International Journal of Biological Sciences
ISSN: 1449-2288
10
Impact Factor
ISSN: 1449-2288
- Current Issue
- Volume 21; 2025
- Volume 20; 2024
- Volume 19; 2023
- Volume 18; 2022
- Volume 17; 2021
- Archive
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Top
Introduction
Results
Discussion
Materials and Methods
Supplementary Material
Acknowledgements
References
Introduction
Results
Discussion
Materials and Methods
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(8):811-818. doi:10.7150/ijbs.24624 This issue Cite
Research Paper
Integrated multifactor analysis explores core dysfunctional modules in autism spectrum disorder
Yan Huang1,*, Zhenghong Chang2,*, Xiaodan Li1, Shuang Liang1 ![]() , Ying Yi2
, Ying Yi2 ![]() , Lijie Wu1
, Lijie Wu1 ![]()
1. Department of Child and Adolescent Health, School of Public Health, Harbin Medical University, Harbin, China;
2. College of Bioinformatics Science and Technology, Harbin Medical University, Harbin, China.
*These authors contributed equally to this work.
Received 2017-12-28; Accepted 2018-3-14; Published 2018-5-22
Citation:
Huang Y, Chang Z, Li X, Liang S, Yi Y, Wu L. Integrated multifactor analysis explores core dysfunctional modules in autism spectrum disorder. Int J Biol Sci 2018; 14(8):811-818. doi:10.7150/ijbs.24624. https://www.ijbs.com/v14p0811.htm
Other stylesAbstract

Autism spectrum disorder (ASD) is a complex neurodevelopmental disease in early childhood, and growing up to be a major cause of disability in children. However, the underlying molecular mechanism of ASD remains elusive. Hence, we represented integrated multifactor analysis exploring dysfunctional modules based on RNA-Seq data from corpus callosum in 6 patients with ASD and 6 normal individuals. According to protein-protein interactions (PPIs) and WGCNA, we performed co-expression modules analysis for ASD-associated genes, and identified 25 modules with differentially expressed genes (DEGs), observing that genes in these modules were significantly involved in various biological processes in nervous system, sensory system, phylogenetic system and variety of signaling pathways. Then, based on transcriptional and post-transcriptional regulations, integrating transcription factor (TF)-target and RNA-associated interactions, significant regulators of co-expression modules were identified as pivot regulators, including 67 pivot TFs, 13 pivot miRNAs and 6 pivot lncRNAs. GO and KEGG pathway enrichment analysis demonstrated that the pivot miRNAs significantly enriched in neural or mental-associated biological progresses. The pivot TFs were mainly involved in various regulation of transcription, immune system and organs development. Finally, our work deciphered a multifactor dysfunctional co-expression subnetwork involved in ASD, helps uncover core dysfunctional modules for this disease and improves our understanding of its underlying molecular mechanism.
Keywords: Autism spectrum disorder, Multifactor analysis, Co-expression, Core dysfunctional module
Introduction
Autism spectrum disorder (ASD), also known as autism, is a representative disease of pervasive neurodevelopmental disorders in children and one of the most rapidly growing diseases in the world [1, 2]. It's a congenital mental disorder that has nothing to do with upbringing, which is mainly characterized by essential defects in social and communication ability, language ability and repetitive patterns of behavior [3]. In addition to these core symptoms, there are also some peripheral symptoms, such as senses interference caused by the digestive system, immune system and sensory system problems. They also exhibit neurobehavioral problems such as learning disabilities, cognitive deficits, anxiety, and hyperactivity. The symptoms of children with ASD are much characterized by social communication disorders, so many early researches focused on social cognition to find the cause of autism. But in recent years, researchers mainly focused on the medical and biology point of view, and made some progress. For example, it's validated that Taar7h and Taar7b in neuroactive ligand-receptor interaction pathway were downregulated in ASD [4]. The study by Rikki Hullinger et.al indicated that an autistic-like phenotype will be caused by the upregulation of AT-1, which affecting key neuronal metabolic pathways [5]. IGF-1 (insulin-like growth factor-1) has been verified related to multi neuropsychiatric disorders, such as depression, Alzheimer's disease and ASD [6].
