Impact Factor ISSN: 1449-2288
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Resources of plant genomes
Resources of miRNAs
Resources of lncRNAs
A usage guide of web-based ncRNA...
Genome browser
Conclusions
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(8):819-832. doi:10.7150/ijbs.24593 This issue Cite
Review
A comprehensive review of web-based resources of non-coding RNAs for plant science research
Peiran Liao1,*, Shipeng Li1,*, Xiuming Cui1,3, Yun Zheng2 ![]()
1. Faculty of Life Science and Technology, Kunming University of Science and Technology, Kunming, Yunnan, 650500,China;
2. Yunnan Key Laboratory of Primate Biomedical Research, Institute of Primate Translational Medicine, Kunming University of Science and Technology, Kunming, Yunnan, 650500, China;
3. Yunnan key laboratory of Panax notoginseng, Kunming, Yunnan, 650500, China.
*These authors contributed equally to the work.
Received 2017-12-27; Accepted 2018-3-14; Published 2018-5-22
Abstract

Non-coding RNAs (ncRNAs) are transcribed from genome but not translated into proteins. Many ncRNAs are key regulators of plants growth and development, metabolism and stress tolerance. In order to make the web-based ncRNA resources for plant science research be more easily accessible and understandable, we made a comprehensive review for 83 web-based resources of three types, including genome databases containing ncRNA data, microRNA (miRNA) databases and long non-coding RNA (lncRNA) databases. To facilitate effective usage of these resources, we also suggested some preferred resources of miRNAs and lncRNAs for performing meaningful analysis.
Keywords: Non-coding RNA (ncRNA), microRNA (miRNA), long non-coding RNA (lncRNA), web-based resources, plant
Introduction
Non-coding RNAs (ncRNAs) are transcribed from genome but will not be translated into proteins [1]. Some of ncRNAs play key roles in plants growth and development, metabolism and stress tolerance [2, 3]. At present, the research of plant non-coding RNA mainly focuses on two areas: small RNAs with lengths of less than 50 nucleotides (nt), including microRNAs (miRNAs) and small interfering RNAs (siRNAs), and long non-coding RNAs (lncRNAs) normally with lengths of more than 200 nt, including long intergenic non-coding RNAs (lincRNAs), long intronic non-coding RNAs, circular RNAs (circRNAs), and circular intronic RNAs (ciRNAs) [4].
Up till now, more than 100 plant genomes have been sequenced, including more than 60 crop plants and several model plants [5]. Most of them are provided with genome databases, such as Arabidopsis [6, 7], wheat [8], rice [9, 10], soybean [11] and so on. Most ncRNAs of crops or model plants could be found by using these databases. And genome databases are useful for exploring the functions of ncRNAs and for searching their genomic contents [12, 13]. Thus genome databases are widely used as references in ncRNA studies.
MicroRNAs (miRNAs) are a class of endogenous single-strand ncRNAs sized from 18 to 25 nt, which participate in the regulation of gene expression at post transcriptional level in plants [14]. Plant miRNAs generally bind to the coding regions of the genes through intensively matched complementary sites that induce cleavage or translational repression of specific protein-coding genes [15]. They affect many biological processes, such as cell cycle, differentiation, and metabolism [2, 3]. And the increasing evidence indicates that miRNAs play key roles in developments of organs and stress responses [16].
Long non-coding RNA (lncRNA) is a class of endogenous ncRNAs with lengths of more than 200 nt but do not have coding potential [17, 18]. Though lncRNAs were ignored in early studies since their low expression and sequence conservation, emerging evidence indicates that many lncRNAs are important in diverse biological processes, such as regulation of flowering time, gene silencing, root organogenesis, seedling photomorphogenesis, and reproduction [19-22].
In the following parts of the work, we will review some web-based resources of genomes, miRNAs and lncRNAs. Then we briefly describe the analysis methods of ncRNAs, and recommend some preferred resources for the related analyses. Finally, we briefly introduce a useful web-based plant comprehensive database, the Joint Genome Institute (JGI) Genome Portal [23, 24].
Resources of plant genomes
As listed in Table 1, we will first introduce 25 resources for genomic information.
Web-based resource for plant genomes
| Database/tool | URL | Description | Ref. |
|---|---|---|---|
| TAIR | http://www.arabidopsis.org/ | A resource for Arabidopsis genome information | [6, 7] |
| Wheat | http://www.wheatgenome.org/ | A resource for wheat genome information | [8] |
| Rice | http://rice.plantbiology.msu.edu/ | A resource for rice genome information | [9, 10] |
| MaizeGDB | https://www.maizegdb.org/ | A database for Maize genetics and genomics | [25, 26] |
| SoyBase | http://soybase.org/ | A database for the USDA-ARS soybean genetics and genomics | [27] |
| funRiceGenes | https://funricegenes.github.io/ | A comprehensive database of functionally characterized rice genes | [28] |
| GO | http://geneontology.org/ | A web for Gene Ontology resource | [29, 30] |
| KEGG | http://www.kegg.jp/ | A Synthesis web for Kyoto Encyclopedia of Genes and Genomes | [31] |
| KOBAS | http://kobas.cbi.pku.edu.cn/ | A wed for gene/protein functional annotation (Annotate module) and functional gene set enrichment(Enrichment module) | [32, 33] |
| RiceWiki | http://ricewiki.big.ac.cn/ | A wiki-based, publicly editable and open-content platform for community curation of genes and Omics Knowledge in rice | [34-37] |
| BRAD | http://brassicadb.org/ | A database for genome scale genetic and genomic data of Brassica crops | [38, 39] |
| MIPS PlantsDB | http://mips.helmholtz-muenchen.de/plant/genomes.jsp/ | A database for comparative plant genome research. | [40, 41] |
| SorGSD | http://sorgsd.big.ac.cn/ | A sorghum genome SNP database | [42] |
| MtGEA | http://bioinfo.noble.org/gene-atlas/ | A web server for analyzing the Medicago transcriptome | [43] |
| DsTRD | http://bi.sky.zstu.edu.cn/DsTRD/home.php/ | A comprehensive resource for the basic and applied for Salvia Miltiorrhiza researchers | [44] |
| SoyFN | http://nclab.hit.edu.cn/SoyFN/ | A database for soybean functional gene networks and miRNA functional networks | [11] |
| GraP | http://structuralbiology.cau.edu.cn/GraP/ | A database for functional annotation, gene family classifications, protein-protein interaction networks, co-expression networks and microRNA-target pairs of Gossypium raimondii | [45] |
| STPD | http://me.lzu.edu.cn/stpd/ | A comprehensive database for salt-tolerant adaption and poplar genomics of tree | [12] |
| TFGD | http://ted.bti.cornell.edu/ | A database provides a comprehensive resource to store, query, mine, analyze, visualize and integrate large-scale tomato functional genomics data sets | [13] |
| LegumeIP | http://plantgrn.noble.org/LegumeIP/ | An integrative database for comparative genomics and transcriptomics of model legumes | [46] |
| Plant Reactome | http://plantreactome.gramene.org/ | A freely accessible database of plant metabolic and regulatory pathways | [47-49] |
| Plant-PrAS | http://plant-pras.riken.jp/ | A database of physicochemical and structural properties and novel functional regions in plant proteomes | [50, 51] |
| PlantGDB | http://www.plantgdb.org/ | A resource for comparative plant genomics | [52] |
| CuGenDB | http://www.icugi.org | A genomic database for Cucurbit species | [53] |
| Ensembl Plants | http://plants.ensembl.org | An integrative resource for sequenced plant species (currently 38) | [54] |
The Arabidopsis Information Resource (TAIR) [6, 7], International Wheat Genome Sequencing Consortium (IWGSC) [8], the Institute for Genomic Research (TIGR)/Michigan State University (MSU) Rice Annotation Database [9, 10], MaizeGDB [25, 26], SoyBase [27] are databases for Arabidopsis, wheat, rice, maize, and soybean genome, respectively. Users can download the genome sequences, annotation of genes and other information from these databases.
The funRiceGenes collected about 2,800 functional enrichment rice genes and about 5,000 members of different gene families [28].This database provides a more detailed data set to rice gene functions for researchers, including biological pathway, gene functional annotation.
The Gene Ontology (GO) is a cluster and correlation analysis database for describing gene products which was built in 1998. With development of sequencing study, increasingly number of databases has been incorporated in the GO Consortium (GOC), including several of green plant, animal, and microbial genomes. The GO has three structured categories that describe gene products in terms of their associated biological processes, cellular components and molecular functions [29, 30]. In ncRNA study, GO could be used to annotate ncRNAs and their cellular function analysis.
Kyoto Encyclopedia of Genes and Genomes (KEGG) is a network resource database for understanding the functions of proteins and utilities of the biological system, such as cell, organism and ecosystem. One of the features of the KEGG database is that the genetic catalogue from the genome which has been sequenced and linked to higher levels of cell, species and ecosystem level [31]. This database is always used to annotate targets of ncRNAs and perform metabolic pathway analysis.
KOBAS includes 1,327 species from 5 pathway databases (KEGG PATHWAY, PID, BioCyc, Reactome and Panther) and 5 human disease databases (OMIM, KEGG DISEASE, FunDO, GAD and NHGRI GWAS Catalog) [32, 33]. In plant non-coding RNA study, it could be used to annotate target gene of non-coding RNA function, including GO and KEGG annotation. Furthermore, KOBAS using statistical methods to identify metabolic pathways significantly enriched [32, 33].
