, Madison, WI, USA). The cDNA was synthesized by using the PrimeScript RT Perfect Real Time reaction kit (TaKaRa Biotechnology, Dalian, China). Quantitative RT-PCR was performed with the SYBREx Tag II premix (TaKaRa Biotechnology) in the Bio-Rad CFX96 TM Real-Time System. The housekeeping gene actin was used to normalize the expression level of related genes.
Intercellular ATP level determination
The cells were cultivated in 6-well plates at 37 °C for 24 h. Next, the cells were treated with DMAE-CB (0-0.01-0.05 mM) in the presence or absence of BSO or OTC at 37 °C for 24 h as described before. ATP level was determined using a firefly luciferase based ATP assay kit (Beyotime Biotechnology, China) according to the manufacturer's protocol.
Measurement of mitochondrial membrane potential (MMP)
The cells were cultivated in 6-well plates at 37 °C for 24 h. Next, the cells were treated with DMAE-CB (0-0.01-0.05 mM) in the presence or absence of BSO or OTC at 37 °C for 24 h as described above. Subsequently, disruption of mitochondrial membrane potential was analyzed using the fluorescent dye JC-1 (Beyotime Biotechnology, China) according to the manufacturer's protocol. For each sample 10,000 cells were analyzed.
Transmission Electron Microscopy (TEM)
The cells were cultivated in 6-well plates at 37 °C for 24 h. Next, the cells were treated with 0.01 mM DMAE-CB in the presence or absence of BSO or OTC at 37 °C for 24 h. Subsequently, the cells were fixed with 2.5% glutaraldehyde in 0.1 M cacodylate buffer for 2 h, and then post fixed with a solution of 1% osmium tetroxide in 0.1 M cacodylate buffer. Thereafter, after a graded-ethanol serial dehydration step, the cells were embedded in epoxy resins. The embedded cells were sectioned into ultrathin slices and stained by uranyl acetate solution and lead citrate. The samples were observed with a transmission electron microscope Tecnai G2 Spirit BioTWIN electron microscope (FEI Company, Eindhoven, The Netherlands).
Immunofluorescence (IF)
The cells were cultivated on glass-bottomed cell culture dishes (Nest Biotechnology, China) at 37 °C for 24 h. Next, cell cultures were subsequently preincubated with either 50 μM BSO or 5 mM OTC for 20 h prior to exposure to DMAE-CB. Then, the cells were treated with 0.01 mM DMAE-CB in the presence or absence of BSO or OTC at 37 °C for 24 h. The cells were then fixed with a modified Zamboni's fixative (4% paraformaldehyde and 0.19% picric acid in PBS, pH 7.4) for 30 min at room temperature. Cells were washed twice with PBS, preincubated in the same buffer containing 0.3% Triton X-100 (Amresco, USA), and blocked with 0.1% BSA (Amresco, USA) in PBS for 1 h. The cells were incubated with primary antibodies (rabbit anti-Bax, 1:200, Beyotime Biotechnology, China; and mouse anti-Cytochrome C, 1:50, Boster Biotechnology, China) overnight at 4°C. After three washes with PBS, cells were incubated with secondary antibodies (Alexa Fluor 594 donkey-rabbit, 1:400; and Alexa Fluor 488 donkey-mouse, 1:200; Life Technologies, Carlsbad, CA) for 30 min at 37°C. Cells were then washed in PBS and further incubated with 10 mg/ml DAPI (Sigma) at room temperature for 10 min. Fluorescent images were obtained by laser scanning confocal microscopy (Keyence Co., Osaka, Japan).
Primer sequences.
| Gene | Primer sequence | Product size (bp) |
|---|---|---|
| ATM | Forward: 5'-TGGTGCTATTTACGGAGCTG-3' | 20 |
| Reverse: 5′-TTCGAAAGTTGACAGCCAAA-3' | 20 | |
| P21 | Forward: 5′-GTTCCTTGTGGAGCCGGAGC-3′ | 20 |
| Reverse: 5′-GGTACAAGACAGTGACAGGTC-3′ | 21 | |
| TP53 | Forward: 5'-CGGAGGTCGTGAGACGCTG-3' | 19 |
| Reverse: 5'-CACATGTACTTGTAGTGGATGGTGG-3' | 25 | |
| BCL-2 | Forward: 5'-CCTGTGGATGACTGAGTACCTGAAC-3' | 25 |
| Reverse: 5'-CAGAGTCTTCAGAGACAGCCAGGA-3' | 24 | |
| BAX | Forward: 5'-CAGGATGCGTCCACCAAGAA-3' | 20 |
| Reverse: 5'-GCAAAGTAGAAGAGGGCAACCAC-3' | 23 | |
| CASPASE-3 | Forward: 5'-GGAGCAGCTTTGTGTGTGTGATTC-3' | 24 |
| Reverse: 5′-TCCATCCTTTGACTCTGCTCATGG-3′ | 24 | |
| ACTIN | Forward: 5'-AGGGTGTGATGGTGGGAA-3' | 18 |
| Reverse: 5′-CATCTGCTGGAAGGTGGA-3' | 18 |
Enzyme-linked immunosorbent assay (ELISA)
The cells were seeded into a 96-well plate at 37 °C for 24 h. Next, cell cultures were subsequently preincubated with either 50 μM BSO or 5 mM OTC for 20 h prior to exposure to DMAE-CB. Then, the cells were treated with 0.01 mM DMAE-CB in the presence or absence of BSO or OTC at 37 °C for 24 h and lysed in RIPA buffer. Subsequently, cells were resuspended in 1.5 mL of cold Mito-Cyto Buffer (Applygen Biotechnology, China). The supernatant was centrifuged at 12,000 g for 10 min in order to separate mitochondria (in pellet) from the cytoplasm (in supernatant). Cytosolic Cytochrome C (Cyto C) in the supernatant was quantified with an ELISA kit (Westang Biotechnology, China), using Cyto C (0-2000 pg/mL) as a standard. Data are expressed as the total amount of intracellular solubilized Cyto C. Cytosolic Cyto C levels were normalized to protein contents measured in a BCA Protein Assay Kit (Beyotime Biotechnology, China).
Statistical analysis
Results were plotted as the mean ± standard deviation (SD) and all experiments were repeated three times. Data was analyzed by one-way analysis of variance (ANOVA) with SPSS 18.0 software (SPSS Inc., Chicago, IL, USA). The statistical significance level was set at P=0.05 for all tests.