Studies have demonstrated that ASD are caused by a combination of genetic, epigenetics and environmental factors [7, 8]. There are more than 300 autism-associated genes identified at the human genome level, however, most of them have no clear genetic causes. Regulation of gene expression includes transcriptional levels, post-transcriptional levels, and translational levels. Transcription regulation refers to changing the level of gene expression by changing the transcription rate, which plays an important role in the accuracy and diversity of the transmission of genetic information [9, 10]. Transcriptional regulation of eukaryotes includes various forms [11-13], such as DNA methylation, histone modification, chromatin remodeling, transcription factors (TFs), and so on. Transcription of eukaryotic genes takes place in the nucleus and translation takes place in the cytoplasm. Therefore, post-transcriptional regulation is another important aspect of gene expression regulation [14-20], including alternative splicing of RNA, RNA methylation, and various regulatory RNAs (miRNAs, lncRNAs) are involved in post-transcriptional regulation. The in-depth study of post-transcriptional and post-translational regulatory mechanisms of genes is of great significance in revealing the nature of life rhythmic activities, the molecular basis of biological evolution, and exploring new fields in genetics research.
To further explore the role of genomic in autism, we performed a systematic integrated strategy constructing multifactor regulatory network to identify meaningful gene modules underlying ASD. Combining RNA-Seq data, protein-protein interactions (PPIs), RNA-associated interactions and co-expression analysis, we first identified gene co-expression modules, observing that genes in these modules were significantly involved in various biological processes in nervous system and sensory system and variety of signaling pathway, and four core modules were identified in which genes significantly involved in ASD symptoms-associated biological processes and pathways. Then, based on transcriptional and post-transcriptional regulations, we identified pivot regulators for each module. And, the relationships between DEGs in the four core dysfunctional modules were further analysis. The results indicated that, in addition to the disorder of the genes within the core modules, the regulation of genes by pivot regulators will also play a key role in the occurrence and development of ASD. In a word, our multifactor co-expression network analysis, not only help to explore the relationship of gene modules and ASD, but also provide a novel direct for biologists to further design their researches.
Results
Identification of gene co-expression modules
We downloaded the RNA-seq data (GSE62098) from the NCBI Gene Expression Omnibus (GEO) database, referring to 6 normal individuals and 6 patients with autism spectrum disorder, and FastQC was performed on the quality control of the data. The filtered reads were aligned to the human genome reference (GRCh38). Based on gene level raw counts, we identified 502 differentially expressed protein-coding genes (DEGs), 24 miRNAs (DEMs) and 139 lncRNAs (DELncRNAs), respectively (|FC| > 1.5, p value < 0.05, Supplementary Table S1).
From the point of view of a single gene, gene module represents a series of highly correlated genes, and the genes in the same module may have similar biological function. And from the perspective of system biology, the search for a gene module with potential function is actually a bridge to understand the function of a single gene and the characteristics of the global network. Hence, identifying gene functional modules is a key step for understanding molecular mechanisms of diseases. We first extracted a protein-protein interaction (PPI) subnetwork (Figure 1), which consisting of DEGs and their 2847 interactors from the human PPI network (see Materials and Methods). Then, based on WGCNA, we performed co-expression modules analysis for these genes in the network. 30 modules were identified (Figure 2) and 25 of these modules containing DEGs were kept for further analysis (Supplementary Table S2). The functional enrichment analysis revealed that the GO biological processes and KEGG pathway involved in nervous system, sensory system and behavioral control tended to be enriched by its dysregulated genes in the 25 modules (Supplementary Table S3), for example, “neuroactive ligand-receptor interaction”, “sensory perception” and “behavioral fear response”, which are autisms symptoms-associated functions. In fact, it is no doubt that those genes also involved in the brain development, like “forebrain development” and “pituitary gland development”. There were 196 genes (18 DEGs) in 25 modules have been reported associated with ASD susceptibility exacted from the SFARI Gene (https://gene.sfari.org/autdb/).
Identification of pivot regulators
Transcriptional and post-transcriptional regulations have long been involved in the initiation and progression of various diseases in human. However, the intricate regulatory mechanisms underlying ASD still remain ambiguous. Integrating TF-target, miRNA- and lncRNA-associated interactions, we constructed multifactor-mediated regulation network for co-expression modules. Finally, pivot regulators significantly regulating the co-expression modules were identified (hypergeometric test, p < 0.05, see Materials and Methods), including 67 pivot TFs, 13 pivot miRNAs and 6 pivot lncRNAs (LINC00668, APCDD1L-AS1, ESRG, LINC01121, LINC01749 and LINC00466) for 20 modules (Supplementary Table S4).
