Impact Factor ISSN: 1449-2288
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Present Status of Bioinformatics...
The Future Perspectives on...
Conclusions
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(8):920-929. doi:10.7150/ijbs.24622 This issue Cite
Review
Translational Bioinformatics for Cholangiocarcinoma: Opportunities and Challenges
Fuliang Qian1, Junping Guo2, Zhi Jiang 1, Bairong Shen1,3,4 ![]()
1. Center for Systems Biology, Soochow University, Suzhou 215006, China;
2. The Affiliated Yixing Hospital of Jiangsu University, Yixing, 214200, China.
3. Guizhou University School of Medicine, Guiyang, 550025, China
4. Institute for Systems Genetics, West China Hospital, Sichuan University, Chengdu, 610041, China
Received 2017-12-28; Accepted 2018-2-2; Published 2018-5-22
Abstract

Translational bioinformatics is becoming a driven force and a new scientific paradigm for cancer research in the era of big data. To promote the cross-disciplinary communication and research, we take cholangiocarcinoma as an example to review the present status and the future perspectives of the bioinformatics models applied in cancer study. We first summarize the present application of computational methods to the study of cholangiocarcinoma ranged from pattern recognition of biological data, knowledge based data annotation to systems biological level modeling and clinical translation. Then the future opportunities and challenges about database or knowledge base building, novel model developing and molecular mechanism exploring as well as the intelligent decision supporting system construction for the precision diagnosis, prognosis and treatment of cholangiocarcinoma are discussed.
Keywords: cholangiocarcinoma, bioinformatics model, precision diagnosis and prognosis, biomarkers discovery
Introduction
Cholangiocarcinoma (CCA) is a relative rare but the second most common type of primary liver tumor [1]. It is an aggressive malignancy of the biliary epithelium with poor prognosis since it is often diagnosed with clinical symptoms presented at a late stage. The treatment options for this disease are very limited. CCA is heterogeneous and it now can be grouped based on their intrahepatic, perihilar, and distal extrahepatic locations. These subtypes are different in tumor biology, genetics and epigenetics or environmental carcinogens [2].
The pathogenic factors for CCA genesis could be genetic alterations, negative lifestyles, environmental exposures or their interactions [3]. Many genetic mutations in genes like TP53, KRAS, SMAD4, ABCB11, ARID1A, ATP8B, BRAF, IDH1/2, MLL3 and PBRM1 were reported to be associated with the pathogenesis and progression of CCA [4-7]. The liver fluke infestation and nitrosamine exposure are the two main exposure risk factors identified associated with patients in northeastern Thailand where CCA is mostly found [8]. Risk factors for the fluke-negative CCA could be HBV/HCV virus infection, chronic biliary and liver diseases such as, primary sclerosing cholangitis, biliary stone disease, congenital biliary malformations and cirrhosis [8-11]. Diabetes was reported contribute to the risk of intrahepatic CCA (iCCA) [12]. The negative lifestyle for CCA could be alcohol consumption, raw freshwater fish and beef sausage, tobacco use, and so on [13, 14]. The CCA associated environmental toxins include dioxin, vinyl chloride and others [15, 16]. The diversity of these risk factors at all the above levels makes the CCA very heterogeneous. The mutational landscape, the cellular origins and the carcinogenesis of the different CCA subtypes are still unclear.
For the diagnosis and treatment of CCA, it is very challenging to classify the subtypes of CCA, or to make a distinction between CCA and other diseases. For example, it is difficult to distinguish iCCA from metastatic adenocarcinoma, especially gastric adenocarcinoma and pancreatic adenocarcinoma [17]. Since the evolutionary origins of CCA are not clearly identified and the heterogeneity of CCA can be caused by the dynamic interaction between many risk factors, no efficient therapeutic targets are screened at present [18-23]. Many open questions are thereby needed to be investigated for the future precision medicine of CCA. In the post-genome era, it becomes cheaper than before to sequence cancer genomes and to measure the alteration in gene expression. Many bio-techniques like deep sequencing, single cell sequencing, CRISPR, etc.[24-26] are developed as efficient tools for the deep investigation of the complex mechanisms of complex disease like CCA. The accumulation of big sequencing data and other clinical data provides the opportunity, but also the challenge, to build bioinformatics models for early diagnosis and personalized treatment, even prevention of cancers like CCA and others.
