International Journal of Biological Sciences
ISSN: 1449-2288
10
Impact Factor
ISSN: 1449-2288
- Current Issue
- Volume 21; 2025
- Volume 20; 2024
- Volume 19; 2023
- Volume 18; 2022
- Volume 17; 2021
- Archive
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Top
Introduction
Implementation
Results and Discussion
Conclusion
Abbreviations
Acknowledgements
References
Introduction
Implementation
Results and Discussion
Conclusion
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(8):843-848. doi:10.7150/ijbs.24626 This issue Cite
Research Paper
GeneFuse: detection and visualization of target gene fusions from DNA sequencing data
Shifu Chen1,2 ![]() , Ming Liu1, Tanxiao Huang1, Wenting Liao1, Mingyan Xu1, Jia Gu2
, Ming Liu1, Tanxiao Huang1, Wenting Liao1, Mingyan Xu1, Jia Gu2
1. HaploX Biotechnology, Shenzhen, China.
2. Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences, Shenzhen, China.
Received 2017-12-28; Accepted 2018-3-6; Published 2018-5-22
Citation:
Chen S, Liu M, Huang T, Liao W, Xu M, Gu J. GeneFuse: detection and visualization of target gene fusions from DNA sequencing data. Int J Biol Sci 2018; 14(8):843-848. doi:10.7150/ijbs.24626. https://www.ijbs.com/v14p0843.htm
Other stylesAbstract

In recent years, gene fusion detection for cancer treatment has become increasingly important since more therapeutic agents have been developed to suppress fusion kinases. Although a number of tools have been developed to detect gene fusions from DNA sequencing data, most of them are not sensitive enough for processing the data from the samples with low tumor DNA composition, like cell-free tumor DNA. In this paper, we will introduce GeneFuse, a tool to detect and visualize gene fusions with high sensitivity and specificity. GeneFuse focuses on the curated gene fusions, which are available in COSMIC (the Catalogue of Somatic Mutations in Cancer) database. For each detected fusion, GeneFuse reports its genome locus, inferred protein forms, and supporting sequencing reads. The fusion detection results are visualized in an HTML page for cloud-friendly validation. GeneFuse is an open source tool available at GitHub:
Keywords: GeneFuse, gene fusion, fusion detection, fusion visualization
Introduction
Fusion genes are chimeras of two different genes that originated from genomic structural rearrangements, transcription read-through, or abnormal RNA splicing [1-3]. In recent years, the landscape of gene fusions in many cancer types has been elucidated with the development of next generation sequencing (NGS). The gene fusion events are quite common in prostate, lymphoid, soft tissue, breast, gastric, and lung cancers [4-10]. Many of them are associated with oncogenesis and tumor progression, and some even are identified as driver mutations among several cancer types [11-12]. The enrichment of knowledge in this aspect further facilitates clinical cares. Some fusion genes are good biomarkers for cancer diagnosis, prognosis, and treatment guidance. The BCR-ABL, TMPRSS2-ERG, EML4-ALK, and KIF5B-RET are good examples [13-16].
To date, several algorithms and tools have been developed to detect gene fusions from NGS data. Some of them rely on the entire or targeted genome sequencing method, while others depend on the RNA-seq [17-19]. Both of them need the alignment step, where sequencing reads are aligned to a reference using mapping tools such as Burrows Wheeler Alignment (BWA) [20] and Bowtie [21]. For example, FACTERA [18] first discovers improperly paired reads from the alignment result, then clusters the closet exons of discordant reads into distinct gene-gene groups, and finally finds the breakpoints to locate gene fusions. DELLY [22] is another structural variant detector that can discover gene fusions from BAM files. First, it implements paired-end mapping analysis from the alignment result to find read pairs with abnormal orientation or insert size. Secondly, the identified paired-end clusters are interpreted as breakpoint-containing genomic intervals, which are screened for split-read support to map the genomic rearrangements at single-nucleotide resolution. Finally DELLY will merge the supporting read pairs and annotate them against the reference genome. The mapping-based gene fusion detectors have several advantages. For example, they can scan for all possible gene fusions and are able to detect novel ones.