Figure 1
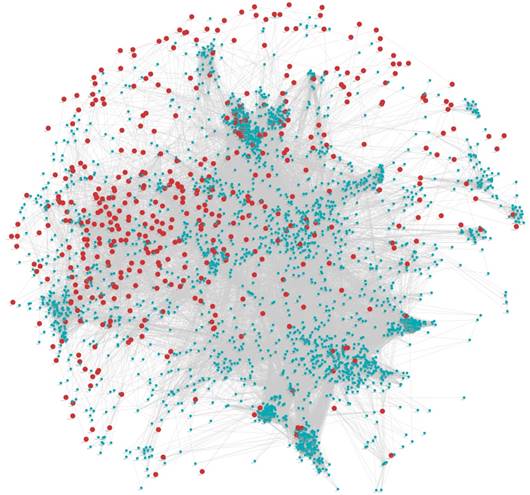
Protein-protein interaction network. Nodes in red represent DEGs and blue represent interactors of these DEGs extracted from STRING.
Figure 2
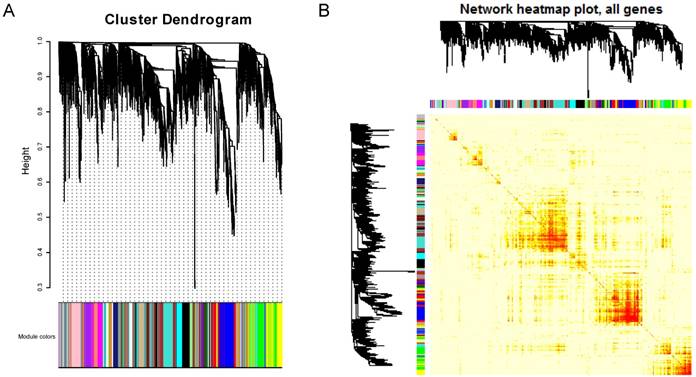
Visualization of WGCNA results. A. Clustering dendrogram of genes. A total of 31 colors corresponding to 30 modules and a gene set containing genes are not included in any module (grey). X-axis represents gene and y-axis represents the height of the gene tree. B. Heatmap plot of the gene network. The heatmap depicts the Topological Overlap Matrix (TOM) among all genes in the analysis. Light color represents low overlap and progressively darker red color represents higher overlap. Blocks of darker colors along the diagonal are the modules.
Similarly, the functional enrichment was performed for the pivot TFs and miRNAs, respectively. The results demonstrated that the pivot TFs were mainly involved in biological processes in transcriptional regulation and organs development (Figure 3A); the pivot miRNAs significantly enriched in neural or mental-associated biological progresses, for example, “neurotrophin TRK receptor signaling pathway” (p=6.16E-19), “nervous system development” (p =1.43E-4), “long-term depression” (p =2.37E-2) (Figure 3B). Moreover, some pivot lncRNAs have been reported to be directly or indirectly related to ASD. The lncRNA LINC00668 is located downstream of gene LAMA1 involved in cerebellum and retinal development [21] suggesting that it may implicated autism etiology. LINC01749 is validated associated with Major Depressive Disorder (MDD) [22], and so on.
Figure 3
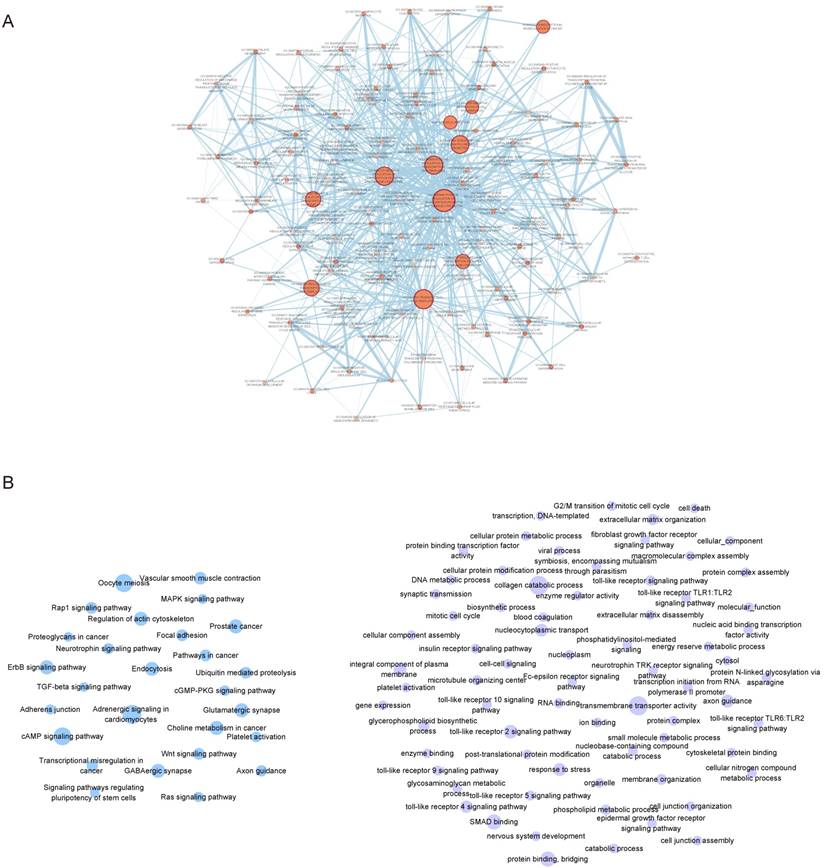
Functional enrichment results. A. Enrichment results of pivot TFs. The color depth represents p-value, the number of genes represented by the node size B. Enrichment results of pivot miRNAs. Nodes in blue represent KEGG pathway enrichment results and in purple represent GO functional enrichment results. The size of nodes represents the number of genes.