Bioinformatics is an interdisciplinary field matured with the initialization of human genome project. It plays key roles in the analysis of high throughput data, such as genome data for computational gene prediction and the microarray data for gene expression profiles. Bioinformatics is now facing many new challenges in the translational research. Translational bioinformatics was proposed and developed as a new field recently [27-29]. Big data driven knowledge discovery is the new paradigm for translational medicine [30]. Biological and medical data are characterized with diversity, dynamics, association, inter-connection and evolution. The analyzing of these data is cross-disciplinary and often needs domain knowledge from basic life science to clinical medicine. Furthermore, the informatics skills such as, computational programming, database building, statistics inference, mathematics simulation and complex model developing are required. The omics data at the genomic, epigenomic, transcriptomic, and proteomic as well as metabolomic levels can only be rationally analyzed in-depth with the cooperation between biologists and data scientists. The translation from the biological findings to the clinical applications is still a big challenge since most of the biological findings are based on the study of cell-lines or animal models. When applied these findings to heterogeneous patients, it is often not successful [31, 32]. Comparing to the animal models, the molecular mechanisms for different patients are very different and diverse caused by their different genetics, lifestyles and environmental exposures. It is clear that the deep cooperation between bioinformatics, biologists and clinical doctors is necessary to make the progress of big-data driven precision medicine.
We here take the CCA as an example and summarize the current applications of bioinformatics to the understanding of CCA in all aspects from basic research to clinical applications. In addition, we discuss the future perspectives on the application of bioinformatics to CCA study, such as the actionable risk factors identification, the systems biological level pattern recognition, the model based high-risk population screening and the biomarker discovery for precision classification and therapeutics of CCA.
Present Status of Bioinformatics Applications in Cholanogiocarcinoma
The general pipeline of bioinformatics applications in the complex disease investigation is scratched in Figure 1. The first step for bioinformatics study is to collect related data based on the concrete biological question. Sometimes a database or knowledge base building is necessary. As shown in Figure 1, the biological data types are diverse and they could be all kinds of molecular omics data or clinical phenotypic data. These data need to be cleaned, standardized for the storage and next step analysis. If the data need to be further shared in a community for cohort analysis, the ontologies are often developed for the widely sharing and cooperation. The data sharing and standardization are often complex especially data are generated from different research groups with different technologies. Markup languages are often developed for standardization and sharing of data to the research society. Systems biology markup language (SBML), microarray gene expression markup language (MAGE-ML) and carbohydrate sequence markup language (CabosML) etc. have been developed and widely applied to the bioinformatics researches [33-36].
The pipeline of bioinformatics application: from biological data to biomedical discovery
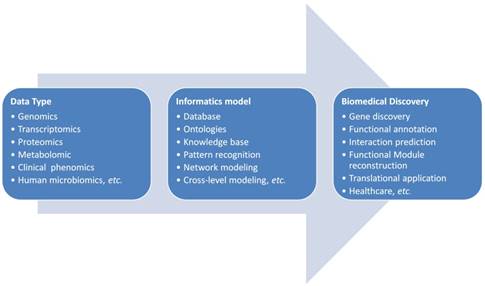
After the data collection and standardization, a biological specific model will be the next step to discover the knowledge in this data. In the past two decades, many models were developed and applied to the complex disease studies. We here summarize these applications to different categories according to the principle of informatics models.
Biological data analyses based on pattern recognition
Biological data have many types, such as DNA/RNA and protein that are often represented with strings formed by four nucleotides or twenty amino acids, the gene expression is described with a series of real values and the proteomics or metabolism data can be presented with mass spectrometry signals. All these data could have specific data patterns which can be extracted and used by biologists to answer special biological questions. Table 1 lists some of the examples applied in CCA studies, which use the pattern recognition principle as the model for biology discovery.