RiceWiki [34-37] is a community-curated resource of rice knowledge. More than 1000 manually curated genes are validated as high quality in RiceWiki. Gene Expression Profiles retrieved from the Information Commons for Rice (IC4R) [35, 37] are integrated in RiceWiki.
The Brassica Database (BRAD) is a genomic database of Brassica rapa and Arabidopsis thaliana [38]. And BRAD v2.0 [39] has integrated 11 more Brassicaceae genomes, including Arabidopsis lyrata, Aethionema arabicum, Brassica oleracea, Brassica napus and so on. This database provides some conserved genomic information about Brassicaceae species, from the molecular level to the genomic level. And a module of Generic Synteny Browser (GBrowse_syn) provides search related functions for seeking syntenic and non-syntenic orthologs and their annotation and sequences [38, 39].
Munich Information Center for Protein Sequences PlantsDB (MIPS PlantsDB) focuses on the bioinformatics analysis methods of plant genomes [40, 41]. It aims to provide some related resource for each plant species. Meanwhile this database provides a search platform for plant genome-related research. Plant Genome and Systems Biology PlantsDB (PGSB PlantsDB) is a database for framework to facilitate comparative plant genome research. It provides services of keyword search options and BLAST sequence search function. It mainly includes complex Triticum aestivum L. genome data, especially for wheat, rye, and barley [41].
SorGSD is a database for sorghum genome SNP [42]. SorGSD contains 62 million SNPs information from 48 groups of different sorghum varieties. Using web interface, it provides an easy way to efficiently browse, search, and analyze SNP. SorGSD allows users to retrieve the SNPs information and their relevant annotations for each sorghum genome SNP.
The Medicago truncatula Gene Expression Atlas (MtGEA) aims to analyze the Medicago transcriptome, and it also offers search and analysis tools on the web platform for Medicago transcriptome [43].
Danshen Transcriptional Resource Database (DsTRD) provides information on transcript sequences and functional annotations including protein-coding RNAs, lncRNAs, other ncRNA, miRNAs and phasiRNAs [44].
SoyFN is a resource about soybean miRNA functional networks and functional gene networks [11]. It also can be used to seek, download, and analyze the functional networks of soybean miRNAs and functional genes, including their KEGG pathways, GO annotations and 3'-UTR sequences. In addition, it provides some useful tools, such as including SoySearch, promoter motif scanning, Genome Browser, eFP Browser and ID mapping [11].
Platform of Functional Genomics Analysis in Gossypium raimondii (GraP) is a genome database for Gossypium raimondii which is one of the most importance natural fiber and oil crops [45]. This database is constructed to provide visualization tools, integration and multi-dimensional analysis which provide service of gene family classifications, functional annotation, microRNA-target pairs, co-expression networks and protein-protein interaction networks [45].
The Salinity Tolerant Poplar Database (STPD) contains data of genomic sequences, genes and their functional information, single nucleotide polymorphisms information, transposable elements, simple sequence repeats and ncRNA sequences from Populus euphratica, gene expression data between Populus euphratica and Populus tomentosa, and whole-genome alignments between Populus trichocarpa, Populus euphratica and Salix suchowensis [12]. And this database provides data mining and useful searching tools, such as genome browser, BLAST servers and genome alignments viewer and so on. These tools can be used to identify similar sequences, visualize genome alignments, and browse genome regions [12].
Tomato Functional Genomics Database (TFGD), a tomato functional genomics database, which collects RNA-seq data, microarrays, small RNA data, metabolite of tomatoes and can be used to store, retrieve, analyze, visualize and integrate large-scale tomato functional annotation genomics data sets, as well as to download all the analysis results [13].
LegumeIP [46] is for research genome evolution and gene functions in legumes. The database includes three model legumes: Medicago truncatula, Glycine max, Lotus japonicus and two reference plant species, Arabidopsis thaliana and Populus trichocarpa. LegumeIP also provides visualization and comprehensive search tools that enable flexible queries based on gene annotation, relative gene expression, and gene family [46].
Plant Reactome is a free open-source curated plant pathway database, including 1,025 reactions associated with 1,173 proteins, 222 rice pathway terms, 907 small molecules [47]. The database provides bioinformatics tools for the visualization, analysis and interpretation of pathway knowledge to support genome analysis, genome annotation and basic research [47-49].
The Plant Protein Annotation Suite database (Plant-PrAS) includes 6 typical plant species, Arabidopsis thaliana, Glycine max (soybean), Populus trichocarpa (poplar), Oryza sativa (rice), Physcomitrella patens (moss) and Cyanidioschyzon merolae (alga) [50, 51]. Plant-PrAS provides multiple physicochemical and secondary structural parameters of protein sequences about these species [50, 51].
PlantGDB is a genome database encompassing sequence data for green plants (Viridiplantae). It provides transcript assemblies annotated for more than 100 plant species [52].
Cucurbit Genomics Database (CuGenDB) is a database for several important crops of Cucurbit species, including cucumber, watermelon, melon and so on [53]. This database not only provides genomic sequences, EST and RNA-seq of Cucurbit species, but also provides tools of searching gene information, BLAST, JBrowse, Pathway and GO enrichment and so on, as well as functions for downloading genome and annotation, genome resequencing, marker, EST, unigene and pathway [53].
Ensembl Plants is an integrative resource for sequenced plant species (with current version 38) [54]. This database provides information of genome sequence, functional annotation, gene models and polymorphic loci. In this database, various kinds of additional information are provided which including individual genotypes, population structure, phenotype data and linkage. And it also provides comparative analyses of whole genome and protein sequences; and service for building genome alignments and gene trees. This database is updated 4-5 times a year as well as added release genomes and information of new species [54].
Resources of miRNAs
We will next introduce 34 resources of miRNAs listed in Table 2.
Web-based resources for miRNAs
| Database/tool | URL | Description | Ref. |
|---|---|---|---|
| miRBase | http://www.mirbase.org/ | The official database of miRNAs in different species | [55-58] |
| Rfam | http://rfam.xfam.org/ | Non-coding RNA genes, structured cis-regulatory elements and self-splicing RNAs | [59] |
| miRTarBase | http://miRTarBase.mbc.nctu.edu.tw/ | A database for experimentally validated microRNA-target | [60-63] |
| mirPub | http://www.microrna.gr/mirpub/ | A database for searching microRNA publications | [64] |
| PatScan | http://www.mcs.anl.gov/home/overbeek/PatScan/HTML/pats~dn.html/ | A Web-based system enablesusers to search for nucleotide sequences for secondary structure (hairpins, pseudoknots, hammerheads and so on) and protein sequences for known motifs | [65] |
| PsRobot | http://omicslab.genetics.ac.cn/psRobot/ | A web-based easy-to-use tool dedicated to the identification of smRNAs with stem-loop shaped precursors (such as microRNAs and short hairpin RNAs) and their target genes/transcripts | [66] |
| psRNATarget | http://plantgrn.noble.org/psRNATarget/ | A plant small RNA target analysis server | [67] |
| CleaveLand | http://www.bio.psu.edu/people/faculty/Axtell/AxtellLab/Software.htm/ | A pipeline for using degradome data to find cleaved small RNA targets | [68] |
| miRNApath | http://lgmb.fmrp.usp.br/mirnapath/ | A database of miRNAs, target genes and metabolic pathways | [69] |
| TarBase | http://microrna.gr/tarbase/ | A database for microRNA targets in several animal species of central scientific interest, plants and viruses | [70, 71] |
| PeTMbase | http://petmbase.org/ | A Database of Plant Endogenous Target Mimics (eTMs) | [72] |
| AtmiRNET | http://AtmiRNET.itps.ncku.edu.tw/ | A resource for reconstructing regulatory networks of Arabidopsis miRNAs | [73] |
| PmiRExAt | http://pmirexat.nabi.res.in/ | A database and web applications for plant miRNA expression atlas | [74] |
| tasiRNAdb | http://bioinfo.jit.edu.cn/tasiRNADatabase/ | A resource for the sequences of ta-siRNA regulatory pathway-related microRNAs, TASs, ta-siRNAs and ta-siRNA targets, and for the cascading relations among them | [75] |
| comTAR | http://rnabiology.ibr-conicet.gov.ar/comtar/ | A web tool for the prediction and characterization of conserved miRNA targets in plants | [76] |
| miR-PREFeR | https://github.com/hangelwen/miRPREFeR/ | A web tool for plant miRNA prediction by using small RNA-Seq data | [77] |
| ASRP | http://asrp.cgrb.oregonstate.edu/ | A database for microRNAs and short-interfering RNAs (siRNAs) of Arabidopsis | [78] |
| PMRD | http://bioinformatics.cau.edu.cn/PMRD/ | A database of plant miRNA data deposited in public databases, gleaned from the recent literature, and data generated in-house | [79] |
| starBase | http://starbase.sysu.edu.cn/ | A database for developing to facilitate the comprehensive exploration of miRNA-target interaction maps from CLIP-Seq and Degradome-Seq data | [80] |
| miRNEST | http://mirnest.amu.edu.pl/ | A database for integrative approach in microRNA search and annotation | [81, 82] |
| siRNAdb | http://siRNA.cgb.ki.se/ | A database of siRNA sequences | [84] |
| pssRNAMiner | http://bioinfo3.noble.org/pssRNAMiner/. | A plant short small RNA regulatory cascade analysis server | [85] |
| TAPIR | http://bioinformatics.psb.ugent.be/webtools/tapir/ | A web server for the prediction of plant microRNA targets | [86] |
| RNA-hybrid | http://bibiserv.techfak.uni-bielefeld.de/rnahybrid/ | A program for predicts multiple potential binding sites of miRNAs in large target RNAs | [87] |
| PMTED | http://pmted.agrinome.org/ | A database for retrieving and analysis expression profiles of miRNA targets represented | [88] |
| PASmiR | http://pcsb.ahau.edu.cn:8080/PASmiR/ | A database for miRNA molecular regulation in plant response to abiotic stress | [89] |
| AHD | http://ahd.cbi.pku.edu.cn/help.php | A database of collection of hormone related genes of the model organism Arabidopsis thaliana | [90, 91] |
| miRFANs | http://www.cassavagenome.cn/mirfans/ | An integrated database for Arabidopsis thaliana microRNA function annotations | [92] |
| MPSS | http://mpss.udel.edu/ | A resource for mRNA and small RNA of Arabidopsis, rice, grape and Magnaporthe grisea and the rice blast fungus | [93] |
| plant_snoRNA DB | http://www.scri.sari. ac.uk/plant_snoRNA/ | A database for information of small nucleolar RNAs from Arabidopsis and 18 other plant species | [94] |
| miSolRNA | http://www.misolrna.org/ | A database for defining physiological modes of action of regulatory process underlying the metabolism of the tomato fruit | [95] |
| PmiRKB | http://bis.zju.edu.cn/pmirkb/ | A database including four major functional modules: SNP, Pri-miR, MiR-Tar and Self-reg module. | [96] |
| PlantDARIO | http://plantdario.bioinf.uni-leipzig.de/ | A web server for qualitative analysis of small RNA-seq data in plants | [98] |
| OmiRas | http://tools.genxpro.net/omiras/ | A Web server for the annotation, comparison and visualization of interaction networks of differential expression non-coding RNAs | [99] |
The miRBase reports the miRNAs in all species officially, including 4,886 plant species [55, 56]. Up to now, the miRBase has been updated to release 21 [57].We can obtain the sequences of pre-miRNAs and mature miRNAs, the secondary structures of pre-miRNAs, and related literature from the miRBase [58].