Profiling core dysfunctional modules
Based on the functional enrichment results, we focused on 4 core dysfunctional modules, including blue, yellow, green and darkred module. The enrichment analysis observed that all of the four modules were significantly enrichment for “neuroactive ligand-receptor interaction”; various G-protein coupled receptor signaling pathway enriched by blue, yellow and green module; “neuropeptide signaling pathway” by yellow, green and darkred module. But beyond that, each module also had own characteristic functions (Supplementary Table S3). The blue module consisting of 314 genes (176 DEGs) was involved in biological progresses of sensory system, such as gustation, smell and optesthesia. The yellow module containing 257 genes (16 DEGs) was observed significantly enrichment for neural system, various synapses, forebrain development and learning. These 228 genes in green module were significantly enrichment for long-term depression, sensory perception, behavioral, learning, cognition and central nervous system neuron development. And 47 genes in darkred module were observed significantly enriched on the biological processes of regulation of behavior. In total, there were 75 overlapping genes with SFARI Gene, including 13 in blue, 37 in yellow and 27 in green.
Functional enrichment showed that, pivot miRNAs (hsa-miR-640 and hsa-miR-1262 for blue module, has-let-7d-5p and hsa-miR-5190 for yellow module) were significantly enrichment for neurotrophin-related signaling pathways and nervous system development, like “neurotrophin TRK receptor signaling pathway” (p=6.160E-19) , “Neurotrophin signaling pathway” (p=7.314E-03) and “nervous system development” (p= 1.431E-04). Has-let-7d-5p have been validated for its role in Alzheimer's disease [23, 24] and brain cancer [25], suggesting that its potential function in ASD. 13 pivot TFs of the four core module, including HNF4A, HNF1A, POU2F1, POU5F1, TBX21, GATA1 (for blue), FOXM1, HNF1A (for yellow), STAT6, HMGA1, ATF3, PAX5, MAZ (for green), POU2F1 and ESR1 (for darkred) were mainly involved in transcription regulation and organ morphogenesis (Figure 4A). And HNF1A, POU2F1 significantly regulated blue and yellow, blue and darkred, respectively. It has been showed that HNF1A is associated with neuropsychiatric and neuropsychological characteristics [26, 27]. The alternative promoter usage and differential expression of POU2F1 transcript variants has an impact on cerebellar development [28], lens and olfactory placode development [29]. ESR1 is also with evident role for autism [30, 31]. Embryonic stem cell related gene (ESRG) is an lncRNA downregulated during the transition from induced pluripotent stem cell (iPSCs) to neural progenitor cells (NPCs) [32].
Considering each of these four core modules corresponded to the most typical features of autisms, we further analyzed the regulation subnetwork and interactions between differentially expressed genes in the four modules (Figure 4B, C). Analysis results presented the interactions among has-let-7d-5p, ESR1 and POU2F1, indicating they can regulate the modules genes together to affect autism. In addition, it was found that the differentially expressed genes in yellow and green are tightly linked with genes in blue, modules are tightly linked. In short, dysregulated genes in one module may directly regulated the expression of genes in another, and then pivot regulators can not only directly regulate module genes, but also indirectly regulate other functional modules through interaction with other pivot regulators of the functional modules.
Discussion
Autism is a complex disease with a strong genetic basis, including a wide range of neurodevelopmental disorders in clinical and etiology [33, 34]. Today, most of our knowledge on ASD genetics has been obtained from the genetic linkage of large ASD patient cohorts or exome sequencing analysis [35], which provides us an opportunity to observe the molecular basis of this disease. However, a complete picture of this disease may require the integration of ASD gene data from different dimensions. Transcription and post-transcription regulation is closely related to various human diseases[20, 36, 37], including brain diseases [38]. In this study, we performed an integrated multifactor analysis to identified core dysfunctional modules on RNA-seq data.