However, the mapping-based detectors also have disadvantages since their detection results are heavily dependent on the alignment results output by the sequence aligner. If the aligner cannot detect accurate clips and chimeras, the mapping-based fusion detection algorithms may not work properly. However, misalignments can happen often for the reads containing fusions. On the other hand, clips and chimeras can also happen often for the normal reads that don't contain any fusions. These factors can affect the sensitivity and specificity of these fusion callers. False positives can happen often at repetitive regions. Meanwhile false negatives can also happen often when they process data from the samples with low tumor DNA composition, like cell-free tumor DNA.
For clinical applications, instead of finding a lot of gene fusions with unknown clinical significance and large uncertainty, it is better to search for gene fusions known to be responsive for clinical treatments. Motivated by the need to detect clinical significant gene fusions with high sensitivity and specificity, we developed GeneFuse, which can directly detect gene fusions from the raw FASTQ files to eliminate the affect of alignment result. GeneFuse only focuses on the fusion genes with known clinical significance, which can be found from the COSMIC (the Catalogue of Somatic Mutations in Cancer) database.
GeneFuse is also able to visualize the detected fusions by rendering them with the supporting reads and inferred fusion protein structures. The novel fusion visualization can improve the interpretability of the results, and is important for experienced bioinformatician and data interpreter to manually validate the fusions. For each detected fusion, GeneFuse reports its genome locus, inferred protein forms, and supporting sequencing reads. The fusion detection results are visualized in an HTML page for cloud-friendly validation.
Implementation
The basic idea of GeneFuse is to search for the reads that can be well mapped to two different genes for its left part and right part, but cannot be entirely mapped to any position of the whole reference genome. A read that matches the two genes of a fusion at its fusion point is called a supporting read, and the duplicated supporting reads of each fusion will be grouped as a single unique supporting read. A fusion will be qualified if it has enough unique supporting reads. The overall design and algorithm components will be presented in following sections.
Overall Design
The program flow of GeneFuse can be divided into four major steps: indexing, matching, filtering, and reporting. Fig. 1 demonstrates how GeneFuse works.
Indexing
A CSV file, which lists the genome regions of target fusion genes and their exons, is needed to extract gene sequences from reference genome. GeneFuse has provided two CSV files giving the curated gene fusion lists from COSMIC database for hg19/GRCh37 and hg38/GRCh38 respectively. COSMIC is one of the most complete databases, providing almost all of the validated human cancer gene fusions. Although COSMIC database is sufficient for most clinical research, GeneFuse still provides a gene list generation utility to customize the target fusion genes.
GeneFuse will extract the sequences from the reference genome within the fusion gene regions. A k-mer (all possible substrings of length k, k=16 in this implementation) of all these sequences will be computed, and each element of the k-mer is associated with a list of genome coordinates that it matches. A hashmap will be used to store the association between k-mer and the genome coordinate, and it will be used for mapping a read to the target genes.
Matching
In the matching step, a set of sequences is computed for each read by collecting its all subsequences with a length of k. Then the associated genes of this read can be found by mapping the subsequences to the genome coordinate using the index computed in the last step. If the left part and right part of a read can be mapped to two different genes, the read will be segmented to two regions. The read will be considered as a match candidate if its left region and right region are both long enough (Tregion = 20 by default), and simultaneously meets such condition that the bases that are out of both regions are less than a certain threshold (Tummapped = 10 by default). All of the fusion match candidates will be stored in a list and will be filtered in the next step.
Sequence length is also a factor that affects mutation detection. To obtain a longer sequence, GeneFuse tries to merge each pair of reads for paired-end sequencing data. For a read pair R1 and R2, rcR2 is computed as the reverse complement of R2. The merging algorithm searches for the largest overlap of R1 and rcR2, while their overlapped subsequences are entirely identical. If the overlapped region is longer than a threshold (by default, Tlen = 30 bp), we consider them as overlapped and merge them to a single read. We can obtain longer sequences after merging read pairs, and continue the matching process even if the mutation point locates on the edge of reads. If one pair of reads cannot be merged, GeneFuse will process them. Although a sequencing library with large insert sizes would prohibit the overlap of read pairs, it will not cause a significant impact on performance since GeneFuse can individually process a read pair as two single-end reads.
Filtering
Once the fusion match candidate list is prepared, enumerating all subsequences of the reads supporting the fusions forms a new k-mer. Then the entire reference genome will be scanned for searching the same k-mer elements, and the matched genome coordinates will similarly be stored to build a new global index G. For each read in the fusion match candidate list, it will be mapped to G to check whether it can be well aligned to a reference genome. If a read could be mapped to a reference genome, it is removed from the fusion match candidate list.