Rfam is a database of RNA families which collected a multiple sequence alignment, a covariance model (CM) and a consensus secondary structure. Rfam provides three broad functional classes including structured cis-regulatory elements, non-coding RNA genes and self-splicing RNAs. Users can use it to identify ncRNA family, including families of miRNAs [59].
The miRTarBase has accumulated more than 360,000 miRNA-target interactions (MTIs) [60]. By providing experimentally validated interaction for miRNA-target, miRTarBase aims to elucidate the role of miRNAs in different conditions and in different species including Arabidopsis thaliana, Oryza sativa [60-63].
The mirPub [64] can be used for searching microRNA publications. Because man-made causes, miRNA always named in the literature of deviation from the standard naming convention, even the official nomenclature is also developing. This database provides powerful search service to help users search relevant miRNA literature [64].
PatScan is one of the earliest programs used for searching complementary target genes [65]. PatScan can also be used to search the complementary matching sequences that allow GU wobble, mismatch and bulged nucleotide (insertion/deletion) between miRNAs and target sites [65].
PsRobot is a web-based plant small RNA meta-analysis toolbox which can be used to solve problems partially in plant miRNA and target prediction [66].
The psRNATarget is a server for analyzing plant small RNA target, containing recent study in plant miRNA target recognition [67]. The psRNATarget server is an efficient distributed computing back-end pipeline that runs on a Linux cluster which is designed for high-throughput analysis of next-generation data [67].
CleaveLand is designed to detect cleaved miRNA targets from degradome data. CleaveLand takes small RNAs, degradome sequences and an mRNA database as input and outputs small RNA targets [68].
The miRNApath is an online database for linking miRNAs to metabolic pathways using miRNA: target relations [69]. Users can enter information to search for relevant target genes and metabolic pathways [69].
In the TarBase [70] and TarBase 5.0 [71], the results are associated with pathway and GO terms, together with regulating miRNAs predicted by different computational methods and the target site information [70, 71].
Plant Endogenous Target Mimicry (PeTMbase) aims to provide a method for endogenous miRNA target mimics (eTM) in plants. It is a detectable database with 2,728 eTM sequences predicted computationally from eleven different green plant species [72].
AtmiRNET is a database of Arabidopsis miRNAs with emphasis on miRNA target recognition, promoter identification, functional enrichment of target genes, and detection of cis- and trans-elements [73]. It also supports the analysis of elucidating of functional enrichment analysis and correlations between the expression levels of miRNAs and their targets [73].
Plant miRNA Expression Atlas (PmiRExAt) is a web-based resource comprises of miRNA expression profile and searching tool for 1,859 wheat, 2,330 rice and 283 maize miRNAs [74]. The database interface offers open and easy access to miRNA expression profiles and helps to identify miRNAs expressing [74].
TasiRNAdb is a database for storing the sequences of ta-siRNA regulatory pathway-related microRNAs, ta-siRNA, TASs and ta-siRNA targets [75]. This database included 583 pathways which are regulated by ta-siRNA from 18 species. TasExpAnalys in this database is used for mapping submitted small RNAs and degradome libraries to input or TasiRNAdb's TAS sequences and performing sRNA phasing analysis and TAS cleavage analysis [75].
Conserved microRNA targets (ComTAR) is a network resource tool for the prediction of miRNA targets [76]. This database includes transcript data of 33 angiosperms which are designed to create data sets of potential miRNA targets. This database includes describing miRNA target information, functions and evolutionary conservation [76].
The miRNA PREdiction From small RNA-Seq data (MiR-PREFeR) is a plant microRNA prediction tool [77]. It can analyze miRNAs from small RNA-Seq data by utilizing expression modes of miRNAs. It follows the criteria for plant microRNA annotation to precisely predict plant miRNAs from small RNA-Seq data samples of the same species [77].
Arabidopsis Small RNA Project (ASRP) is a search tool of Arabidopsis small RNA which can describe and analyze the major classes of endogenous small RNAs in Arabidopsis. It offers sequences of small RNAs cloned from multiplicate Arabidopsis genotypes, including 1,920 unique sequences of miRNA and siRNA [78].
The Plant miRNA Database (PMRD) is a plant miRNA database which including 8,433 miRNAs assembled from 121 plant species and integrate plant miRNA data from public databases, recent researches [79]. This database contains small RNA sequence information, target sites, secondary structure, a genome browser and expression profiles [79].
StarBase is a database which collects extensive researches of miRNA-target interaction maps from Degradome-Seq and CLIP-Seq data, including high-throughput sequencing data originated from 10 degradome-Seq and 21 CLIP-Seq experiments from 6 organisms [80]. More than 2 million cleaved target clusters of plants and more than 1 Ago-binding clusters of animal are identified. From these clusters, 400,000 and 66,000 miRNA-target regulatory relationships from Degradome-Seq and CLIP-Seq data respectively are identified. This database also can be used to seek novel miRNA target sites from Degradome-Seq and CLIP-Seq data and to annotate their targets by GO terms and pathways [80].
MiRNEST is an integrative miRNAs resource which integrates external miRNA data from 13 databases and related researches, which includes small RNA sequencing data, miRNA sequences, polymorphisms, expression and targets data as well as associates to external miRNA resources from 544 species, including animals, plants and viruses [81]. The miRNA predictions performed on Expressed Sequence Tags of 202 plant and 225 animal species are the core of this database [81]. MiRNEST 2.0 added new miRNA predictions program from deep sequencing libraries [82], results of pre-miRNA classification with HuntMi [83], data from the analyses of plant degradome, and miRNA splice sites information and functions of download/upload options. Furthermore MiRNEST 2.0 improved the user interface to make it more convenient to browse through miRNA records [81, 82].
The siRNA Database (SiRNAdb) provides a gene-centric comment of siRNA experimental data, including siRNAs predicted to be of high efficacy and siRNAs of known efficacy by a combination of methods [84]. The database is developed for evaluating a siRNA's potential for non-specific and inhibition effects [84].
PssRNAMiner is designed to identify both the potential phase-initiator and the clusters of phased small RNAs [85]. To identify phase-initiators, the user should submit no less than one small RNA sequence as candidate phase initiator. The pssRNAMiner can be used to identify phased small RNA clusters as ta-siRNA candidates by evaluating the P-values of hypergeometric distribution [85].
TAPIR is performed for the prediction of plant microRNA targets [86]. The server provides a fast and precise algorithm to retrieve plant miRNA target sites. The precise option guarantees to find less perfectly paired miRNA-target duplexes though it is much slower [86].
RNA-hybrid is used for finding the minimum free energy hybridization of ncRNA [87]. The tool is primarily considered as a means for microRNA target prediction [87].
Plant MiRNA Target Expression Database (PMTED) is a database for retrieving and analyzing the expression of miRNA target genes based on the miRNA data in existing literatures [88]. Function for miRNAs and their target sequences, and differential expression profiles can be easily obtained by using this database. And it can be used to predict conserved and novel miRNAs and their targets with their corresponding expression profile retrieval. It also provides service of browsing functions in a global Meta-network among bioprocesses, species, conditions, and miRNAs, meta-terms collected from well annotated microarray experiments, which are displayed by a Cytoscape Web-based graphical interface [88].
PASmiR is a database for miRNA molecular regulation network in different plant under abiotic stresses [89]. It contains more than two hundred published studies, representing more than one thousand regulatory relationships between 35 abiotic stresses and 682 miRNAs in 33 plant species. Users can search the miRNA-stress regulatory entries with keywords of plant species, abiotic stress, and miRNA identifier. In this database, users are provided with detailed regulation information of a specific miRNA, including miRNA expression pattern, the name of species, stress name, miRNA identifier, detection method for miRNA expression, a reference literature, and target gene(s) of the miRNA extracted from the corresponding reference or the miRBase [89].