Results
DMAE-CB induced oxidative stress in human dental pulp cells
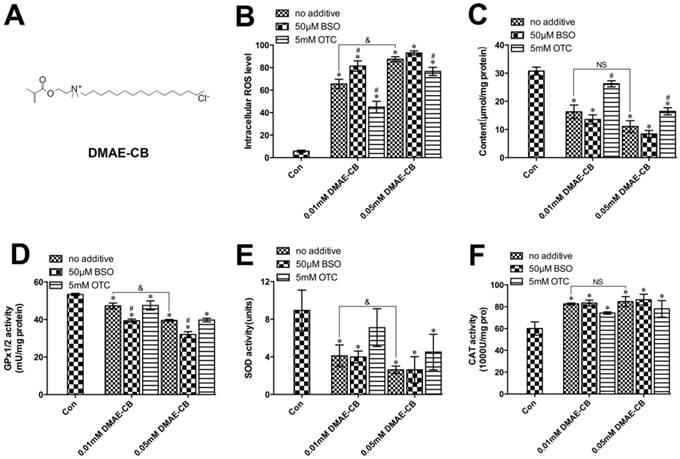
The formation of ROS in hDPCs after DMAE-CB treatment in the presence or absence of BSO or OTC was evaluated to identify differential effects of the GSH-modifying substances. As shown in Fig. 1B, a concentration-dependent increase in ROS level was observed after DMAE-CB treatment. Moreover, for cells treated with both DAME-CB and the GSH-synthesis inhibitor BSO, the formation of ROS was further enhanced. Although not completely inhibited, the amount of ROS remained at a low level for cells treated with DMAE-CB and OTC. As shown in Fig.1C, a decrease in GSH level was observed in cells exposed to DMAE-CB. The reduced GSH content was only slightly amplified by the presence of BSO. As expected, the presence of OTC replenished the depleted GSH pool after DMAE-CB treatment.
Next, we analyzed the adaptive and coordinated activities of various antioxidative enzymes. The activity of GPx1/2 was dose-dependently reduced in cell cultures exposed to DMAE-CB compared with control group (P < 0.05, Fig.1D), and lowered by BSO even more. However, an increase was observed in GPx1/2 activity by OTC in cell cultures exposed to DMAE-CB. A similar dose-dependent reduction in the SOD activity was observed after exposure to DMAE-CB (P < 0.05 as compared with untreated cultures, Fig.1E). The decreased SOD activity was only slightly enhanced by BSO. In contrast, OTC drastically increased the levels of SOD in cells exposed to DMAE-CB. The activity of CAT was significantly enhanced in cell cultures exposed to DMAE-CB, compared with the control group (P < 0.05, Fig.1F). In contrast, although the presence of BSO had only little effect on CAT activity, the activity of CAT was slightly reduced by OTC in cells exposed to DMAE-CB.
DMAE-CB induced oxidative stress in hDPCs. A. Chemical structure of DMAE-CB. B. Intracellular ROS levels in hDPCs exposed to DMAE-CB in the absence or presence of BSO or OTC for 6 h. C-F. Contents of GSH (C), activity of GPx1/2 (D), SOD (E), and CAT (F), in hDPCs exposed to DMAE-CB in the absence or presence of BSO or OTC for 24 h. Data represent the mean ± SD of three independent experiments (n = 3). *P < 0.05 vs. control group; #P < 0.05 vs. DMAE-CB-treated cells; &P < 0.05 between cells treated with 0.01 mM DMAE-CB and 0.05 mM DMAE-CB by one-way ANOVA. NS, no significance.
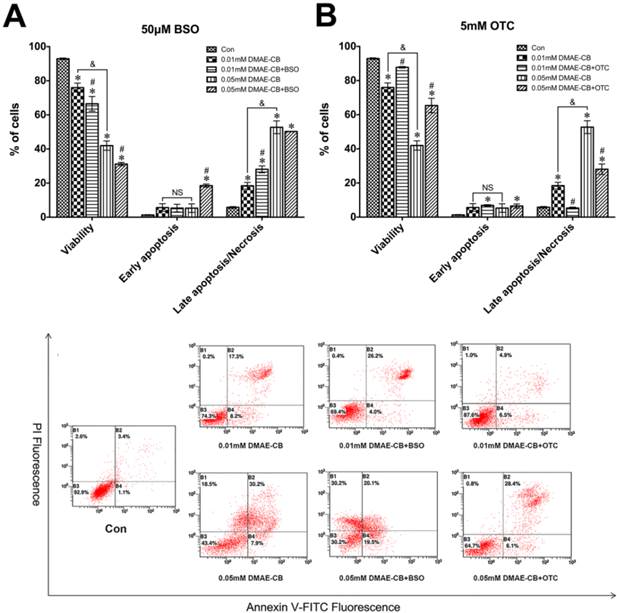
DMAE-CB induced apoptosis in human dental pulp cells
As shown in Fig. 2, cell viability was reduced to 76% (0.01 mM) or 42% (0.05 mM) by DMAE-CB compared with untreated cultures (93%), whereas the percentage of cells distributed in late apoptosis/necrosis has increased simultaneously. Moreover, the presence of BSO further induced an increase in the percentage of cells distributed in late apoptosis/necrosis, and meanwhile decreased the percentage of viable cells to 31% in cells exposed to 0.05 mM DMAE-CB (Fig. 2A). Quite in contrast, the presence of OTC in 0.05 mM DMAE-CB-treated cell cultures significantly raised the percentage of viable cells from 42% to more than 65%, and the percentage late apoptosis/necrosis of hDPCs was reduced from 53% to 28% in parallel (Fig. 2B).
DMAE-CB induced DNA damage in dental pulp cells
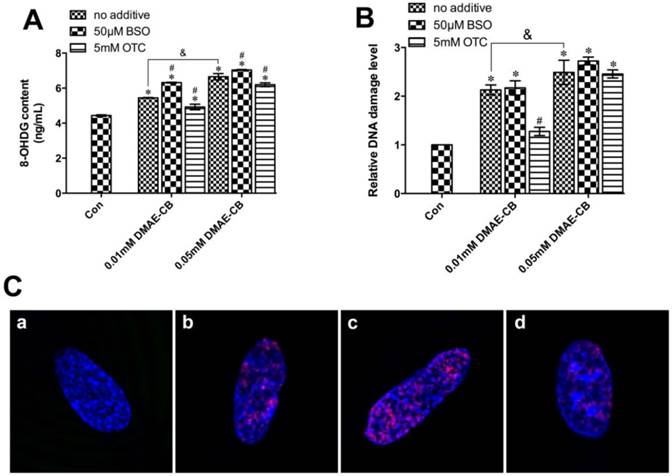
As shown in Fig. 3A, DMAE-CB significantly increased the intracellular 8-OHdG content in hDPCs cultures in a concentration-dependent manner after 24 h treatment. The generation of 8-OHdG by DMAE-CB was further enhanced by BSO, whereas reduced in the presence of OTC.