Other filters - like low complexity filter and match quality filter - will also be applied to eliminate false callings. Furthermore, if one read is mapped to two segments of one single gene, it will be treated as a deletion, and removed if the deletion length is too short.
Fig 1
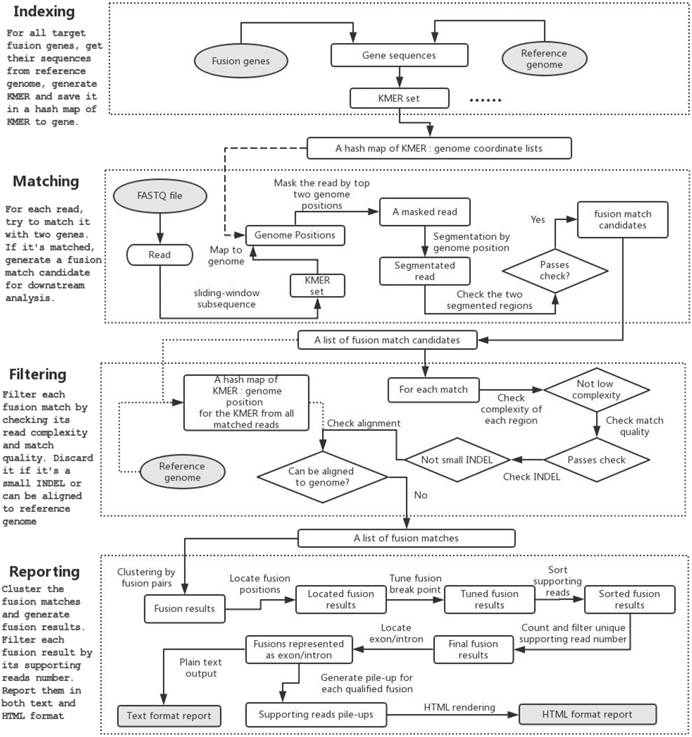
The program flow of GeneFuse. Four steps are included in the workflow: indexing, matching, filtering, and reporting. In the indexing step, a hashmap of mapping to genes is computed. In the matching step, reads are mapped to the genes using the computed hashmap, and those that can be mapped to two genes are saved to fusion matches. In the filtering step, each fusion match is filtered by its read complexity, match quality, and other factors. Finally, in the reporting step, the detected fusions are validated, and the supporting reads for each fusion are piled up and rendered to an HTML page. The input and output files are then highlighted in grey.
Reporting
In the reporting step, the fusion matches will first be clustered as fusion results by the fusion points. For each fusion result, its fusion position will be located and its breakpoint will be tuned to find a better separation of fused reads. The supporting reads for each fusion result will then be sorted and the duplicated reads will be grouped as a single unique read. Each fusion result's unique supporting read number will be compared to a threshold (Tunique = 2 by default), and the ones passing the filter will be considered as qualified fusions, for which the exon or intron of the fusion breakpoint will be located.
GeneFuse provides reports in two formats: plain text report and HTML report. The text format report is a standard FASTA file, which provides the fusion information in the comment lines and the read sequences in the sequence lines. This FASTA file is very convenient for further validation using sequence-searching systems like BLAST. For HTML report, all the supporting reads of each fusion are piled up and all the fusions are rendered in a single HTML page. Fig. 2 demonstrates a result of a CD74-ROS1 fusion.
Results and Discussion
We conducted some experiments to compare GeneFuse with FACTERA and DELLY since they are two widely used gene fusion callers.
Sensitivity and specificity
To evaluate the performance of GeneFuse, we applied it to 10 NSCLC cell-free DNA samples covering our 1.6 Mb custom panel, six of which harboring known rearrangements (EML4:exon6-ALK exon20; EML4:exon13-ALK exon20) confirmed by digital droplet PCR (ddPCR), and GeneFuse was able to detect all of them. On the contrary, none could be detected in the four ALK wild-type samples, yielding a sensitivity and specificity of 100 % for both in detecting the ALK fusion events. We tested the same dataset with FACTERA v1.4.4 and DELLY v0.7.6. The results are shown in Table 1.