To determine the role of dysfunctional gene set in the occurrence and development of ASD, we first identified the differentially expressed genes based on RNA-seq data. Then, combining protein-protein interactions and WGCNA, 25 co-expression modules with differentially expressed genes were exacted, and observed genes in these modules were significantly involved in various biological processes in nervous system, sensory system, phylogenetic system and variety of signaling pathways. In addition to, four core dysfunction modules were identified for further analysis, which mainly enriched in gene ontology (GO) biological progresses or KEGG pathways closely related with the core symptoms of autisms, respectively. Finally, to explored whether transcripttional and post-transcriptional regulation influence the development of ASD by regulating the genes in the four core dysfunction modules, significant regulators of co-expression modules were identified as pivot regulators by integrating TF-targets and RNA-associated interactions, including TFs, miRNAs and lncRNAs. These pivot regulators were also mainly involved in a variety of developmental, neural or mental-associated biological progresses, have been reported to be more or less directly or indirectly related to neurological diseases [32, 39-41], suggesting their vital role in the regulation subnetwork.
Finally, we deciphered several core dysfunctional modules for this disease, and improved our understanding of its underlying molecular mechanism further. Experts believe that autism is also a neuronal synaptic disease which closely related with neuronal development, but its molecular mechanism needs further study. From a molecular neurobiology point of view, the functions of dendrites, axons-related proteins and non-coding genes during of the neuro-synapse are helpful to understand their role in brain development and mature, and reveal the autism pathogenesis.
Figure 4
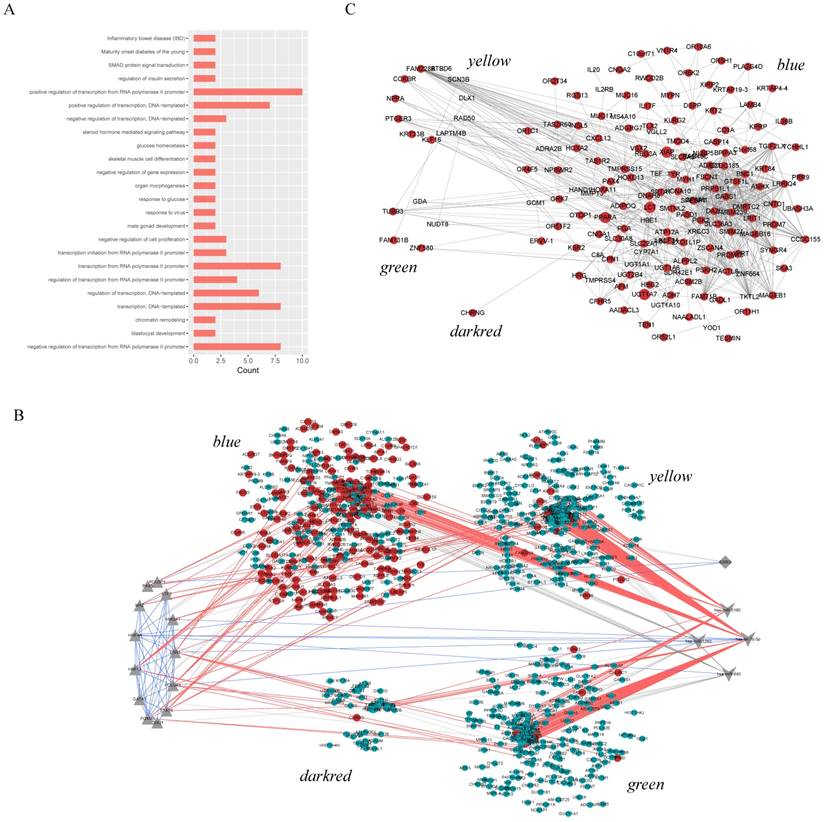
A. The biological processes enriched by 13 pivot TFs of 4 core modules. B. The transcriptional and post-transcriptional regulation for core modules. Circles in red represent DEGs. Triangles, diamonds and “V” in gray represent significant relationship between pivot regulators and genes in module. And the blue lines are interactions among these pivot regulators. C. DEG-DEG interactions between modules. The size of nodes represents the degree.
In summary, our work detailed multifactor-mediated core dysfunctional modules under ASD, which may also contribute to the discovery of more detailed molecular mechanisms and provided a rich resource of potential candidates for future experimental validations and a theoretical guidance for biological research in the future.