Next, we used the a commercial in situ DNA damage assay kit to assess and quantify whether DSBs were formed in hDPCs after 24 h treated with DMAE-CB in the absence or presence of BSO or OTC. It was shown that DMAE-CB dose-dependently enhanced H2AXser139 phosphorylation (P < 0.05, Fig. 3B). Such effect was further amplified by BSO. OTC significantly reduced H2AXser139 phosphorylation when the dose of DMAE-CB was 0.01 mM (P < 0.05), however, such protective effect was not observed when the dose of DMAE-CB was 0.05 mM. Similar results were revealed by immunofluorescence observation. It was found that the red fluorescence of γ-H2AX was readily discernible in hDPCs nuclei by immunofluorescence using γ-H2AX-specific antibodies (Fig. 3C). Microscopic enumeration of red fluorescence of γ-H2AX revealed that DMAE-CB treatment induced H2AXser139 phosphorylation, and BSO co-treatment further exaggerated this trend. The presence of OTC weakened the red fluorescence of γ-H2AX, indicating that OTC could partly restore DNA damage caused by DMAE-CB.
DMAE-CB induced apoptosis in hDPCs. A-C. Apoptosis of hDPCs after exposure to DMAE-CB in the absence or presence of BSO (A) or OTC (B) for 24 h, as assayed by Annexin V and PI double staining. *P < 0.05 vs. control group; #P < 0.05 vs. dental monomer-treated cells; &P < 0.05 between cells treated with 0.01 mM DMAE-CB and 0.05 mM DMAE-CB by one-way ANOVA. NS, no significance.
DMAE-CB induced DNA damage in dental pulp cells. A-B. 8-OHdG content (A) and the phosphorylation of H2AXser139 (γ-H2AX)(B) in hDPCs exposed to DMAE-CB in the absence or presence of BSO or OTC for 24 h. *P < 0.05 vs. control group; #P < 0.05 vs. DMAE-CB-treated cells; &P < 0.05 between cells treated with 0.01 mM DMAE-CB and 0.05 mM DMAE-CB by one-way ANOVA. C. Representative images of immunofluorescent staining for γ-H2AX in hDPCs exposed to DMAE-CB in the absence or presence of BSO or OTC for 24 h. Hochest 33342 (blue fluorescence) is a marker for DNA and stains the whole nucleus of a cell, while the red fluorescence indicates γ-H2AX-specific foci. (a) A nucleus of hDPCs without foci is typically seen in most of untreated cells. (b) A nucleus of hDPCs with typical γ-H2AX-specific foci can be observed in DMAE-CB-treated cells. (c) The presence of BSO enhanced the γ-H2AX-specific foci in the nucleus of hDPCs exposed to DMAE-CB. (d) OTC attenuated the red γ-H2AX-specific foci in the nucleus of hDPCs exposed to DMAE-CB.
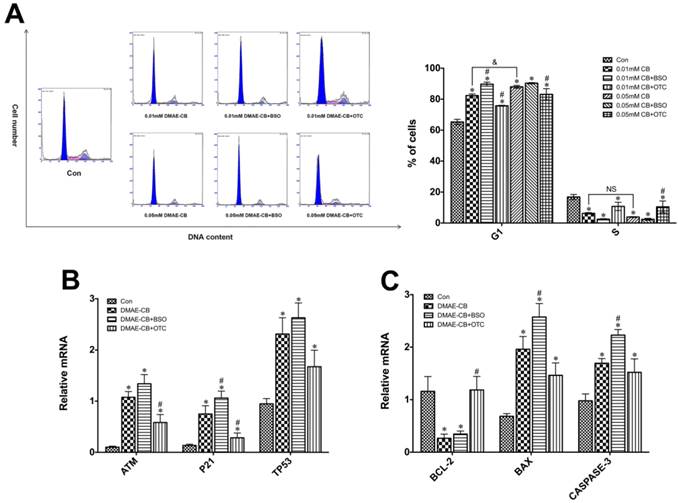
DMAE-CB induced cycle arrest in dental pulp cells
It was observed that DMAE-CB induced a concentration-related delay of the cell cycle in G1 phase (Fig.4A). The percentage of cells in G1 phase greatly increased from 65% in control cultures to 82% in cultures exposed to 0.01 mM and 88% treated with 0.05 mM DMAE-CB (P < 0.05 as compared with untreated cells). BSO co-treatment further increased the percentage of cells distributed in G1 phase to 89.7% after 0.01 mM DMAE-CB treatment. However, for 0.05 mM DMAE-CB, BSO showed no significant effects on the percent of cells residing in G1 phase. OTC partially reversed the DMAE-CB induced G1 arrestment. The percentage of cells residing in S phase was significantly reduced after DMAE-CB treatment (P < 0.05), whereas OTC restored the effects to some extent. The percentage of cells found in G2 phase under the current conditions followed the changes found in G1 and S phase (not shown).
Effect of DMAE-CB on expression of cell-cycle-related genes
As shown in Fig.4B, the hDPCs exposed to DMAE-CB exhibited significantly enhanced expression levels of the cell-cycle-related genes (ATM, P21 and TP53) compared with untreated cells (P < 0.05). BSO further amplified DMAE-CB-induced up-regulation of ATM, P21 and TP53 expression, while OTC showed the opposite effects.
Effect of DMAE-CB on expression of pro- and anti-apoptotic genes
According to our previous studies [11, 12], the cytotoxicity of DMAE-CB was related to the intrinsic mitochondrial apoptotic pathway. Thus, we used qRT-PCR to analyze the expression levels of pro- and anti-apoptotic proteins of the Bcl-2 family, as well as caspase-3 (Fig. 4C). DMAE-CB reduced the expression of BCL-2, regardless of the absence or presence of BSO compared with control group (P < 0.05). On the other hand, DMAE-CB induced higher expression of both BAX and CASPASE-3 as compared with control group (P < 0.05). This trend was further enhanced in the presence of BSO, and partially reversed in the presence of OTC.
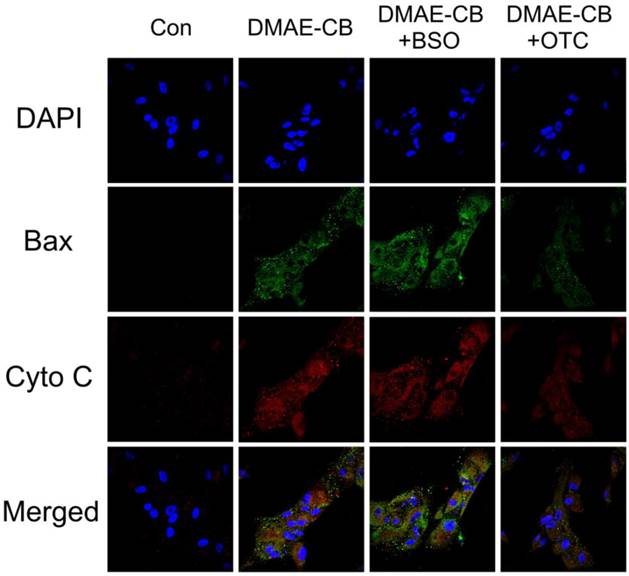
Immunofluorescence (IF) analysis revealed that in untreated cells Bax was nearly undetectable and Cyto C was retained in the mitochondria (Fig. 5). The green fluorescence of Bax was obvious after treatment with DMAE-CB, regardless of the absence or presence of BSO, and the dot-like staining on the mitochondria was enhanced. Meanwhile, the red fluorescence of Cyto C was diffused in the cytoplasm. Quite the reverse, the presence of OTC weakened the green fluorescence of Bax, which was diffused in the cytoplasm. And for red fluorescence of Cyto C, except for weak signal diffused in the cytoplasm, most of the strong spot signal was retained in the mitochondria.