Speed evaluation
To evaluate the speed of GeneFuse, we conducted an experiment with 13 different FASTQ paired-end files. Since FACTERA and DELLY require sorted BAM files as input, we also recorded the alignment time and BAM sorting time, which were performed with BWA and Picard, respectively. GeneFuse was run with almost all the druggable gene fusions including the major forms of ALK, ROS1, RET, NTRK1, NTRK3, and BCR-ABL1 fusions. The result showed that GeneFuse took much less time than (BWA + Picard + FACTERA) or (BWA + Picard + DELLY). The results are shown in Table 2.
Table 1
The results of GeneFuse in detecting the EML4-ALK fusion events in 10 cfDNA samples compared to DELLY and FACTERA. With the ddPCR result as the golden standard, it was observed that GeneFuse had the highest sensitivity.
| Sample ID | Fusion type | ddPCR | GeneFuse | DELLY | FACTERA |
|---|---|---|---|---|---|
| cfDNA_001 | EML4:exon6-ALK exon20 | detected | detected | detected | detected |
| cfDNA_001 | EML4:exon13-ALK exon20 | detected | detected | detected | detected |
| cfDNA_002 | Wild Type | Not detected | Not detected | Not detected | Not detected |
| cfDNA_003 | Wild Type | Not detected | Not detected | Not detected | Not detected |
| cfDNA_004 | Wild Type | Not detected | Not detected | Not detected | Not detected |
| cfDNA_005 | Wild Type | Not detected | Not detected | Not detected | Not detected |
| cfDNA_006 | EML4:exon6-ALK exon20 | detected | detected | detected | detected |
| cfDNA_006 | EML4:exon13-ALK exon20 | detected | detected | Not detected | detected |
| cfDNA_007 | EML4:exon6-ALK exon20 | detected | detected | detected | detected |
| cfDNA_007 | EML4:exon13-ALK exon20 | detected | detected | Not detected | detected |
| cfDNA_008 | EML4:exon6-ALK exon20 | detected | detected | detected | detected |
| cfDNA_008 | EML4:exon13-ALK exon20 | detected | detected | Not detected | detected |
| cfDNA_009 | EML4:exon6-ALK exon20 | detected | detected | Not detected | detected |
| cfDNA_009 | EML4:exon13-ALK exon20 | detected | detected | Not detected | Not detected |
| cfDNA_010 | EML4:exon6-ALK exon20 | detected | detected | detected | detected |
| cfDNA_010 | EML4:exon13-ALK exon20 | detected | detected | detected | detected |
Table 2
The speed evaluation result of GeneFuse against FACTERA and DELLY. The file size in the first column is the sum of read1 and read2 base numbers. BWA-MEM was run with 4 threads, while GeneFuse was also run with 4 threads. The druggable targets can be found from the GeneFuse github repository.
| File size (bases) | BWA MEM | Picard sort | FACTERA step | DELLY step | BWA+Picard+FACTERA | BWA+Picard+DELLY | GeneFuse with druggable targets |
|---|---|---|---|---|---|---|---|
| 6.48 G | 0:28:44 | 0:11:15 | 0:05:38 | 0:04:52 | 0:45:37 | 0:45:51 | 0:05:30 |
| 7.26 G | 0:30:10 | 0:11:59 | 0:06:00 | 0:05:09 | 0:48:09 | 0:48:18 | 0:05:39 |
| 9.13 G | 0:49:37 | 0:17:51 | 0:05:43 | 0:08:23 | 1:13:11 | 1:16:51 | 0:08:26 |
| 7.48 G | 0:41:32 | 0:13:54 | 0:06:47 | 0:11:18 | 1:02:13 | 1:07:44 | 0:07:25 |
| 7.33 G | 0:40:52 | 0:14:04 | 0:06:46 | 0:10:59 | 1:01:42 | 1:06:55 | 0:07:34 |
| 7.19 G | 0:43:11 | 0:14:46 | 0:03:35 | 0:05:22 | 1:01:32 | 1:04:19 | 0:07:00 |