Materials and Methods
Data resources and Differentially expression analysis
The RNA-seq data (GSE62098 [42]) was collected from the NCBI Gene Expression Omnibus [43] (GEO) database, which contained 6 patients with autism spectrum disorders and 6 normal individuals (tissue: corpus callosum). First, the quality control of the downloaded data was performed by FastQC (Version 0.11.5). Then, the filtered reads were used to mapped to the hg38 genome reference genome (GRCh38) using HISAT2 (version 2.1.0) [44] with default parameters. The reference genome and gene annotation file (.gtf format) was downloaded from GENCODE Release 27 (http://www.gencodegenes.org/). Third, FPKM (Fragments Per Kilobase transcriptome per Million reads) values were calculated using StringTie (v1.3.3 release) [45] with parameters “--known-splicesite-infile”. Fourth, gene level raw counts were calculated by the preDE.py script within StringTie. Finally, based on raw counts, we identified differentially expressed protein-coding genes, miRNAs and lncRNAs using DEseq2 (|fold change (FC)| > 1.5 and p-value < 0.05) for further analysis.
Generating gene co-expression modules
The WGCNA R software package [46] was performed to identify gene co-expression modules. We firstly extracted a protein-protein physical interaction (PPI) subnetwork (combined_score > 900) in human from the STRING [47] database (v10.5), containing 9637 proteins and 170987 interactions. Then, DGEs and genes interacted with these DEGs in the PPI subnetwork were identified to construct weighted gene correlation network. Finally, the expression profile of genes in the weighted gene correlation network was input to WGCNA for co-expression modules. The parameters (minModuleSize = 20; minimum height = 0.15) were set to cut tree.
Identifying pivot regulators
For each gene co-expression module, we explored transcriptional and post-transcriptional regulations. From AnimalTFDB2.0 [48] and TRRUST v2 [49], we recruited 3632 regulatory relationships in human, referring to 393 TFs and 1463 target genes. As for post-transcriptional regulations, 18294 differentially expressed miRNA (DEM)-association interacttions were collected from RAID v2.0 [50] database. In addition to, we also collected 321 differentially expressed lncRNA (DELncRNA)-associated interacttions from RAID v2.0.
If a regulator i)  2 relationships between module and the regulator; and (ii) the number of its targets significant enriched for per module (hypergeometric test, p value < 0.05) [51], we defined pivot regulator as the regulator significantly regulated the module.
2 relationships between module and the regulator; and (ii) the number of its targets significant enriched for per module (hypergeometric test, p value < 0.05) [51], we defined pivot regulator as the regulator significantly regulated the module.
Gene ontology and KEGG pathway enrichment analysis
Significant gene ontology (GO) biological processes and KEGG pathway enrichment analysis were performed on DAVID [52]. And miRNAs function was deciphered by DIANA-miRPath v3.0 [53].
Supplementary Material
Supplementary Table S1.
Supplementary Table S2.
Supplementary Table S3.
Supplementary Table S4.
Acknowledgements
This project was supported by a Grant from the National Natural Science Foundation of China (81202221), and the Education Department of Heilongjiang Province (12521232).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Parikshak NN, Swarup V, Belgard TG, Irimia M, Ramaswami G, Gandal MJ. et al. Genome-wide changes in lncRNA, splicing, and regional gene expression patterns in autism. Nature. 2016;540:423-7
2. Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S. et al. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011;474:380-4
3. Lord C, Bishop SL. Recent advances in autism research as reflected in DSM-5 criteria for autism spectrum disorder. Annu Rev Clin Psychol. 2015;11:53-70
4. Huang JY, Tian Y, Wang HJ, Shen H, Wang H, Long S. et al. Functional Genomic Analyses Identify Pathways Dysregulated in Animal Model of Autism. CNS Neurosci Ther. 2016;22:845-53
5. Hullinger R, Li M, Wang J, Peng Y, Dowell JA, Bomba-Warczak E. et al. Increased expression of AT-1/SLC33A1 causes an autistic-like phenotype in mice by affecting dendritic branching and spine formation. J Exp Med. 2016;213:1267-84