DMAE-CB impacts the morphologies and functions of mitochondria
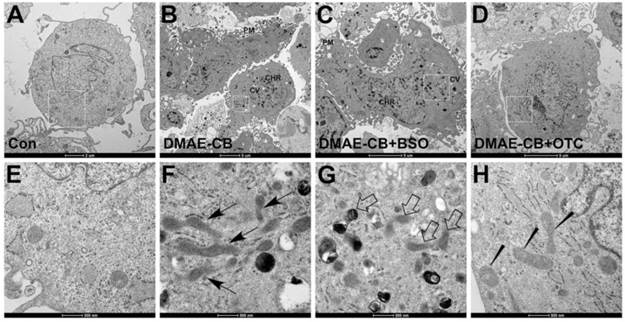
We further examined mitochondria morphology in cells exposed to DMAE-CB, in the absence or presence of BSO or OTC, by using TEM. It was observed that control cells showed the normal morphology with healthy mitochondria (Fig. 6AE). After DMAE-CB treatment, nuclear membrane condensation, cytoplasmic vacuolization, increased lysosome organelles and severe plasma membrane blebling could be observed, which suggested apoptosis might occur [34]. In addition, some mitochondria became larger and elongated with a reduced number of cristae and deformed cristae (Fig. 6BF). The presence of BSO even enhanced the destructive effects of DMAE-CB on mitochondria. Some mitochondria became swollen and showed disorganized cristae and altered membrane structural integrity (Fig. 6CG). Quite in contrast, OTC restored the cells exposed to DMAE-CB. Nearly no cytoplasmic vacuolization and lysosome organelles were observed, and mitochondria with relatively normal appearance were detected (Fig. 6DH).
DMAE-CB induced cell cycle arrest, cell-cycle-related gene and pro- and anti-apoptotic gene expression in dental pulp cells. A. Cell cycle analyses of hDPCs after exposure to DMAE-CB in the absence or presence of BSO or OTC for 24 h. The percentages of cells found in G2 phases (not shown) varied according to the changes observed with the number of cells in G1 and S. B-C. Gene expression of ATM, P21, TP53(B) BCL-2, BAX and CASPASE-3(C) in hDPCs exposed to DMAE-CB in the absence or presence of BSO or OTC for 24 h. *P < 0.05 vs. control group; #P < 0.05 vs. dental monomer-treated cells; &P < 0.05 between cells treated with 0.01 mM DMAE-CB and 0.05 mM DMAE-CB by one-way ANOVA. NS, no significance.
Double Immunofluorescence staining for Bax and Cyto C in dental pulp cells exposed to DMAE-CB. Green fluorescence represents Bax, whereas red green fluorescence represents Cyto C. Yellow color in the overlay of these two images indicates co-localization of Bax and Cyto C (presumably in mitochondria). In control group, Bax was nearly undetectable and Cyto C was retained in the mitochondria. However, upon DMAE-CB regardless of in the absence or presence of BSO, Bax was obvious and dot-like staining on the mitochondria. Meanwhile, Cyto C was diffused in the cytoplasm. The presence of OTC weakened Bax, which was diffused in the cytoplasm. And Cyto C was weaker and retained in the mitochondria.
Changes of mitochondrial morphology observed by transmission electron microscopic (TEM). (A, E) Representative images of untreated control cells. Healthy cell morphology and preserved mitochondria in the cytoplasm were observed. (B, F) Representative images of DMAE-CB-treated cells. Apoptosis signs such as plasma membrane (PM) blebing, condensed/peripheralized nuclear chromatin (CHR) condensation, and cytoplasmic vacuolization (CV) were observed. Some mitochondria were larger and elongated with reduced number of cristae and deformed cristae (black arrow). (C, G) Some mitochondria became swollen with disorganized cristae with altered membrane structural integrity (disorganized inner and outer membranes) in the presence of BSO in cells exposed to DMAE-CB (white arrow). (D, H) The presence of OTC restored cell and mitochondrial morphology, although pycnosis, swelling and less number of cristae or deformed cristae were observed in some mitochondria (wedged).
DMAE-CB impaired the functions of mitochondria in dental pulp cells. A-C. ATP level (A), MMP (B) and levels of cytosolic Cyto C (C) of hDPCs after exposure to DMAE-CB in the absence or presence of BSO or OTC for 24 h, as assayed by Annexin V and PI double staining. *P < 0.05 vs. control group; #P < 0.05 vs. dental monomer-treated cells; &P < 0.05 between cells treated with 0.01 mM DMAE-CB and 0.05 mM DMAE-CB by one-way ANOVA.
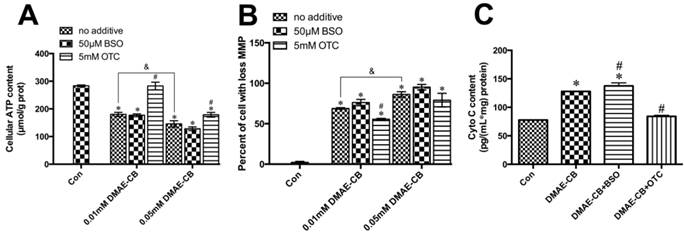
The ATP levels of hDPCs exposed to DMAE-CB, in the absence or presence of BSO or OTC, were demonstrated in Fig. 7A. After 24 h of DMAE-CB treatment, ATP level was significantly lower than that in control cultures both in 0.01 mM or 0.05 mM DMAE-CB (P < 0.05). The presence of BSO further decreased ATP levels to some extent. When the concentration of DMAE-CB was 0.01 mM, OTC restored cellular ATP level to almost normal level (p>0.05). For higher dose of DMAE-CB (0.05 mM), OTC still exhibited protective effects and partly recovered the ATP levels. Next, we analyzed the effects of DMAE-CB on the MMP, which is an important biomarker of intrinsic mitochondrial pathway [35]. DMAE-CB induced a dose-dependent MMP depolarization after a 24 h exposure period as compared with untreated cells (P < 0.05, Fig.7B). Disruption of MMP by DMAE-CB was slightly amplified in the presence of BSO, though the effects were not statistically significant. The presence of OTC partially reversed DMAE-CB-induced disruption of MMP. Then, we measured the cytosolic Cyto C level using an ELISA kit (Fig.7C). The cytosolic Cyto C level was greatly elevated after cells were exposed to DMAE-CB for 24 h, compared with control group (P < 0.05). These increases were further enhanced by BSO, whereas completely blocked by OTC (P > 0.05 as compared with untreated cells).