| 7.38 G | 1:01:12 | 0:13:39 | 0:09:51 | 0:10:50 | 1:24:42 | 1:26:41 | 0:09:47 |
| 7.80 G | 1:00:45 | 0:14:46 | 0:06:42 | 0:07:17 | 1:22:13 | 1:23:48 | 0:10:07 |
| 7.46 G | 0:54:54 | 0:14:08 | 0:06:34 | 0:08:45 | 1:15:36 | 1:18:47 | 0:09:50 |
| 8.14 G | 1:05:04 | 0:14:56 | 0:08:20 | 0:08:42 | 1:28:20 | 1:29:42 | 0:10:45 |
| 8.53 G | 0:52:06 | 0:15:58 | 0:03:43 | 0:03:19 | 1:11:47 | 1:12:23 | 0:07:57 |
| 9.75 G | 0:48:30 | 0:18:04 | 0:04:27 | 0:04:11 | 1:11:01 | 1:11:45 | 0:08:55 |
| 9.42 G | 0:47:52 | 0:17:46 | 0:06:03 | 0:04:25 | 1:11:41 | 1:11:03 | 0:09:27 |
Fig 2

A screenshot of a GeneFuse's pile-up result. The demonstrated fusion is CD74-ROS1, which is an important druggable target for lung cancer. From the title, we can find that it is the third detected fusion in this report. The inferred fusion protein below the title shows it has 3 exons from CD74 gene, and 33 exons from ROS1 gene. The supporting reads are presented in a table, and the fusion breakpoint is given in the first row of the table, while the reference sequences are given in the second row. For each supporting read, the color of its bases indicates the quality score (green and blue indicate high quality, red indicates low quality). An online report can be found at http://opengene.org/GeneFuse/report.html
Conclusion
In the clinical application of analyzing cancer sequencing data, it is essential to detect druggable mutations and fusions with low MAF from ultra-deep sequencing data. The existing tools, like DELLY and FACTERA, are not sensitive enough, and are lacking the function of visualizing detected fusions. As a fast and lightweight tool aimed at detecting target gene fusions from raw FASTQ data, GeneFuse has high sensitivity and can visualize detected fusions by generating HTML-based read pile-up visualizations. It will further put the gene fusion testing to clinical applications.
Abbreviations
k-mer: all possible substrings of length k; ctDNA: circulating tumor DNA; NGS: next-generation sequencing; INDEL: insertion and deletion; CD74: gene name for HLA-DR antigens-associated invariant chain or CD74 (Cluster of Differentiation 74); ROS1: a receptor tyrosine kinase (RTK) of the insulin receptor family; MFH: Malignant Fibrous Histiocytoma; ALK: the anaplastic lymphoma kinase; EML4: echinoderm microtubule-associated protein-like 4; COSMIC: the Catalogue Of Somatic Mutations In Cancer.
Acknowledgements
This study was partially financed by the National Science Foundation of China (NSFC Project No. 61472411), Special Funds for Future Industries of Shenzhen (Project No. JSGG20160229123927512) and the SZSTI Entrepreneurship Funds of Shenzhen (Project No. CYZZ20150527145115656).
Authors' Contributions
Shifu Chen developed this tool and wrote the paper; Ming Liu did the background survey about similar tools; Tanxiao Huang, Wenting Liao, and Mingyan Xu performed the experiments; Jia Gu co-supervised this project.
Availability of Data and Materials
The code project of GeneFuse is available at: https://github.com/OpenGene/GeneFuse and the dataset to test GeneFuse can be downloaded from: http://opengene.org/dataset.html
Competing Interests
The authors have declared that no competing interest exists.