6. Zegarra-Valdivia JA. Insulin-like growth factor type 1 and its relation with neuropsychiatric disorders. Medwave. 2017;17:e7031
7. Iossifov I, O'Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D. et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515:216-21
8. Parikshak NN, Luo R, Zhang A, Won H, Lowe JK, Chandran V. et al. Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell. 2013;155:1008-21
9. Wray GA, Hahn MW, Abouheif E, Balhoff JP, Pizer M, Rockman MV. et al. The evolution of transcriptional regulation in eukaryotes. Mol Biol Evol. 2003;20:1377-419
10. Levine M, Tjian R. Transcription regulation and animal diversity. Nature. 2003;424:147-51
11. Lubin FD, Roth TL, Sweatt JD. Epigenetic regulation of BDNF gene transcription in the consolidation of fear memory. J Neurosci. 2008;28:10576-86
12. Zhang Y, Reinberg D. Transcription regulation by histone methylation: interplay between different covalent modifications of the core histone tails. Genes Dev. 2001;15:2343-60
13. Chen K, Rajewsky N. The evolution of gene regulation by transcription factors and microRNAs. Nat Rev Genet. 2007;8:93-103
14. Franks A, Airoldi E, Slavov N. Post-transcriptional regulation across human tissues. PLoS Comput Biol. 2017;13:e1005535
15. Friedman RC, Farh KK, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92-105
16. Hu W, Coller J. What comes first: translational repression or mRNA degradation? The deepening mystery of microRNA function. Cell Res. 2012;22:1322-4
17. Yue Y, Liu J, He C. RNA N6-methyladenosine methylation in post-transcriptional gene expression regulation. Genes Dev. 2015;29:1343-55
18. Boutz PL, Stoilov P, Li Q, Lin CH, Chawla G, Ostrow K. et al. A post-transcriptional regulatory switch in polypyrimidine tract-binding proteins reprograms alternative splicing in developing neurons. Genes Dev. 2007;21:1636-52
19. Shi X, Sun M, Wu Y, Yao Y, Liu H, Wu G. et al. Post-transcriptional regulation of long noncoding RNAs in cancer. Tumour Biol. 2015;36:503-13
20. Tang W, Wan S, Yang Z, Teschendorff AE, Zou Q. Tumor origin detection with tissue-specific miRNA and DNA methylation markers. Bioinformatics. 2018;34:398-406
21. Giordano M, Muratore V, Babu D, Meazza C, Bozzola M. A 18p11.23-p11.31 microduplication in a boy with psychomotor delay, cerebellar vermis hypoplasia, chorioretinal coloboma, deafness and GH deficiency. Mol Cytogenet. 2016;9:89
22. Gedik H. Investigation on Genetic Modifiers of Age at Onset of Major Depressive Disorder. 2017.
23. Kumar P, Dezso Z, MacKenzie C, Oestreicher J, Agoulnik S, Byrne M. et al. Circulating miRNA biomarkers for Alzheimer's disease. PLoS One. 2013;8:e69807
24. Tan L, Yu JT, Tan MS, Liu QY, Wang HF, Zhang W. et al. Genome-wide serum microRNA expression profiling identifies serum biomarkers for Alzheimer's disease. J Alzheimers Dis. 2014;40:1017-27
25. Saydam O, Senol O, Wurdinger T, Mizrak A, Ozdener GB, Stemmer-Rachamimov AO. et al. miRNA-7 attenuation in Schwannoma tumors stimulates growth by upregulating three oncogenic signaling pathways. Cancer Res. 2011;71:852-61
26. Verhoeven WM, Egger JI, van den Bergh JP, van Beek R, Kleefstra T, de Leeuw N. A 12q24.31 interstitial deletion in an adult male with MODY3: neuropsychiatric and neuropsychological characteristics. Am J Med Genet A. 2015;167A:169-73
27. Nevado J, Mergener R, Palomares-Bralo M, Souza KR, Vallespin E, Mena R. et al. New microdeletion and microduplication syndromes: A comprehensive review. Genet Mol Biol. 2014;37:210-9
28. Pal S, Gupta R, Kim H, Wickramasinghe P, Baubet V, Showe LC. et al. Alternative transcription exceeds alternative splicing in generating the transcriptome diversity of cerebellar development. Genome Res. 2011;21:1260-72
29. Donner AL, Episkopou V, Maas RL. Sox2 and Pou2f1 interact to control lens and olfactory placode development. Dev Biol. 2007;303:784-99
30. Wang X, Liang S, Fujisawa TX, Nishitani S, Tomoda A, Zou M. et al. Association of estrogen receptor alpha polymorphisms with symptoms of autism among Chinese Han children. Neuro Endocrinol Lett. 2016;37:439-44
31. Crawley JN, Heyer WD, LaSalle JM. Autism and Cancer Share Risk Genes, Pathways, and Drug Targets. Trends Genet. 2016;32:139-46
32. Chen J, Lin M, Foxe JJ, Pedrosa E, Hrabovsky A, Carroll R. et al. Transcriptome comparison of human neurons generated using induced pluripotent stem cells derived from dental pulp and skin fibroblasts. PLoS One. 2013;8:e75682