Arabidopsis Hormone Database (AHD) provides a comprehensive and systematic view of genes participating in plant hormonal regulation, and describes morphological phenotypes controlled by plant hormones [90]. AHD 2.0 adds predicted miRNA complementary site(s) and integrated genes that genetically interact with each Arabidopsis thaliana hormone related genes by literatures mining. This database is useful for studying miRNA mediated hormone regulation in plants [90, 91].
MiRFANs is a database for Arabidopsis thaliana miRNA function annotations, including transcription factor (TF), miRNA-target interactions and their target sites, expression profiles, pathways and genomic annotations [92]. It also can be used to predict miRNA target by its various types of tools. Each miRNA target is related to pathway and GO terms and is associated with regulating miRNAs predicted information [92].
Massively Parallel Signature Sequencing (MPSS) is a signature-based transcriptional resource for mRNA and small RNA of four species (Arabidopsis, rice, grape and Magnaporthe grisea) [93]. It can measure the expression levels of most genes under defined conditions and it provides information about potentially novel transcripts [93].
Plant snoRNA DB provides information on small nucleolar RNAs from Arabidopsis and 18 other plant species, including their polycistronic, intronic and the number of gene variants and many snoRNA genes from alignments of orthologous genes and gene variants from different plant species [94].
MiSolRNA collects data of tomato for integrating genetic map sites of miRNA-targeted genes, their relations with quantitative fruit metabolic loci and their expression profiles and yield associated traits [95]. And it provides a metadata source to facilitate the construction of hypothesis aimed at defining physiological modes of action of regulatory process underlying the metabolism of the tomato fruit [95].
Plant miRNA knowledge base (PmiRKB) is a database mainly for miRNA information of Arabidopsis (Arabidopsis thaliana) and rice (Oryza sativa), including single nucleotide polymorphism (SNP) data to inspect the SNPs within pre-miRNAs (precursor microRNAs) and miRNA—target RNA duplexes, investigating the tissue-specific, transcriptional contexts of pre- and pri-miRNAs (primary microRNAs) and validating miRNA and target pairs [96].
PlantDARIO is a database for processing and analyzing of plant-specific small non-coding RNAs (sncRNAs) [97, 98]. PlantDARIO has diverse functions, such as small RNA-seq quality control, analyses of mapping files, expression analyses of annotated sncRNAs, as well as the prediction of novel miRNAs and snoRNAs from expression analysis of user-defined loci and unknown expressed loci [98].
OmiRas is an online server to analyze differential expression of miRNAs derived from small RNA-Seq data [99]. OmiRas has two main functions. The first function is static annotation, including length distribution, alignments mapping statistics, and quantification tables for each library and analysis on different expression of ncRNAs between treatment and conditions. And the second function is an interactive network of miRNAs and their target genes that are analyzed by the combination of several miRNA-mRNA interaction databases [99].
Resources of lncRNAs
In this section, we introduce 24 resources for lncRNAs, as listed in Table 3, and a new database for plant circRNAs.
Web-based resources for lncRNAs
| Database/tool | URL | Description | Ref. |
|---|---|---|---|
| lncRNAdb | http://www.lncrnadb.org/ | A database for regulation mRNA and lncRNAs that have or associate with biological functions in eukaryotes | [100, 101] |
| NONCODE | http://www.noncode.org/ | A database of expression and biological functions of lncRNAs | [102-106] |
| CPC | http://cpc2.cbi.pku.edu.cn/ | Calculate protein-coding potential of lncRNAs and other RNAs | [107, 108] |
| NPInter | http://www.bioinfo.org/NPInter/ | Functional interactions between ncRNAs and biomolecules | [109, 110] |
| CNCI | http://www.bioinfo.org/software/cnci/ | A tool for classify protein-coding or non-coding transcripts | [111] |
| COME | https://github.com/lulab/COME | A tool for identification and characterization of coding potential lncRNAs | [112] |
| CPAT | http://code.google.com/p/cpat/ | A tool for extracting coding and non-coding transcripts from raw transcriptome sequencing data by using logistic regression model | [113] |
| Pfamscan | https://www.ebi.ac.uk/Tools/pfa/pfamscan/ | A tool for protein functional analysis | [114] |
| phyloCSF | https://github.com/mlin/PhyloCSF/wiki/ | A tool for distinguish protein coding and non-coding regions | [115] |
| PLNlncRbase | http://bioinformatics.ahau.edu.cn/PLNlncRbase/ | A database for lncRNAs that collected 1,187 plant lncRNAs in 43 plant species | [116] |
| PLncDB | http://chualab.rockefeller.edu/gbrowse2/homepage.html | A database of plant long non-coding | [117] |
| PNRD | http://structuralbiology.cau.edu.cn/PNRD/ | A comprehensive integrated web resource for ncRNA searching, browsing, predicting, visualizing and downloading | [118] |
| LncReg | http://bioinformatics.ustc.edu.cn/lncreg/ | A database developed by collecting 1,081 validated lncRNA-associated regulatory entries | [119] |
| Mfoldweb server | http://www.bioinfo.rpi.edu/applications/mfold/ | A web server for nucleic acid folding and hybridization prediction | [120] |
| RNAsoft | http://www.RNAsoft.ca/ | A suite of RNA secondary structure prediction and design software tools | [121] |
| RNA shapes | http://bibiserv.cebitec.uni-bielefeld.de/rnashapesstudio/ | A tool for RNA structure analysis | [122] |
| RNA Movies 2 | http://bibiserv.techfak.uni-bielefeld.de/rnamovies/ | A powerful visualization tool for RNA secondary structure analysis | [123] |
| RNAz | http://www.rna.tbi.univie.ac.at/RNAz/ | A web for RNA secondary structure prediction | [123, 124] |
| Infernal | http://infernal.janelia.org/ | A tool for search and align homologous RNAs | [126] |
| PlantcircBase | http://ibi.zju.edu.cn/plantcircbase/ | A database of plant circRNAs which collect reported and unpublished circRNAs of five model plants and provides services of predicting circRNAs | [127] |
| PlantCircNet | http://bis.zju.edu.cn/plantcircnet/index.php | A visualized web-based database of plant circRNA, miRNA and mRNA regulatory networks in eight model plants | [128] |
| PlantNATsDB | http://bis.zju.edu.cn/pnatdb/ | A web database which includes high-throughput small RNA sequencing data investigate the biological function of natural antisense transcripts (NATs) | [129]. |
| RNAcentral | http://rnacentral.org | A database for integrating ncRNA from most organisms and providing text, sequence similarity and genome browsing functionality searching service | [130, 131] |
| ncRNAdb | http://biobases.ibch.poznan.pl/ncRNA/ | A database for collecting ncRNA except microRNAs, snoRNAs and housekeeping transcripts in animals, plants, eukaryotes, eubacteria and archaea. | [132, 133] |
The lncRNAdb is a database of lncRNAs which have or suspected to have biological functions in eukaryotes [100]. There are many referenced information of RNA in each entry, including sequences, structural information, genomic context, expression, subcellular localization, conservation, functional evidence and other relevant information. The lncRNAdb can be searched by keywords, including published RNA names and aliases, sequences, species and associated protein-coding genes and so on. The lncRNAdb v2.0 [101] provides reference database of more than 280 eukaryotic lncRNAs from scientific literature and add new characteristics in lncRNAdb contain a BLAST search tool and easy export of content via direct download or a REST API, the integration of Illumina Body Atlas expression profiles, nucleotide sequence information [100, 101].
NONCODE is an integrated knowledge database dedicated to non-coding RNAs, including non-coding RNAs of plants [102]. This database provides the detailed information of lncRNAs, which strand, exon number, length and sequence. In addition, it also provides advanced information, which include the expression profiles, exosome expression profiles, conservation information, predicted functions and disease relations [102-106].
Coding Potential Calculator (CPC) is an available mobile-friendly web server which can be used to assess the potential protein-coding transcripts by using sequence features and provide vector machine [107]. CPC2 added the function of downloadable standalone package and is much faster and more accurate [107, 108].
NPInter is a database for functional interactions between non-coding RNAs (except tRNAs and rRNAs) and biomolecules (proteins, RNAs and DNAs) [109]. It also provides a convenient search tool option which is allow users seek the interactions, related publications and other information [109, 110].
Coding-Non-Coding Index (CNCI) is a tool for assortment with protein-coding or non-coding transcripts. It can use incomplete transcripts and antisense transcripts to effectively predict non-coding RNA [111].
Coding potential calculator based on multiple evidences (COME) is an accurate and stable tool for identification and characterization of coding potential lncRNAs. And this tool integrates multiple sequence-derived and experiment-based features using a method of decomposition and combination. COME can greatly improve the consistency of predication results from other coding potential calculators. In addition, COME can annotates and characterizes each predicted lncRNA transcripts with multiple lines of supporting evidence, rather than other tools provided. [112].
Coding-Potential Assessment Tool (CPAT) is a high accuracy and fast tool for extracting coding and non-coding transcripts from raw transcriptome sequencing data by using logistic regression model which base on four sequence features: open reading frame size, open reading frame coverage, Fickett TESTCODE statistic and hexamer usage bias. It also can be used online and allows users to submit FASTA or BED formatted data files of sequences [113].
Pfamscan is a tool for protein functional analysis. The possible protein sequences in each transcript will be searched homologous to the database to eliminate the possibilities of being coding genes for putative lncRNAs [114].