References
1. Weckselblatt B, Rudd M K. Human structural variation: mechanisms of chromosome rearrangements[J]. Trends in Genetics. 2015;31(10):587-599
2. Nacu S, Yuan W, Kan Z. et al. Deep RNA sequencing analysis of readthrough gene fusions in human prostate adenocarcinoma and reference samples[J]. BMC medical genomics. 2011;4(1):11
3. Jividen K, Li H. Chimeric RNAs generated by intergenic splicing in normal and cancer cells[J]. Genes, Chromosomes and Cancer. 2014;53(12):963-971
4. Yoshihara K, Wang Q, Torres-Garcia W. et al. Mertens F, Johansson B, Fioretos T, et al. The emerging complexity of gene fusions in cancer[J]. Nature Reviews Cancer. 2015;15(6):371-381
5. Lilljebjörn H, Ågerstam H, Orsmark-Pietras C. et al. RNA-seq identifies clinically relevant fusion genes in leukemia including a novel MEF2D/CSF1R fusion responsive to imatinib[J]. Leukemia. 2014;28(4):977
6. Mertens F, Antonescu C R, Mitelman F. Gene fusions in soft tissue tumors: recurrent and overlapping pathogenetic themes[J]. Genes, Chromosomes and Cancer. 2016;55(4):291-310
7. Nam R K, Sugar L, Yang W. et al. Expression of the TMPRSS2: ERG fusion gene predicts cancer recurrence after surgery for localised prostate cancer[J]. British journal of cancer. 2007;97(12):1690-1695
8. Robinson D R, Kalyana-Sundaram S, Wu Y M. et al. Functionally recurrent rearrangements of the MAST kinase and Notch gene families in breast cancer[J]. Nature medicine. 2011;17(12):1646-1651
9. Iwakawa R, Takenaka M, Kohno T. et al. Genome-wide identification of genes with amplification and/or fusion in small cell lung cancer[J]. Genes, Chromosomes and Cancer. 2013;52(9):802-816
10. Kim H P, Cho G A, Han S W. et al. Novel fusion transcripts in human gastric cancer revealed by transcriptome analysis[J]. Oncogene. 2014;33(47):5434-5441
11. Latysheva N S, Babu M M. Discovering and understanding oncogenic gene fusions through data intensive computational approaches[J]. Nucleic acids research. 2016;44(10):4487-4503
12. Stransky N, Cerami E, Schalm S. et al. The landscape of kinase fusions in cancer[J]. Nature communications. 2014;5:4846
13. Bleckmann K, Alten J, Moericke A. et al. Clinical Implication of BCR/ABL Fusion Transcript Monitoring in Addition to Ig/TCR Gene Rearrangement-Based Minimal Residual Disease in Philadelphia Chromosome-Positive Childhood Acute Lymphoblastic Leukemia[J]. 2014.
14. Leyten G H J M, Hessels D, Jannink S A. et al. Prospective multicentre evaluation of PCA3 and TMPRSS2-ERG gene fusions as diagnostic and prognostic urinary biomarkers for prostate cancer[J]. European urology. 2014;65(3):534-542
15. Seo S, Woo C G, Lee D H. et al. The clinical impact of an EML4-ALK variant on survival following crizotinib treatment in patients with advanced ALK-rearranged non-small-cell lung cancer[J]. Annals of Oncology. 2017;28(7):1667-1668
16. Drilon A, Wang L, Hasanovic A. et al. Response to Cabozantinib in patients with RET fusion-positive lung adenocarcinomas[J]. Cancer discovery. 2013;3(6):630-635
17. Fan X, Abbott T E, Larson D. et al. BreakDancer: Identification of Genomic Structural Variation from Paired-End Read Mapping[J]. Current Protocols in Bioinformatics. 2014:15 6. 1-15.6. 11
18. Newman A M, Bratman S V, Stehr H. et al. FACTERA: a practical method for the discovery of genomic rearrangements at breakpoint resolution[J]. Bioinformatics. 2014;30(23):3390-3393
19. Beccuti M, Carrara M, Cordero F. et al. The structure of state-of-art gene fusion-finder algorithms[J]. Genome Bioinformatics. 2013;1(1):2
20. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform[J]. Bioinformatics. 2009;25(14):1754-1760
21. Langmead B, Salzberg S L. Fast gapped-read alignment with Bowtie 2[J]. Nature methods. 2012;9(4):357-359
22. Rausch T. et al. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics. 2012;28:333-339
Author contact
![]() Corresponding author
Corresponding author
Citation styles
APA
Chen, S., Liu, M., Huang, T., Liao, W., Xu, M., Gu, J. (2018). GeneFuse: detection and visualization of target gene fusions from DNA sequencing data. International Journal of Biological Sciences, 14(8), 843-848. https://doi.org/10.7150/ijbs.24626.
ACS
Chen, S.; Liu, M.; Huang, T.; Liao, W.; Xu, M.; Gu, J. GeneFuse: detection and visualization of target gene fusions from DNA sequencing data. Int. J. Biol. Sci. 2018, 14 (8), 843-848. DOI: 10.7150/ijbs.24626.
NLM
Chen S, Liu M, Huang T, Liao W, Xu M, Gu J. GeneFuse: detection and visualization of target gene fusions from DNA sequencing data. Int J Biol Sci 2018; 14(8):843-848. doi:10.7150/ijbs.24626. https://www.ijbs.com/v14p0843.htm
CSE
Chen S, Liu M, Huang T, Liao W, Xu M, Gu J. 2018. GeneFuse: detection and visualization of target gene fusions from DNA sequencing data. Int J Biol Sci. 14(8):843-848.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.