Discussion
The recent findings strongly suggest that antibacterial monomers, such as DMAE-CB induce adverse biological effects in vitro in various cell types [11, 12, 14, 15, 36]. However, the underlying mechanism is still not clear. Based on our previous findings that DMAE-CB could induce oxidative stress in vitro, we hypothesized that DNA damage, cell cycle arrest and apoptosis might subsequently occur as a consequence of ROS over-production. Furthermore, GSH synthesis-modulating substances, such as GSH synthesis inhibitor BSO and GSH synthesis promoter OTC, were used to investigate whether GSH played a key role in the DMAE-CB-induced cytotoxicity.
Oxidative stress induced by increasing concentrations of DMAE-CB was indicated here by the overproduction of ROS, depletion of GSH, and the differential activities of anti-oxidative elements. In accordance with previous studies [12], DMAE-CB could dose-dependently increase ROS formation, whereas decrease GSH level in parallel. Functionally, GSH is the most important redox-regulating non-enzymatic thiol in the anti-oxidative defense system. GSH can directly scavenge ROS or act as a substrate of GPx1/2, which catalyzes the reduction of H2O2 [37]. The GSH synthesis-modulating substances such as BSO and OTC, could alter the GSH level as well as ROS formation here, in cells exposed to DMAE-CB. The enzymatic antioxidative defense system controls the intracellular oxidative state by metabolizing and scavenging diverse ROS, including superoxide anion (O2-), hydroxyl radical (OH-), and hydrogen peroxide (H2O2), aiming at a balanced redox system [38, 39]. Among them, SOD, GPx1/2 and CAT are the key enzymatic antioxidants of this defense system by which help remove the free radicals. SOD catalyzes the breakdown of O2- into O2 and H2O2. GPx1/2 and CAT then decompose H2O2 into H2O and O2 [40-42]. In our study, it was found that DMAE-CB reduced SOD and GPx1/2 activities while at the same time increased CAT activity. Based on recent publication, H2O2 is the major sort of ROS which is over-produced after methacrylic monomer treatment [20]. Because GPx1/2 requires GSH as a substrate to reduce H2O2, the limited source of the substrate GSH as a consequence of DMAE-induced GSH depletion can subsequently restrict the activity of GPx1/2, causing the observed reduction in GPx1/2 activity [19]. The elevated H2O2 formation may lead to the increased CAT activity and decreased SOD activity due to a feedback mechanism [16].
By using BSO and OTC, the key role of GSH in DMAE-CB-induced cytotoxicity was identified. The combination of DMAE-CB with GSH synthesis inhibitor BSO decreased GPx1/2 activity even further. In contrast to BSO, the GSH synthesis supporter OTC partially reversed DMAE-CB-induced differential activation of the investigated anti-oxidative enzymes. These observations showed that the depletion of GSH constitutes a key event during the generation of oxidative stress induced by DMAE-CB. These findings are in accordance with previous reports about conventional methyacrylate monomers [43-45]. Notably, by using GSH synthesis-modulating substances, we also present experimental evidence that the availability of GSH is causally related to DMAE-CB-induced apoptosis. BSO considerably increased the percentage of cells distributed in late apoptosis/necrosis On the other hand, OTC was observed to partially prevent DMAE-CB-induced apoptosis. These data indicated that, similar to other methacrylic monomers [46-51], depletion of GSH may be the origin of oxidative stress and subsequent apoptosis after the treatment of DMAE-CB.
In our previous studies, it has been found that DMAE-CB induced apoptosis through the intrinsic apoptotic pathway [11-13]. Nevertheless, the origin upstream of this signaling pathway still needs to be identified. The formation of oxidative stress beyond the capacities of antioxidant mechanisms can cause oxidative damage to DNA, which may activate DNA repair process, or trigger cellular signaling resulting in apoptotic cell death. Thus, we hypothesized that oxidative DNA damage might be the initial reason for DMAE-CB induced apoptosis. To test our hypothesis, a series of oxidative DNA damage biomarkers was investigated in the present research work. It was found that 8-OHdG, a well-known biomarker of oxidative DNA damage [52, 53], was dose-dependently increased in hDPCs exposed to DMAE-CB. 8-OHdG is a modified base that occurs in DNA, when DNA is attacked by hydroxyl radicals, singlet oxygen or one-electron oxidants in cellular DNA [52, 53]. On the other hand, severe DNA damage might result in the formation of DSBs, structural damage to the DNA molecules, DNA protein crosslinks, and harmful mutations in the genomes [54]. There is increasing evidence that several diseases in humans, including Alzheimer's disease, cancer and rheumatoid arthritis are associated with free radical reactions with DNA [55]. Here, the quantative evaluation of γ-H2AX and immunofluorescent staining revealed that DMAE-CB could induce DSBs. DSBs are particularly hazardous to the cell due to their induction of genome rearrangements [56]. In addition, the formation of DSBs is considered as a activating signal which lead to cell cycle arrest to repair severe DNA damage [57]. Thus, the analysis of the cell cycle was performed. It was found that DMAE-CB greatly increased the percentage of cells distributed in G1 phase and meanwhile reduced the percentage of cells residing in S phase. The combined use of GSH synthesis inhibitor BSO further enhanced DMAE-CB-induced 8-OHdG and DSBs formation, and cell cycle arrest. On the other hand, the GSH synthesis supporter OTC partially reversed DAME-CB-induced DNA damage and cell cycle arrest. These data indicate that DMAE-CB induced oxidative DNA damage and subsequent cell cycle arrest is related to the availability of GSH, at least, to some extent, under the current culture conditions. These observations were also consistent with previous publication which demonstrated that the supplement of GSH could attenuate methacrylic monomer-induced genotoxicity. Lottner et al. found that the addition of NAC could reduce the DSBs induced by methacrylic monomers [25]. Likewise, Schweikl et al. found that the presence of NAC could reduce the formation of micronuclei caused by TEGDMA and HEMA [28].