PhyloCSF is a tool for differentiate protein coding and non-coding regions. It can help distinguish between protein-coding and non-coding RNA in transcripts obtained by sequencing high-throughput transcriptomes [115].
PLNlncRbase is a database which provides detailed information for identified lncRNA and includes 1,187 plant lncRNAs of more than 40 species from nearly 200 published literatures [116]. Users can seek plant lncRNAs by keywords of plant species or lncRNA identifier. The details of lncRNAs include species name, description of the potential biological function, lncRNA sequences and classification, expression patterns of lncRNAs, the tissue, developmental stage and condition for lncRNA expression, detection method for lncRNA expression, a reference literature, and the potential target genes of the lncRNA taken out from the original reference [116].
Plant Long non-coding RNA Database (PLncDB) includes a comprehensive genomic view of Arabidopsis lncRNAs for the plant research community [117].
Plant ncRNA Database (PNRD) contains 25,739 non-coding RNA records, including lncRNAs, tRNA, rRNA, tasiRNA, snRNA and snoRNA, etc [118]. It also provides many functional search and analysis tools, including search of ncRNA keyword, literature-based function and miRNA-targets [118].
LncReg is a database for regulatory relationships of the lncRNAs, including 1,081 reported lncRNA-associated regulatory entries, 258 non-redundant lncRNAs and 571 non-redundant genes [119]. It can be used to seek regulatory networks of lncRNAs and comprehensive data for bioinformatics studies and beneficial to understand the function of lncRNAs [119].
Mfold web server is a tool to predict the secondary structure of single stranded nucleic acids [120]. It provides service for RNA and DNA folding and hybridization software in nucleic acids [120].
RNAsoft is a tool for prediction of the secondary structure of a pair of DNA and RNA molecules, removing unwanted secondary structure and drawing RNA strands that fold to a given input secondary structure [121]. It can be used to analysis of secondary structure of lncRNA.
RNA Shapes allows users to extract several relevant structures from the folding space of RNA sequence with a single structure of minimal free energy [122].
RNA Movies 2, which enables to browse sequential paths through RNA secondary structure landscapes, can be used to visualize structural rearrangement processes of RNA [123].
RNAz is a web database for RNA secondary structure prediction [124, 125]. Infernal is a tool for searching and aligning homologous RNAs [126].
PlantcircBase is a database of plant circRNAs which collect reported and unpublished circRNAs of Oryza sativa, Arabidopsis thaliana, Zea mays, Solanum lycopersicum, and Hordeum vulgare [127]. And this database also provides services of predicting circRNAs from high throughput sequencing profiles input by users, for visualizing the structures of circRNAs, and analysis of functions and biogenesis mechanisms of circRNAs [127].
PlantCircNet is a visualized web-based database of plant circRNA, miRNA and mRNA regulatory networks, which can show querying detailed information of specific plant circRNAs in a user-friendly interface [128]. It contain identified circRNAs in eight model plants and can find significantly overrepresented Gene Ontology categories of miRNA targets by its enrichment analysis tool. It also provides investigation of genomic annotations, sequences and isoforms of circRNAs [128].
Plant Natural Antisense Transcripts Database (PlantNATsDB) is a database for plant natural antisense transcripts (NATs) which are regulated physiological and pathological processes [129]. In order to investigate the biological function of NATs, 2 million NAT pairs in 69 plant species and GO annotation and high-throughput small RNA sequencing data are integrated in this database. Small RNA can get GO annotation and their NATs function analysis in this database and their NATs regulation network could also be generated [129].
RNAcentral is a database which collects and integrates data and information of ncRNA from international consortium of established RNA sequence databases [130]. The initial release contains more than 8.1 million sequences, including all major representative functional group [130]. New release collects data from specialized ncRNA resources and provides a single entry point for all types of ncRNAs' sequences from most organisms, and created new species-specific identifiers of unique RNA sequences of single species. It also provides searching service of text and sequence similarity as well as genome browsing functionality [131].
Noncoding RNAs database (ncRNAdb) collected currently available sequence data on ncRNAs which have regulatory functions in prokaryotic, animal and plant cells, including regulation of chromatin structure, translational gene expression, modulation of protein function and subcellular distribution of RNAs and proteins [132, 133]. New release added ncRNA sequences from eukaryotes, eubacteria and archaea. This database does not collect microRNAs, snoRNAs and housekeeping transcripts [132, 133].
A usage guide of web-based ncRNA resources
Up to now, many online resources for ncRNAs have been reported. To make it easier to use these resources, we will introduce a number of related databases and analysis tools in the general bioinformatics analysis processes of miRNAs and lncRNAs, and suggest some preferred resources.
As shown in Figure 1a, is the general process of miRNAs analysis. The first step is to obtain the required data from sequencing companies or public database such as NCBI GEO or SRA databases, and then filter the raw data to get high quality reads. There are some tools for filtering low quality reads and removing adapters in raw reads, such as Fastx-toolkit (http://hannonlab.cshl.edu/fastx_toolkit/), Trimmomatic [134], cutadapt [135]. Fastx-toolkit is developed for short-reads FASTA/FASTQ files preprocessing, primarily including identical reads collapsing, read length trimming, adapter removing and so on. Fastx-toolkit is a preferred tool to filter low quantity reads and remove adapters. The second step is to align reads to mRNA databases to remove sequences from protein-coding genes, known miRNAs, other ncRNAs and repeats. Several databases such as miRBase [56], RepBase [136], and TIGR Plant Repeat Database [137] and Rfam [59] are recommended in this step. The cDNAs of the species under investigation should also be used to remove sRNAs originated from coding genes in this step. After that, in the third step, the remaining reads were aligned to the genome database. Since there could be many reads in the sequencing libraries, Bowtie [138] is suggested in this step. In the fourth step, the secondary structures of the flanking sequences of the matched loci should be predicted. Two RNA secondary structure prediction tools, Mfold [120] and RNAfold [139] are preferred in this step. Next, in the fifth step, the sequenced sRNAs from the hair-pin structures were aligned to the miRBase to examine whether the identified miRNAs were conserved or novel with BLASTN. In the sixth step, to investigate the putative functions of conserved and novel miRNAs, their target genes could be predicted using target prediction tools, such as psRobot [66], psRNATarget [67], CleaveLand [68], SeqTar [140]. SeqTar [140] and PsRNATarget [67] are preferred for predicting plant miRNA target genes with and without degradome profiles, respectively. Finally, the seventh step is to predict potential functions of miRNAs. There are several tools for functional annotations, such as starBase [80], miRTarBase [60], and KOBAS [32]. KOBAS is a preferred for functional annotation [32, 33] since it includes GO enrichment, and KEGG pathway, and provides a temporary storage space for user analysis. The miRTarBase is a preferred tool to search experimentally validated miRNA targets by using miRNA, target gene, pathway, validation method, disease and literature. Normally, the putative targets of miRNAs are used in functional annotation of miRNAs because miRNAs realize their functions through their regulations on target mRNAs.
The general bioinformatics analysis processes of miRNAs and lncRNAs, and their corresponding computational resources. The arrows indicate the suggested steps when performing miRNAs and lncRNAs bioinformatics analysis. The corresponding computational resources for the analysis steps are shown in the right panels within the dashed-line boxes. Part a and b are the general analysis processes of miRNAs and lncRNAs, respectively. The resources in bold are recommended in the corresponding analysis steps.
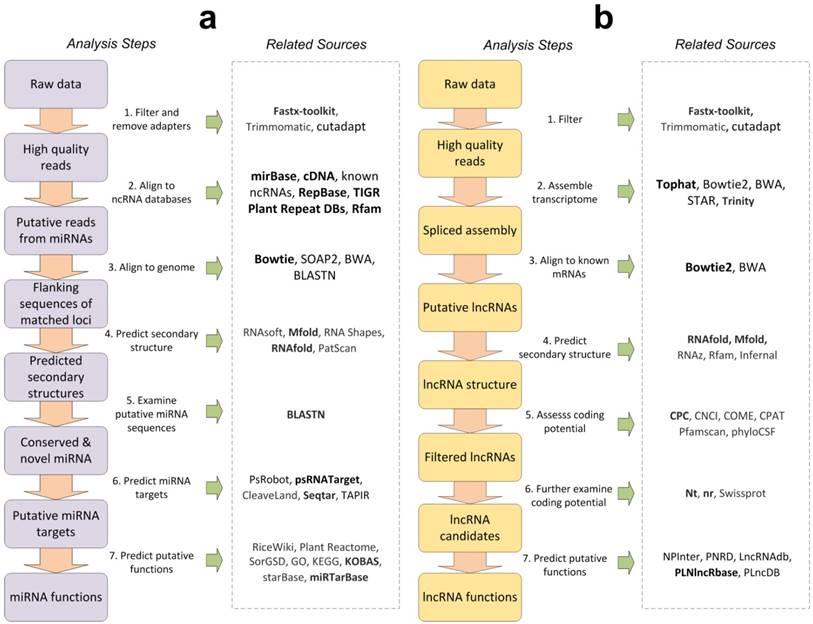
Figure 1b is the general analysis process of lncRNAs. Similar to miRNA analysis workflow, the first step is to filter low-quality reads from raw data. Fastx-toolkit is a preferred tool in this step. The second step is to construct spliced transcript assembly from filtered high quality reads. TopHat [141], Bowtie [138, 142], STAR [143] are preferred tools for transcript assembling with reference gene annotation and another tool, Trinity [144], is suggested for assembly without reference. In the third step, the assembled transcripts are aligned to known mRNAs to remove transcripts from known coding genes by Bowtie 2 or BWA tools. In the fourth step, the secondary structures of the remaining transcripts were predicted. RNAfold [139] and Mfold [120] are recommended for secondary structure prediction. The fifth step is to use coding potential assessment tools, such as CPC [107], CNCI [111], COME [112], CPAT [113], Pfamscan [114], phyloCSF [115], to further predict coding potentials of putative lncRNAs. CPC2 is preferred tools to predict coding potentials of lncRNAs, since CPC2 is available mobile-friendly web server and as a downloadable standalone package [107, 108]. The sixth step is to examine coding potential for filtered lncRNAs by mapping them to different databases. The NCBI nt and nr database is the preferred mapping database in this step since they include sequences for all kinds of species. The seventh step is to predict potential functions of lncRNAs. NPInter [109], PNRD [118], LncRNAdb [100], PLNlncRbase [116], PLncDB [117] could be used in this step. PLNlncRbase is preferred because it is an easy-to-use database which provides detailed information for experimentally identified plant lncRNAs.