Pattern recognition in biological data
| Application | Method | Discovery | Ref. |
|---|---|---|---|
| miRNA-lncRNA interaction | Base pairing based prediction of miRNA-lncRNA interaction | LncRNA -CCAT1 inhibits miR152 and acts as an oncogene in intra-hepatic cholangiocarcinoma (iCCA). | [37] |
| Base pairing based prediction of miRNA-lncRNA interaction | LncRNA -PCAT1 inhibits miR122 and regulates WNT/β-catenin signaling pathway extra-hepatic cholangiocarcinoma (eCCA). | [38] | |
| miRNA-mRNA interaction | miRNA target prediction based on several database | miR-26a is a potential tumor suppressor of CCA via regulating of KRT19 which is the key biomarkers for distinguishing CCA and hepatocellular carcinoma | [39] |
| miRNA target prediction and signal pathways analysis | miRNA-410 directly targets the X-linked inhibitor of apoptosis protein (XIAP) and acts as an anti-apoptotic regulator of CCA. | [40] | |
| miRNA target prediction | Targets of miR-101 were predicted and validated as a tumor suppressor for CCA | [41] | |
| lncRNA-mRNA interaction | Base pairing based prediction of lncRNA- mRNA interaction and co-expression analysis | CPS1 and lncRNA (CPS1-IT1) may be potential prognostic indicators for patients with ICC. | [42] |
| Mass spectrometry data analysis | Proteomic mass spectrometry pattern analysis with MASCOT | Increased EXT1 expression in plasma is found associated with CCA genesis | [43] |
| HBV integration into cellular genome of hilar CCA (HCCA) | DNASIS MAX is used to sequence analysis of viral-host junction | HBV integration is highly detected in the cancer related genes of HCCA and indicate that HBCV infection may be related to HCCA pathogenesis | [44] |
In the early stage of human genome project implementation, the gene finding is often based on the genetic patterns hidden in the genomics, and then when microRNAs are found as an important family for controlling of biological processes, the microRNA-mRNA complementary patterns are used to predict the interaction between microRNA and other types of RNAs. The principle can be also used for peptide vaccine design and neo-epitope prediction based on the patterns in sequence of virus proteins [45, 46]. Huge amount of bioinformatics analyses are pattern recognition in all kinds of biological data. The applications of this principle for modeling of CCA could be more in the future when the clinical scientists can map the idea to their biological data for specific questions.
Knowledge-based model for biological function annotation
Knowledge based model is the most successful one applied to the complex disease study. Two knowledge bases are often used in the enrichment analysis of biological data. One is the Kyoto Encyclopedia of Genes and Genomes (KEGG), which can be used to identify pathways important to a concrete biological system to understand the biologyical functions [47]. Another one is the gene ontology (GO) database, which is useful to the understanding of biological processes, molecular functions and sub-cellular localization of the studied systems [48].
As the complex diseases like cancers are always heterogeneous, the differentially expressed genes (DEG) may be different at the gene level, but the DEG could be enriched in a common pathway or a biological process or a sub-cellular location [49-51]. The enrichment analysis is very powerful to the exploring of gene set level mechanisms, such as the pathway, module or network at the systems level.
Connectivity Map (CMap) is another useful knowledge base for pharmacogenomics and drug design. It links together the small molecules, gene expression and diseases. CMap could be used to explore the molecular pathways of a specific drug functions on a disease. The connective information is also useful to the discovery of new molecules as putative drugs if it can reverse the altered gene expression in the complex disease to the normal state of expression. As listed in Table 2, this strategy was also applied to CCA drug discovery, and the HSP90 inhibitor, NVP-AUY922 was predicted effective to CCA [52]. Ontology or XML based knowledge base will be very useful to the future personalized medicine. The knowledge base works like a dictionary connecting different genotypes, phenotypes, drugs or lifestyles and could provide a powerful tool for the precision mapping of diseases to specific gene signatures which is important to the diagnosis and treatment of the disease. Some of the examples applied knowledge base to annotation of the CCA data are listed in Table 2.