Next, the signaling pathway involved in DMAE-CB induced apoptosis was investigated. Ataxia telangiectasia mutated (ATM), a member of the phosphoinositide 3-kinase-like family of serine/threonine protein kinases, can be recruited and activated by DNA double-strand breaks [58]. Upon DNA damage, especially the formation of DSBs, the activation of ATM by autophosphorylation on Ser1981 occurs, and it coordinates cycle with DNA repair or apoptosis by the activation of its downstream targets such as p53 [59]. The tumor suppressor protein p53 acts as a crucial regulator of cell cycle checkpoints, DNA repair and apoptosis [60]. The tumor suppressor protein p21 functions as an inhibitor of cell cycle progression at G1 phase. During oxidative stress stimuli, the expression of P21 gene is tightly controlled by p53. Via such a mechanism, protein p53 mediates the p53-dependent cell cycle G1 phase arrest [61]. Thus, we hypothesized ATM signaling pathway might be involved in DMAE-CB-induced DNA damage. Here, we found that the expression of ATM, P21 and TP53 was significantly increased in cell cultures after treatment with DMAE-CB. The results on gene expression presented here suggest a central role of the ATM signaling pathway in DMAE-CB-induced apoptosis. Furthermore, the causal relationship between the generation of oxidative stress induced by DMAE-CB and the activation of ATM became even more apparent, since the expressions of ATM, P21 and TP53 were further modified by GSH-synthesis inhibitor BSO or activator OTC. In summary, DMAE-CB caused the formation of DSBs, and thus resulted in the activation of ATM as well as the activation of its downstream targets such as p53. The activation of tumor suppressor protein p53 upregulated P21 expression and mediated the p53-dependent cell cycle arrest for DNA repair.
In response to DNA damage, p53 can initiate apoptosis through the intrinsic apoptosis pathway [62, 63]. Thereafter, Bcl-2 family proteins change their usual localization as well as targeting patterns, trigger mitochondrial dysfunction, Cyto C release and caspase activation, and eventually lead to cell death via apoptosis [64]. Our qRT-PCR analyses showed a decrease in BCL-2 expression, as well as increases in both BAX and CASPASE-3 expression after 24 h treatment of DMAE-CB. Indeed, our immunofluorescence analysis revealed that after DMAE-CB treatment, Cyto C was released from the mitochondrial membrane into the cytoplasm, while Bax translocated from cytoplasm to mitochondria.
Functionally, mitochondria also act as sensors and amplifiers in the execution of cell programmed death [65]. Here, we found that DMAE-CB had very destructive effects on mitochondria by TEM observation. Pycnosis, swelling and less number of cristae as well as deformed cristae were observed. Damage and subsequent dysfunction in mitochondria may influence cell metabolism. It was found that the changes in mitochondrial morphology were accompanied with a decreased level of ATP. Furthermore, disruption of the mitochondrial membrane integrity inevitably resulted in depolarization of MMP and subsequent Cyto C release from mitochondrial membrane into the cytoplasm. We also found that the presence of BSO further enhanced the destructive effects of DMAE-CB, while OTC partially restored the morphology and function of mitochondria. These findings indicated that DMAE-CB-caused damage on mitochondria was correlated the formation of oxidative stress in DMAE-CB-treated cells.
Conclusions
With the limitations of the present in vitro study, it might be concluded that DMAE-CB induced oxidative stress, and thus triggered DNA damage and apoptosis by the modification of signal transduction pathways. DMAE-CB caused disturbance of intracellular redox balance, characterized by depletion of GSH, over-production of ROS, and differential changes of anti-oxidative enzymes. The formation of oxidative stress beyond the capacities of antioxidant mechanisms could cause oxidative DNA damage, induce cell cycle arrest and thus trigger the activation of ATM signaling, the intrinsic apoptosis pathway. By using substances that can modulate GSH synthesis, we found that GSH played a key role in cell reaction in response to DMAE-CB.
Abbreviations
ATM: ataxia telangiectasia-mutated gene; BAX: Bcl2-associated X; BCL-2: B-cell lymphoma-2; BSO: buthionine sulfoximine; CAT: catalase; Cyto C: Cytochrome C; DMAE-CB: Methacryloxylethyl cetyl ammonium chloride; DSBs: DNA double-strand breaks; ELISA: enzyme-linked immunosorbent assay; hDPCs: human dental pulp cells; H2O2: hydrogen peroxide; GPx1/2: glutathione peroxidase; GSH: glutathione; HEMA: 2-hydroxy ethyl methacrylate; H2O2: hydrogen peroxide; IF: Immunofluorescence; MMP: mitochondrial membrane potential; NAC: N-acetyl cysteine; OH-: hydroxyl radical; OTC: 2-oxo-4-thiazolidine-carboxylic acid; O2-: superoxide anion; PI: Propidium Iodide; RT-PCR: reverse transcription-polymerase chain reaction; ROS: reactive oxygen species; SD: standard deviation; SOD: superoxide dismutase; TEM: transmission electron microscopy; TEGDMA: triethylene glycol dimethacrylate; TP53: tumor protein p53; 8-OHdG: 8-Oxo-2'-deoxyguanosine.
Acknowledgements
This study was financially supported by grant 2015JQ8301 (principal investigator Sai Ma) from the Natural Science Foundation of Shaanxi Province, grant 81130078 (principal investigator Jihua Chen), grant 81300927 (principal investigator Sai Ma), and grant 81272072 (principal investigator Lequn Shan) from the National Nature Science Foundation of China, and Program No. IRT13051 from the Program for Changjiang Scholars and Innovative Research Team in University (PCSIRT). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The authors also thanked the editors and anonymous reviewers for their comments and critique.
Conflicts of interest
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
References
1. Bagramian RA, Garcia-Godoy F, Volpe AR. The global increase in dental caries. A pending public health crisis. Am J Dent. 2009;22:3-8