Although we introduced some related databases and tools in the analysis miRNAs and lncRNAs as shown in Figure 1, researchers can choose some other tools based on their own needs.
Genome browser
The Department of Energy (DOE) Joint Genome Institute (JGI) Genome Portal [23, 24] (http://genome.jgi.doe.gov/) is a comprehensive website that stores the high-throughput sequencing information for diverse species, including green plant, microbes, fungi, and some biological information analysis tools. JGI Phytozome (https://phytozome.jgi.doe.gov/pz/portal.html) is its sub website which is a comparative database for genomes of green plant and gene family data and analysis. There are three functional modules where users can choose and set, i.e., Species, Tools, and Download. In the module of Species, the website provides the genome of most plant species, e.g., Arabidopsis thaliana TAIR10 (Thale cress), Oryza sativa (Rice), Medicago truncatula (Barrel medic), Triticum aestivum (Common wheat), Sorghum bicolor (Cereal grass) and so on. The Tools module includes Keyword search, BLAST, BLAT, JBrowse, PhytoMine, and BioMart. Users can download a different version of plant genome sequencing information through the Download module. JGI Phytozome is updated regularly. All genomes in JGI Phytozome have been annotated with KOG, KEGG, ENZYME, Pathway, InterPro [145]. In JBrowse option, users can browse the details of listed genomes, including gene site, length, and annotation.
Conclusions
NcRNAs play important regulatory roles in plants, affecting plant growth and development, disease resistances, stress tolerances, biosynthesis of secondary metabolites and many other aspects [2]. With the development of modern high-throughput sequencing technology, more and more ncRNA has been reported. We summarize the web-based resources of ncRNAs and provide some preferred tools for different analysis tasks.
Acknowledgements
The research was supported in part by two grants (No. 31460295 and 31760314) of National Natural Science Foundation of China (http://www.nsfc.gov.cn/) and a grant (No. SKLGE-1511) of the Open Research Funds of the State KeyLaboratory of Genetic Engineering, Fudan University, China, to YZ, and grants (No. 2013FA031 and 2014FA003) of the Science and Technology Bureau of Yunnan Province to XC. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Kapranov P, Cheng J, Dike S, Nix DA, Duttagupta R, Willingham AT. et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science. 2007;316:1484-8
2. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281-97
3. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215-33
4. St Laurent G, Wahlestedt C, Kapranov P. The Landscape of long noncoding RNA classification. Trends in genetics: TIG. 2015;31:239-51
5. Michael TP, VanBuren R. Progress, challenges and the future of crop genomes. Current opinion in plant biology. 2015;24:71-81
6. Berardini TZ, Reiser L, Li D, Mezheritsky Y, Muller R, Strait E. et al. The Arabidopsis information resource: Making and mining the "gold standard" annotated reference plant genome. Genesis. 2015;53:474-85
7. Lamesch P, Berardini TZ, Li D, Swarbreck D, Wilks C, Sasidharan R. et al. The Arabidopsis Information Resource (TAIR): improved gene annotation and new tools. Nucleic acids research. 2012;40:D1202-10
8. International Wheat Genome Sequencing C. A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science. 2014;345:1251788
9. Ouyang S, Zhu W, Hamilton J, Lin H, Campbell M, Childs K. et al. The TIGR Rice Genome Annotation Resource: improvements and new features. Nucleic acids research. 2007;35:D883-7
10. Kawahara Y, de la Bastide M, Hamilton JP, Kanamori H, McCombie WR, Ouyang S. et al. Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice. 2013;6:4
11. Xu Y, Guo M, Liu X, Wang C, Liu Y. SoyFN: a knowledge database of soybean functional networks. Database: the journal of biological databases and curation. 2014;2014:bau019
12. Ma Y, Xu T, Wan D, Ma T, Shi S, Liu J. et al. The salinity tolerant poplar database (STPD): a comprehensive database for studying tree salt-tolerant adaption and poplar genomics. BMC genomics. 2015;16:205
13. Fei Z, Joung JG, Tang X, Zheng Y, Huang M, Lee JM. et al. Tomato Functional Genomics Database: a comprehensive resource and analysis package for tomato functional genomics. Nucleic acids research. 2011;39:D1156-63
14. Iwakawa HO, Tomari Y. The Functions of MicroRNAs: mRNA Decay and Translational Repression. Trends in cell biology. 2015;25:651-65
15. Ling H, Fabbri M, Calin GA. MicroRNAs and other non-coding RNAs as targets for anticancer drug development. Nature reviews Drug discovery. 2013;12:847-65
16. Ferdous J, Hussain SS, Shi BJ. Role of microRNAs in plant drought tolerance. Plant biotechnology journal. 2015;13:293-305
17. Wang H, Chung PJ, Liu J, Jang IC, Kean MJ, Xu J. et al. Genome-wide identification of long noncoding natural antisense transcripts and their responses to light in Arabidopsis. Genome research. 2014;24:444-53
18. Zhang YC, Liao JY, Li ZY, Yu Y, Zhang JP, Li QF. et al. Genome-wide screening and functional analysis identify a large number of long noncoding RNAs involved in the sexual reproduction of rice. Genome biology. 2014;15:512
19. Chekanova JA. Long non-coding RNAs and their functions in plants. Current opinion in plant biology. 2015;27:207-16
20. Bardou F, Ariel F, Simpson CG, Romero-Barrios N, Laporte P, Balzergue S. et al. Long noncoding RNA modulates alternative splicing regulators in Arabidopsis. Developmental cell. 2014;30:166-76
21. Wang Y, Fan X, Lin F, He G, Terzaghi W, Zhu D. et al. Arabidopsis noncoding RNA mediates control of photomorphogenesis by red light. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:10359-64
22. Mattick JS, Rinn JL. Discovery and annotation of long noncoding RNAs. Nature structural & molecular biology. 2015;22:5-7
23. Grigoriev IV, Nordberg H, Shabalov I, Aerts A, Cantor M, Goodstein D. et al. The genome portal of the Department of Energy Joint Genome Institute. Nucleic acids research. 2012;40:D26-32
24. Nordberg H, Cantor M, Dusheyko S, Hua S, Poliakov A, Shabalov I. et al. The genome portal of the Department of Energy Joint Genome Institute: 2014 updates. Nucleic acids research. 2014;42:D26-31
25. Lawrence CJ, Harper LC, Schaeffer ML, Sen TZ, Seigfried TE, Campbell DA. MaizeGDB: The maize model organism database for basic, translational, and applied research. International journal of plant genomics. 2008;2008:496957
26. Andorf CM, Cannon EK, Portwood JL 2nd, Gardiner JM, Harper LC, Schaeffer ML. et al. MaizeGDB update: new tools, data and interface for the maize model organism database. Nucleic acids research. 2016;44:D1195-201
27. Grant D, Nelson RT, Cannon SB, Shoemaker RC. SoyBase, the USDA-ARS soybean genetics and genomics database. Nucleic acids research. 2010;38:D843-6
28. Yao W, Li G, Yu Y, Ouyang Y. funRiceGenes dataset for comprehensive understanding and application of rice functional genes. GigaScience. 2017
29. Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nature genetics. 2000;25:25-9
30. The Gene Ontology C. Expansion of the Gene Ontology knowledgebase and resources. Nucleic acids research. 2017;45:D331-D8
31. Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M. The KEGG resource for deciphering the genome. Nucleic acids research. 2004;32:D277-80
32. Wu J, Mao X, Cai T, Luo J, Wei L. KOBAS server: a web-based platform for automated annotation and pathway identification. Nucleic acids research. 2006;34:W720-4
33. Xie C, Mao X, Huang J, Ding Y, Wu J, Dong S. et al. KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic acids research. 2011;39:W316-22
34. Zhang Z, Sang J, Ma L, Wu G, Wu H, Huang D. et al. RiceWiki: a wiki-based database for community curation of rice genes. Nucleic acids research. 2014;42:D1222-8
35. Xia L, Zou D, Sang J, Xu X, Yin H, Li M. et al. Rice Expression Database (RED): An integrated RNA-Seq-derived gene expression database for rice. Journal of genetics and genomics = Yi chuan xue bao. 2017;44:235-41
36. Members BIGDC. The BIG Data Center: from deposition to integration to translation. Nucleic acids research. 2017;45:D18-D24
37. Consortium IRP, Hao L, Zhang H, Zhang Z, Hu S, Xue Y. Information Commons for Rice (IC4R). Nucleic acids research. 2016;44:D1172-80
38. Cheng F, Liu S, Wu J, Fang L, Sun S, Liu B. et al. BRAD, the genetics and genomics database for Brassica plants. BMC plant biology. 2011;11:136