33. Abrahams BS, Geschwind DH. Advances in autism genetics: on the threshold of a new neurobiology. Nat Rev Genet. 2008;9:341-55
34. Buxbaum JD. Multiple rare variants in the etiology of autism spectrum disorders. Dialogues Clin Neurosci. 2009;11:35-43
35. Liu G, Zhang F, Hu Y, Jiang Y, Gong Z, Liu S. et al. Genetic Variants and Multiple Sclerosis Risk Gene SLC9A9 Expression in Distinct Human Brain Regions. Mol Neurobiol. 2017;54:6820-6
36. Liu Y, Zeng X, He Z, Zou Q. Inferring microRNA-disease associations by random walk on a heterogeneous network with multiple data sources. IEEE/ACM Trans Comput Biol Bioinform. 2016 doi: 10.1109/TCBB.2016.2550432
37. Zou Q, Li J, Song L, Zeng X, Wang G. Similarity computation strategies in the microRNA-disease network: a survey. Brief Funct Genomics. 2016;15:55-64
38. Jiang Q, Hu Y, Liu G. Association of Alzheimer Disease Susceptibility Variants and Gene Expression in the Human Brain. JAMA Neurol. 2016;73:1255
39. Elsen GE, Choi LY, Prince VE, Ho RK. The autism susceptibility gene met regulates zebrafish cerebellar development and facial motor neuron migration. Dev Biol. 2009;335:78-92
40. Krishnan A, Zhang R, Yao V, Theesfeld CL, Wong AK, Tadych A. et al. Genome-wide prediction and functional characterization of the genetic basis of autism spectrum disorder. Nat Neurosci. 2016;19:1454-62
41. O'Roak BJ, Stessman HA, Boyle EA, Witherspoon KT, Martin B, Lee C. et al. Recurrent de novo mutations implicate novel genes underlying simplex autism risk. Nat Commun. 2014;5:5595
42. Li J, Shi M, Ma Z, Zhao S, Euskirchen G, Ziskin J. et al. Integrated systems analysis reveals a molecular network underlying autism spectrum disorders. Mol Syst Biol. 2014;10:774
43. Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M. et al. NCBI GEO: archive for functional genomics data sets-update. Nucleic Acids Res. 2013;41:D991-5
44. Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12:357-60
45. Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33:290-5
46. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559
47. Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M. et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017;45:D362-D8
48. Zhang HM, Liu T, Liu CJ, Song S, Zhang X, Liu W. et al. AnimalTFDB 2.0: a resource for expression, prediction and functional study of animal transcription factors. Nucleic Acids Res. 2015;43:D76-81
49. Han H, Cho JW, Lee S, Yun A, Kim H, Bae D. et al. TRRUST v2: an expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 2018;46:D380-D6
50. Yi Y, Zhao Y, Li C, Zhang L, Huang H, Li Y. et al. RAID v2.0: an updated resource of RNA-associated interactions across organisms. Nucleic Acids Research. 2017;45:D115-8
51. Ulitsky I, Shamir R. Pathway redundancy and protein essentiality revealed in the Saccharomyces cerevisiae interaction networks. Mol Syst Biol. 2007;3:104
52. Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1-13
53. Vlachos IS, Zagganas K, Paraskevopoulou MD, Georgakilas G, Karagkouni D, Vergoulis T. et al. DIANA-miRPath v3.0: deciphering microRNA function with experimental support. Nucleic Acids Res. 2015;43:W460-6
Author contact
![]() Corresponding authors: Shuang Liang, Email: liangyouyou2004com; Ying Yi, Email: yiyingedu.cn; Lijie Wu, Email: wulijiehydcom
Corresponding authors: Shuang Liang, Email: liangyouyou2004com; Ying Yi, Email: yiyingedu.cn; Lijie Wu, Email: wulijiehydcom
Citation styles
APA
Huang, Y., Chang, Z., Li, X., Liang, S., Yi, Y., Wu, L. (2018). Integrated multifactor analysis explores core dysfunctional modules in autism spectrum disorder. International Journal of Biological Sciences, 14(8), 811-818. https://doi.org/10.7150/ijbs.24624.
ACS
Huang, Y.; Chang, Z.; Li, X.; Liang, S.; Yi, Y.; Wu, L. Integrated multifactor analysis explores core dysfunctional modules in autism spectrum disorder. Int. J. Biol. Sci. 2018, 14 (8), 811-818. DOI: 10.7150/ijbs.24624.
NLM
Huang Y, Chang Z, Li X, Liang S, Yi Y, Wu L. Integrated multifactor analysis explores core dysfunctional modules in autism spectrum disorder. Int J Biol Sci 2018; 14(8):811-818. doi:10.7150/ijbs.24624. https://www.ijbs.com/v14p0811.htm
CSE
Huang Y, Chang Z, Li X, Liang S, Yi Y, Wu L. 2018. Integrated multifactor analysis explores core dysfunctional modules in autism spectrum disorder. Int J Biol Sci. 14(8):811-818.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.