2. Center ADAS. The 1999 survey of dental services rendered: American Dental Association; 2002.
3. Frost PM. An audit on the placement and replacement of restorations in a general dental practice. Prim Dent Care. 2002;9:31-6
4. Jokstad A, Bayne S, Blunck U, Tyas M, Wilson N. Quality of dental restorations. FDI Commission Project 2-95. Int Dent J. 2001;51:117-58
5. Totiam P, Gonzalez-Cabezas C, Fontana MR, Zero DT. A new in vitro model to study the relationship of gap size and secondary caries. Caries Res. 2007;41:467-73
6. Ma S, Niu L, Li F, Fang M, Zhang L, Tay FR. et al. Adhesive Materials with Bioprotective/Biopromoting Functions. Oral Health Rep. 2014;1:213-21
7. Imazato S, Ma S, Chen JH, Xu HH. Therapeutic polymers for dental adhesives: loading resins with bio-active components. Dent Mater. 2014;30:97-104
8. Xiao YH, Ma S, Chen JH, Chai ZG, Li F, Wang YJ. Antibacterial activity and bonding ability of an adhesive incorporating an antibacterial monomer DMAE-CB. J Biomed Mater Res B Appl Biomater. 2009;90:813-7
9. Li F, Li F, Wu D, Ma S, Gao J, Li Y. et al. The effect of an antibacterial monomer on the antibacterial activity and mechanical properties of a pit-and-fissure sealant. J Am Dent Assoc. 2011;142:184-93
10. Kitagawa R, Kitagawa H, Izutani N, Hirose N, Hayashi M, Imazato S. Development of an antibacterial root canal filling system containing MDPB. J Dent Res. 2014;93:1277-82
11. Ma S, Shan L, Xiao Y, Li F, Huang L, Shen L. et al. The cytotoxicity of methacryloxylethyl cetyl ammonium chloride, a cationic antibacterial monomer, is related to oxidative stress and the intrinsic mitochondrial apoptotic pathway. Braz J Med Biol Res. 2011;44:1125-33
12. Jiao Y, Ma S, Li J, Shan L, Wang Y, Tian M. et al. N-Acetyl Cysteine (NAC)-Directed Detoxification of Methacryloxylethyl Cetyl Ammonium Chloride (DMAE-CB). PLoS One. 2015;10:e0135815
13. Jiao Y, Ma S, Wang Y, Li J, Shan L, Chen J. Epigallocatechin-3-Gallate Reduces Cytotoxic Effects Caused by Dental Monomers: A Hypothesis. Med Sci Monit. 2015;21:3197-202
14. Ma S, Imazato S, Takahashi Y, Kiba W, Takeda K, Izutani N. et al. Mechanism of detoxification of the cationic antibacterial monomer 12-methacryloyloxydodecylpyridiniumbromide (MDPB) by N-acetyl cysteine. Dent Mater. 2013;29:1219-27
15. Nishida M, Imazato S, Takahashi Y, Ebisu S, Ishimoto T, Nakano T. et al. The influence of the antibacterial monomer 12-methacryloyloxydodecylpyridinium bromide on the proliferation, differentiation and mineralization of odontoblast-like cells. Biomaterials. 2010;31:1518-32
16. Krifka S, Spagnuolo G, Schmalz G, Schweikl H. A review of adaptive mechanisms in cell responses towards oxidative stress caused by dental resin monomers. Biomaterials. 2013;34:4555-63
17. Schweikl H, Spagnuolo G, Schmalz G. Genetic and cellular toxicology of dental resin monomers. J Dent Res. 2006;85:870-7
18. Jiao Y, Ma S, Wang Y, Li J, Shan L, Liu Q. et al. N-Acetyl Cysteine Depletes Reactive Oxygen Species and Prevents Dental Monomer-Induced Intrinsic Mitochondrial Apoptosis In Vitro in Human Dental Pulp Cells. PLoS ONE. 2016;11:e0147858
19. Fridovich I. Superoxide radical and superoxide dismutases. Annu Rev Biochem. 1995;64:97-112
20. Schweikl H, Petzel C, Bolay C, Hiller KA, Buchalla W, Krifka S. 2-Hydroxyethyl methacrylate-induced apoptosis through the ATM- and p53-dependent intrinsic mitochondrial pathway. Biomaterials. 2014;35:2890-904
21. Eckhardt A, Gerstmayr N, Hiller KA, Bolay C, Waha C, Spagnuolo G. et al. TEGDMA-induced oxidative DNA damage and activation of ATM and MAP kinases. Biomaterials. 2009;30:2006-14
22. Styllou M, Reichl FX, Styllou P, Urcan E, Rothmund L, Hickel R. et al. Dental composite components induce DNA-damage and altered nuclear morphology in gingiva fibroblasts. Dent Mater. 2015;31:1335-44
23. Shehata M, Durner J, Eldenez A, Van Landuyt K, Styllou P, Rothmund L. et al. Cytotoxicity and induction of DNA double-strand breaks by components leached from dental composites in primary human gingival fibroblasts. Dent Mater. 2013;29:971-9
24. Krifka S, Spagnuolo G, Schmalz G, Schweikl H. A review of adaptive mechanisms in cell responses towards oxidative stress caused by dental resin monomers. Biomaterials. 2013;34:4555-63
25. Lottner S, Shehata M, Hickel R, Reichl FX, Durner J. Effects of antioxidants on DNA-double strand breaks in human gingival fibroblasts exposed to methacrylate based monomers. Dent Mater. 2013;29:991-8
26. Styllou M, Reichl FX, Styllou P, Urcan E, Rothmund L, Hickel R. et al. Dental composite components induce DNA-damage and altered nuclear morphology in gingiva fibroblasts. Dent Mater. 2015;31:1335-44
27. Shehata M, Durner J, Eldenez A, Van Landuyt K, Styllou P, Rothmund L. et al. Cytotoxicity and induction of DNA double-strand breaks by components leached from dental composites in primary human gingival fibroblasts. Dent Mater. 2013;29:971-9
28. Schweikl H, Hartmann A, Hiller KA, Spagnuolo G, Bolay C, Brockhoff G. et al. Inhibition of TEGDMA and HEMA-induced genotoxicity and cell cycle arrest by N-acetylcysteine. Dent Mater. 2007;23:688-95
29. Matsuzawa A, Ichijo H. Stress-responsive protein kinases in redox-regulated apoptosis signaling. Antioxid Redox Signal. 2005;7:472-81
30. Krifka S, Hiller KA, Spagnuolo G, Jewett A, Schmalz G, Schweikl H. The influence of glutathione on redox regulation by antioxidant proteins and apoptosis in macrophages exposed to 2-hydroxyethyl methacrylate (HEMA). Biomaterials. 2012;33:5177-86
31. Jiao Y, Ma S, Li J, Shan L, Yang Y, Li M. et al. The influences of N-acetyl cysteine (NAC) on the cytotoxicity and mechanical properties of Poly-methylmethacrylate (PMMA)-based dental resin. PeerJ. 2015;3:e868
32. Jiao Y, Ma S, Wang Y, Li J, Shan L, Liu Q. et al. N-Acetyl Cysteine Depletes Reactive Oxygen Species and Prevents Dental Monomer-Induced Intrinsic Mitochondrial Apoptosis In Vitro in Human Dental Pulp Cells. PLoS One. 2016;11:e0147858