39. Wang X, Wu J, Liang J, Cheng F, Wang X. Brassica database (BRAD) version 2.0: integrating and mining Brassicaceae species genomic resources. Database: the journal of biological databases and curation. 2015:2015
40. Nussbaumer T, Martis MM, Roessner SK, Pfeifer M, Bader KC, Sharma S. et al. MIPS PlantsDB: a database framework for comparative plant genome research. Nucleic acids research. 2013;41:D1144-51
41. Spannagl M, Nussbaumer T, Bader KC, Martis MM, Seidel M, Kugler KG. et al. PGSB PlantsDB: updates to the database framework for comparative plant genome research. Nucleic acids research. 2016;44:D1141-7
42. Luo H, Zhao W, Wang Y, Xia Y, Wu X, Zhang L. et al. SorGSD: a sorghum genome SNP database. Biotechnology for biofuels. 2016;9:6
43. He J, Benedito VA, Wang M, Murray JD, Zhao PX, Tang Y. et al. The Medicago truncatula gene expression atlas web server. BMC bioinformatics. 2009;10:441
44. Shao Y, Wei J, Wu F, Zhang H, Yang D, Liang Z. et al. DsTRD: Danshen Transcriptional Resource Database. PloS one. 2016;11:e0149747
45. Zhang L, Guo J, You Q, Yi X, Ling Y, Xu W. et al. GraP: platform for functional genomics analysis of Gossypium raimondii. Database: the journal of biological databases and curation. 2015;2015:bav047
46. Li J, Dai X, Liu T, Zhao PX. LegumeIP: an integrative database for comparative genomics and transcriptomics of model legumes. Nucleic acids research. 2012;40:D1221-9
47. Naithani S, Preece J, D'Eustachio P, Gupta P, Amarasinghe V, Dharmawardhana PD. et al. Plant Reactome: a resource for plant pathways and comparative analysis. Nucleic acids research. 2017;45:D1029-D39
48. Tello-Ruiz MK, Stein J, Wei S, Preece J, Olson A, Naithani S. et al. Gramene 2016: comparative plant genomics and pathway resources. Nucleic acids research. 2016;44:D1133-40
49. Petryszak R, Keays M, Tang YA, Fonseca NA, Barrera E, Burdett T. et al. Expression Atlas update-an integrated database of gene and protein expression in humans, animals and plants. Nucleic acids research. 2016;44:D746-52
50. Kurotani A, Yamada Y, Shinozaki K, Kuroda Y, Sakurai T. Plant-PrAS: a database of physicochemical and structural properties and novel functional regions in plant proteomes. Plant & cell physiology. 2015;56:e11
51. Kurotani A, Yamada Y, Sakurai T. Alga-PrAS (Algal Protein Annotation Suite): A Database of Comprehensive Annotation in Algal Proteomes. Plant & cell physiology. 2017;58:e6
52. Duvick J, Fu A, Muppirala U, Sabharwal M, Wilkerson MD, Lawrence CJ. et al. PlantGDB: a resource for comparative plant genomics. Nucleic acids research. 2008;36:D959-65
53. Bai Y, Zhang Z, Fei Z. Databases and Bioinformatics for Cucurbit Species; 2016.
54. Bolser D, Staines DM, Pritchard E, Kersey P. Ensembl Plants: Integrating Tools for Visualizing, Mining, and Analyzing Plant Genomics Data. Methods in molecular biology. 2016;1374:115-40
55. Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic acids research. 2006;34:D140-4
56. Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. miRBase: tools for microRNA genomics. Nucleic acids research. 2008;36:D154-8
57. Kozomara A, Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic acids research. 2014;42:D68-73
58. Kozomara A, Griffiths-Jones S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic acids research. 2011;39:D152-7
59. Kalvari I, Argasinska J, Quinones-Olvera N, Nawrocki EP, Rivas E, Eddy SR. et al. Rfam 13.0: shifting to a genome-centric resource for non-coding RNA families. Nucleic acids research. 2017
60. Hsu SD, Lin FM, Wu WY, Liang C, Huang WC, Chan WL. et al. miRTarBase: a database curates experimentally validated microRNA-target interactions. Nucleic acids research. 2011;39:D163-9
61. Hsu SD, Tseng YT, Shrestha S, Lin YL, Khaleel A, Chou CH. et al. miRTarBase update 2014: an information resource for experimentally validated miRNA-target interactions. Nucleic acids research. 2014;42:D78-85
62. Chou CH, Chang NW, Shrestha S, Hsu SD, Lin YL, Lee WH. et al. miRTarBase 2016: updates to the experimentally validated miRNA-target interactions database. Nucleic acids research. 2016;44:D239-47
63. Chou CH, Shrestha S, Yang CD, Chang NW, Lin YL, Liao KW. et al. miRTarBase update 2018: a resource for experimentally validated microRNA-target interactions. Nucleic acids research. 2017
64. Vergoulis T, Kanellos I, Kostoulas N, Georgakilas G, Sellis T, Hatzigeorgiou A. et al. mirPub: a database for searching microRNA publications. Bioinformatics. 2015;31:1502-4
65. Dsouza M, Larsen N, Overbeek R. Searching for patterns in genomic data. Trends in Genetics. 1997;13:497-8
66. Wu HJ, Ma YK, Chen T, Wang M, Wang XJ. PsRobot: a web-based plant small RNA meta-analysis toolbox. Nucleic acids research. 2012;40:W22-8
67. Dai X, Zhao PX. psRNATarget: a plant small RNA target analysis server. Nucleic acids research. 2011;39:W155-9
68. Addo-Quaye C, Miller W, Axtell MJ. CleaveLand: a pipeline for using degradome data to find cleaved small RNA targets. Bioinformatics. 2009;25:130-1
69. Chiromatzo AO, Oliveira TY, Pereira G, Costa AY, Montesco CA, Gras DE. et al. miRNApath: a database of miRNAs, target genes and metabolic pathways. Genetics and molecular research: GMR. 2007;6:859-65
70. Sethupathy P, Corda B, Hatzigeorgiou AG. TarBase: A comprehensive database of experimentally supported animal microRNA targets. Rna. 2006;12:192-7
71. Papadopoulos GL, Reczko M, Simossis VA, Sethupathy P, Hatzigeorgiou AG. The database of experimentally supported targets: a functional update of TarBase. Nucleic acids research. 2009;37:D155-8
72. Karakulah G, Yucebilgili Kurtoglu K, Unver T. PeTMbase: A Database of Plant Endogenous Target Mimics (eTMs). PloS one. 2016;11:e0167698
73. Chien CH, Chiang-Hsieh YF, Chen YA, Chow CN, Wu NY, Hou PF. et al. AtmiRNET: a web-based resource for reconstructing regulatory networks of Arabidopsis microRNAs. Database: the journal of biological databases and curation. 2015;2015:bav042
74. Gurjar AK, Panwar AS, Gupta R, Mantri SS. PmiRExAt: plant miRNA expression atlas database and web applications. Database: the journal of biological databases and curation. 2016:2016
75. Zhang C, Li G, Zhu S, Zhang S, Fang J. tasiRNAdb: a database of ta-siRNA regulatory pathways. Bioinformatics. 2014;30:1045-6
76. Chorostecki U, Palatnik JF. comTAR: a web tool for the prediction and characterization of conserved microRNA targets in plants. Bioinformatics. 2014;30:2066-7
77. Lei J, Sun Y. miR-PREFeR: an accurate, fast and easy-to-use plant miRNA prediction tool using small RNA-Seq data. Bioinformatics. 2014;30:2837-9
78. Gustafson AM, Allen E, Givan S, Smith D, Carrington JC, Kasschau KD. ASRP: the Arabidopsis Small RNA Project Database. Nucleic acids research. 2005;33:D637-40
79. Zhang Z, Yu J, Li D, Zhang Z, Liu F, Zhou X. et al. PMRD: plant microRNA database. Nucleic acids research. 2010;38:D806-13
80. Yang JH, Li JH, Shao P, Zhou H, Chen YQ, Qu LH. starBase: a database for exploring microRNA-mRNA interaction maps from Argonaute CLIP-Seq and Degradome-Seq data. Nucleic acids research. 2011;39:D202-9
81. Szczesniak MW, Deorowicz S, Gapski J, Kaczynski L, Makalowska I. miRNEST database: an integrative approach in microRNA search and annotation. Nucleic acids research. 2012;40:D198-204
82. Szczesniak MW, Makalowska I. miRNEST 2.0: a database of plant and animal microRNAs. Nucleic acids research. 2014;42:D74-7
83. Gudys A, Szczesniak MW, Sikora M, Makalowska I. HuntMi: an efficient and taxon-specific approach in pre-miRNA identification. BMC bioinformatics. 2013;14:83
84. Chalk AM, Warfinge RE, Georgii-Hemming P, Sonnhammer EL. siRNAdb: a database of siRNA sequences. Nucleic acids research. 2005;33:D131-4
85. Dai X, Zhao PX. pssRNAMiner: a plant short small RNA regulatory cascade analysis server. Nucleic acids research. 2008;36:W114-8
86. Bonnet E, He Y, Billiau K, Van de Peer Y. TAPIR, a web server for the prediction of plant microRNA targets, including target mimics. Bioinformatics. 2010;26:1566-8
87. Rehmsmeier M, Steffen P, Hochsmann M, Giegerich R. Fast and effective prediction of microRNA/target duplexes. Rna. 2004;10:1507-17
88. Sun X, Dong B, Yin L, Zhang R, Du W, Liu D. et al. PMTED: a plant microRNA target expression database. BMC bioinformatics. 2013;14:174
89. Zhang S, Yue Y, Sheng L, Wu Y, Fan G, Li A. et al. PASmiR: a literature-curated database for miRNA molecular regulation in plant response to abiotic stress. BMC plant biology. 2013;13:33
90. Peng ZY, Zhou X, Li L, Yu X, Li H, Jiang Z. et al. Arabidopsis Hormone Database: a comprehensive genetic and phenotypic information database for plant hormone research in Arabidopsis. Nucleic acids research. 2009;37:D975-82
91. Jiang Z, Liu X, Peng Z, Wan Y, Ji Y, He W. et al. AHD2.0: an update version of Arabidopsis Hormone Database for plant systematic studies. Nucleic acids research. 2011;39:D1123-9
92. Liu H, Jin T, Liao R, Wan L, Xu B, Zhou S. et al. miRFANs: an integrated database for Arabidopsis thaliana microRNA function annotations. BMC plant biology. 2012;12:68
93. Nakano M, Nobuta K, Vemaraju K, Tej SS, Skogen JW, Meyers BC. Plant MPSS databases: signature-based transcriptional resources for analyses of mRNA and small RNA. Nucleic acids research. 2006;34:D731-5
94. Brown JW, Echeverria M, Qu LH, Lowe TM, Bachellerie JP, Huttenhofer A. et al. Plant snoRNA database. Nucleic acids research. 2003;31:432-5
95. Bazzini AA, Asis R, Gonzalez V, Bassi S, Conte M, Soria M. et al. miSolRNA: A tomato micro RNA relational database. BMC plant biology. 2010;10:240
96. Meng Y, Gou L, Chen D, Mao C, Jin Y, Wu P. et al. PmiRKB: a plant microRNA knowledge base. Nucleic acids research. 2011;39:D181-7
97. Fasold M, Langenberger D, Binder H, Stadler PF, Hoffmann S. DARIO: a ncRNA detection and analysis tool for next-generation sequencing experiments. Nucleic acids research. 2011;39:W112-7
98. Patra D, Fasold M, Langenberger D, Steger G, Grosse I, Stadler PF. plantDARIO: web based quantitative and qualitative analysis of small RNA-seq data in plants. Frontiers in plant science. 2014;5:708
99. Muller S, Rycak L, Winter P, Kahl G, Koch I, Rotter B. omiRas: a Web server for differential expression analysis of miRNAs derived from small RNA-Seq data. Bioinformatics. 2013;29:2651-2
100. Amaral PP, Clark MB, Gascoigne DK, Dinger ME, Mattick JS. lncRNAdb: a reference database for long noncoding RNAs. Nucleic acids research. 2011;39:D146-51
101. Quek XC, Thomson DW, Maag JL, Bartonicek N, Signal B, Clark MB. et al. lncRNAdb v2.0: expanding the reference database for functional long noncoding RNAs. Nucleic acids research. 2015;43:D168-73
102. Liu C, Bai B, Skogerbo G, Cai L, Deng W, Zhang Y. et al. NONCODE: an integrated knowledge database of non-coding RNAs. Nucleic acids research. 2005;33:D112-5
103. He S, Liu C, Skogerbo G, Zhao H, Wang J, Liu T. et al. NONCODE v2.0: decoding the non-coding. Nucleic acids research. 2008;36:D170-2
104. Bu D, Yu K, Sun S, Xie C, Skogerbo G, Miao R. et al. NONCODE v3.0: integrative annotation of long noncoding RNAs. Nucleic acids research. 2012;40:D210-5
105. Xie C, Yuan J, Li H, Li M, Zhao G, Bu D. et al. NONCODEv4: exploring the world of long non-coding RNA genes. Nucleic acids research. 2014;42:D98-103
106. Zhao Y, Li H, Fang S, Kang Y, Wu W, Hao Y. et al. NONCODE 2016: an informative and valuable data source of long non-coding RNAs. Nucleic acids research. 2016;44:D203-8
107. Kong L, Zhang Y, Ye ZQ, Liu XQ, Zhao SQ, Wei L. et al. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic acids research. 2007;35:W345-9
108. Kang YJ, Yang DC, Kong L, Hou M, Meng YQ, Wei L. et al. CPC2: a fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic acids research. 2017
109. Yuan J, Wu W, Xie C, Zhao G, Zhao Y, Chen R. NPInter v2.0: an updated database of ncRNA interactions. Nucleic acids research. 2014;42:D104-8
110. Hao Y, Wu W, Li H, Yuan J, Luo J, Zhao Y. et al. NPInter v3.0: an upgraded database of noncoding RNA-associated interactions. Database: the journal of biological databases and curation. 2016:2016
111. Sun L, Luo H, Bu D, Zhao G, Yu K, Zhang C. et al. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic acids research. 2013;41:e166
112. Hu L, Xu Z, Hu B, Lu ZJ. COME: a robust coding potential calculation tool for lncRNA identification and characterization based on multiple features. Nucleic acids research. 2017;45:e2
113. Wang L, Park HJ, Dasari S, Wang S, Kocher JP, Li W. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic acids research. 2013;41:e74
114. Mistry J, Bateman A, Finn RD. Predicting active site residue annotations in the Pfam database. BMC bioinformatics. 2007;8:298
115. Lin MF, Jungreis I, Kellis M. PhyloCSF: a comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics. 2011;27:i275-82
116. Xuan H, Zhang L, Liu X, Han G, Li J, Li X. et al. PLNlncRbase: A resource for experimentally identified lncRNAs in plants. Gene. 2015;573:328-32
117. Jin J, Liu J, Wang H, Wong L, Chua NH. PLncDB: plant long non-coding RNA database. Bioinformatics. 2013;29:1068-71
118. Yi X, Zhang Z, Ling Y, Xu W, Su Z. PNRD: a plant non-coding RNA database. Nucleic acids research. 2015;43:D982-9
119. Zhou Z, Shen Y, Khan MR, Li A. LncReg: a reference resource for lncRNA-associated regulatory networks. Database: the journal of biological databases and curation. 2015:2015
120. Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic acids research. 2003;31:3406-15
121. Andronescu M, Aguirre-Hernandez R, Condon A, Hoos HH. RNAsoft: A suite of RNA secondary structure prediction and design software tools. Nucleic acids research. 2003;31:3416-22
122. Janssen S, Giegerich R. The RNA shapes studio. Bioinformatics. 2015;31:423-5
123. Kaiser A, Kruger J, Evers DJ. RNA Movies 2: sequential animation of RNA secondary structures. Nucleic acids research. 2007;35:W330-4
124. Washietl S, Hofacker IL, Stadler PF. Fast and reliable prediction of noncoding RNAs. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:2454-9
125. Gruber AR, Neubock R, Hofacker IL, Washietl S. The RNAz web server: prediction of thermodynamically stable and evolutionarily conserved RNA structures. Nucleic acids research. 2007;35:W335-8
126. Nawrocki EP, Eddy SR. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 2013;29:2933-5
127. Chu Q, Zhang X, Zhu X, Liu C, Mao L, Ye C. et al. PlantcircBase: A Database for Plant Circular RNAs. Molecular plant. 2017;10:1126-8
128. Zhang P, Meng X, Chen H, Liu Y, Xue J, Zhou Y. et al. PlantCircNet: a database for plant circRNA-miRNA-mRNA regulatory networks. Database: The Journal of Biological Databases and Curation. 2017:2017
129. Chen D, Yuan C, Zhang J, Zhang Z, Bai L, Meng Y. et al. PlantNATsDB: a comprehensive database of plant natural antisense transcripts. Nucleic acids research. 2012;40:D1187-93
130. Consortium RN. RNAcentral: an international database of ncRNA sequences. Nucleic acids research. 2015;43:D123-9
131. The RC. RNAcentral: a comprehensive database of non-coding RNA sequences. Nucleic acids research. 2017;45:D128-D34
132. Szymanski M, Erdmann VA, Barciszewski J. Noncoding regulatory RNAs database. Nucleic acids research. 2003;31:429-31
133. Szymanski M, Erdmann VA, Barciszewski J. Noncoding RNAs database (ncRNAdb). Nucleic acids research. 2007;35:D162-4
134. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114-20
135. Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet journal. 2011;17:pp. 10-2
136. Bao W, Kojima KK, Kohany O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mobile DNA. 2015;6:11
137. Ouyang S, Buell CR. The TIGR Plant Repeat Databases: a collective resource for the identification of repetitive sequences in plants. Nucleic acids research. 2004;32:D360-3
138. Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome biology. 2009;10:R25
139. Lorenz R, Bernhart SH, Honer Zu Siederdissen C, Tafer H, Flamm C, Stadler PF. et al. ViennaRNA Package 2.0. Algorithms for molecular biology: AMB. 2011;6:26
140. Zheng Y, Li YF, Sunkar R, Zhang W. SeqTar: an effective method for identifying microRNA guided cleavage sites from degradome of polyadenylated transcripts in plants. Nucleic acids research. 2012;40:e28
141. Brueffer C, Saal LH. TopHat-Recondition: a post-processor for TopHat unmapped reads. BMC bioinformatics. 2016;17:199
142. Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nature methods. 2012;9:357-9
143. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15-21
144. Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nature biotechnology. 2011;29:644-52
145. Finn RD, Attwood TK, Babbitt PC, Bateman A, Bork P, Bridge AJ. et al. InterPro in 2017-beyond protein family and domain annotations. Nucleic acids research. 2017;45:D190-D9
Author contact
![]() Corresponding author: YZ (zhengyun5488com)
Corresponding author: YZ (zhengyun5488com)