33. de Souza-Pinto NC, Eide L, Hogue BA, Thybo T, Stevnsner T, Seeberg E. et al. Repair of 8-oxodeoxyguanosine lesions in mitochondrial dna depends on the oxoguanine dna glycosylase (OGG1) gene and 8-oxoguanine accumulates in the mitochondrial dna of OGG1-defective mice. Cancer Res. 2001;61:5378-81
34. Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35:495-516
35. Song X, Zhang J, Wang M, Liu W, Gu X, Lv C. Astaxanthin induces mitochondria-mediated apoptosis in rat hepatocellular carcinoma CBRH-7919 cells. Biol Pharm Bull. 2011;34:839-44
36. Sokolova AS, Yarovaya CO, Shernyukov CA, Pokrovsky CE, Pokrovsky CA, Lavrinenko VA. et al. New quaternary ammonium camphor derivatives and their antiviral activity, genotoxic effects and cytotoxicity. Bioorg Med Chem. 2013;21:6690-8
37. Imlay JA. Cellular defenses against superoxide and hydrogen peroxide. Annu Rev Biochem. 2008;77:755-76
38. Jeeva JS, Sunitha J, Ananthalakshmi R, Rajkumari S, Ramesh M, Krishnan R. Enzymatic antioxidants and its role in oral diseases. J Pharm Bioallied Sci. 2015;7:S331-3
39. Mates JM, Perez-Gomez C, Nunez de Castro I. Antioxidant enzymes and human diseases. Clin Biochem. 1999;32:595-603
40. Pisoschi AM, Pop A. The role of antioxidants in the chemistry of oxidative stress: A review. Eur J Med Chem. 2015;97:55-74
41. Kermanizadeh A, Chauche C, Brown DM, Loft S, Moller P. The role of intracellular redox imbalance in nanomaterial induced cellular damage and genotoxicity: a review. Environ Mol Mutagen. 2015;56:111-24
42. Miller AF. Superoxide dismutases: ancient enzymes and new insights. FEBS Lett. 2012;586:585-95
43. Walther UI, Siagian II, Walther SC, Reichl FX, Hickel R. Antioxidative vitamins decrease cytotoxicity of HEMA and TEGDMA in cultured cell lines. Arch Oral Biol. 2004;49:125-31
44. Stanislawski L, Lefeuvre M, Bourd K, Soheili-Majd E, Goldberg M, Perianin A. TEGDMA-induced toxicity in human fibroblasts is associated with early and drastic glutathione depletion with subsequent production of oxygen reactive species. J Biomed Mater Res A. 2003;66:476-82
45. Volk J, Engelmann J, Leyhausen G, Geurtsen W. Effects of three resin monomers on the cellular glutathione concentration of cultured human gingival fibroblasts. Dent Mater. 2006;22:499-505
46. Lee DH, Lim BS, Lee YK, Ahn SJ, Yang HC. Involvement of oxidative stress in mutagenicity and apoptosis caused by dental resin monomers in cell cultures. Dent Mater. 2006;22:1086-92
47. Demirci M, Hiller KA, Bosl C, Galler K, Schmalz G, Schweikl H. The induction of oxidative stress, cytotoxicity, and genotoxicity by dental adhesives. Dent Mater. 2008;24:362-71
48. Kim NR, Lim BS, Park HC, Son KM, Yang HC. Effects of N-acetylcysteine on TEGDMA- and HEMA-induced suppression of osteogenic differentiation of human osteosarcoma MG63 cells. J Biomed Mater Res B Appl Biomater. 2011;98:300-7
49. Krifka S, Seidenader C, Hiller KA, Schmalz G, Schweikl H. Oxidative stress and cytotoxicity generated by dental composites in human pulp cells. Clin Oral Investig. 2012;16:215-24
50. Gallorini M, Petzel C, Bolay C, Hiller KA, Cataldi A, Buchalla W. et al. Activation of the Nrf2-regulated antioxidant cell response inhibits HEMA-induced oxidative stress and supports cell viability. Biomaterials. 2015;56:114-28
51. Van Landuyt KL, Krifka S, Hiller KA, Bolay C, Waha C, Van Meerbeek B. et al. Evaluation of cell responses toward adhesives with different photoinitiating systems. Dent Mater. 2015;31:916-27
52. Valavanidis A, Vlachogianni T, Fiotakis C. 8-hydroxy-2'-deoxyguanosine (8-OHdG): A critical biomarker of oxidative stress and carcinogenesis. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev. 2009;27:120-39
53. Evans MD, Dizdaroglu M, Cooke MS. Oxidative DNA damage and disease: induction, repair and significance. Mutat Res. 2004;567:1-61
54. Tsunoda M, Sakaue T, Naito S, Sunami T, Abe N, Ueno Y. et al. Insights into the structures of DNA damaged by hydroxyl radical: crystal structures of DNA duplexes containing 5-formyluracil. J Nucleic Acids. 2010;2010:107289
55. Berquist BR, Wilson DM 3rd. Pathways for repairing and tolerating the spectrum of oxidative DNA lesions. Cancer Lett. 2012;327:61-72
56. Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39-85
57. Khanna KK, Jackson SP. DNA double-strand breaks: signaling, repair and the cancer connection. Nat Genet. 2001;27:247-54
58. Cline SD, Hanawalt PC. Who's on first in the cellular response to DNA damage?. Nat Rev Mol Cell Biol. 2003;4:361-72
59. Sancar A, Lindsey-Boltz LA, Ünsal-Kaçmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39-85
60. Stewart GS, Maser RS, Stankovic T, Bressan DA, Kaplan MI, Jaspers NG. et al. The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell. 1999;99:577-87
61. Rodriguez R, Meuth M. Chk1 and p21 cooperate to prevent apoptosis during DNA replication fork stress. Mol Biol Cell. 2006;17:402-12
62. Slee EA, O'Connor DJ, Lu X. To die or not to die: how does p53 decide?. Oncogene. 2004;23:2809-18
63. Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell. 1993;74:609-19
64. Kim JH, Choi YW, Park C, Jin CY, Lee YJ, Park da J. et al. Apoptosis induction of human leukemia U937 cells by gomisin N, a dibenzocyclooctadiene lignan, isolated from Schizandra chinensis Baill. Food Chem Toxicol. 2010;48:807-13
65. Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309-12
Author contact
![]() Corresponding author: Jihua Chen, Ph. D, Professor, State Key Laboratory of Military Stomatology & National Clinical Research Center for Oral Diseases & Shaanxi Key Laboratory of Oral Diseases, Department of Prosthodontics, School of Stomatology, The Fourth Military Medical University, Changle West Road 145, Xi'an 710032, PR China. Tel: 86 29 8477 6329 Fax: 86 29 8477 6329 E-mail: jhchenedu.cn.
Corresponding author: Jihua Chen, Ph. D, Professor, State Key Laboratory of Military Stomatology & National Clinical Research Center for Oral Diseases & Shaanxi Key Laboratory of Oral Diseases, Department of Prosthodontics, School of Stomatology, The Fourth Military Medical University, Changle West Road 145, Xi'an 710032, PR China. Tel: 86 29 8477 6329 Fax: 86 29 8477 6329 E-mail: jhchenedu.cn.
Received 2015-12-2
Accepted 2016-2-17
Published 2016-3-21