Knowledge based analyses of biological data
| Knowledge base | Methods | Discovery | Ref. |
|---|---|---|---|
| Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Ontology (GO) | Enrichment analysis of miRNA targets. | Over-expression of miR-150-5p could inhibit proliferation, migration, and invasion capability of CCA cells and indicate it contributes to the CCA development and progression. | [53] |
| Genome-MuSiC was used to mapping the mutations to the KEGG and NCI pathways and the pathways were clustered and statistically analyzed based on the mutation frequencies. | Several important pathways were identified were identified altered in ICC | [54] | |
| Connectivity map (CMap), it includes the relationship for drugs, genes and diseases. | A candidate drug is supposed to reverse the gene expression signature of CCA | The HSP90 inhibitor, NVP-AUY922, is screened as a putative effective CCA drug. | [52] |
| Interleukin-6 associated genomic signature in Mz-ChA-1 human malignant cholangiocytes was derived and compounds that induced inverse gene changes to the signature were screened. | Nitrendipine, nifedipine and felodipine that are structurally similar compounds were identified cytotoxic to Mz-ChA-1 cells and could be the potential therapeutic use for CCA. | [55] |
Network and integrative systems biology as the paradigm
Since the phenotypes, especially the clinical phenotypes are caused by interaction among different genes, lifestyles and experimental exposures. The network reconstruction for gene regulation [56, 57], protein-protein interaction (PPI), as well as the integration of disease specific risk factors is a systems biological paradigm for the investigation of complex disease. Based on the network analysis, we could screen diagnosis biomarkers, driver genes and mutations for complex diseases [58-61]. Table 3 and Table 4 list the examples of CCA mechanism studies with network and integrative analysis of gene expression data, copy number variation data and other biological information.
Network level discovery and functional investigation
| Network type | Construction method | Discovery | Ref. |
|---|---|---|---|
| lncRNA-miRNA co-expression network | Spearman's correlation calculation for miRNA targeted lncRNA pairs. | The dysregulated network for intra-hepatic cholangiocarcinoma (ICC) was associated with cholesterol homeostasis, insoluble fraction and lipid binding activity.etc. | [62] |
| MicroRNA-gene network | MicroRNA and mRNA associated expression and microRNA target prediction | The hsa-miR-96, hsa-miR-1 and hsa-miR-25 are identified as potential therapeutic targets for ICC treatment | [63] |
| Gene co-expression network | The regulatory network was constructed by mapping the differentially co-expressed genes to known regulation data. | Several important transcription factors such as, FOXC1, ZIC2, NKX2-2 and GCGR were identified for the future target design. | [64] |
| Epithelial-mesenchymal transition (EMT) network in CCA | An extensive overview and summarization of the EMT regulatory network in CCA. | EMT regulatory network from the membrane receptors to the EMT-inducing transcription factors such as, SNAIL, TWIST and ZEB. The plasticity of CCA caused by the redundancies and bypasses of regulating EMT is discussed for the therapeutic challenges. | [65] |
Integrative analyses for the heterogeneous cholangiocarcinoma
| Integrative data | Description | Discovery | Ref. |
|---|---|---|---|
| Genetic alterations, gene expression and epigenetics | The samples are clustered to 4 subtypes based on their molecular landscapes which are associated with etiology. | The CCAs could be grouped to fluke-negative or positive clusters and new CCA driver genes/mutations and structural variants were discovered. | [2] |
| Copy number, DNA methylation, somatic mutations and RNA expression | Multiple omics data from TCGA are analyzed with cluster ensembles method (CLUE R package). | IDH-mutant enriched subtype has distinct molecular profiles | [70] |
| Transcriptional and copy number variation (CNV) data | Reconstruction of CCA associated transcriptional regulatory network, then integrate the CNV to the network and selected the CVN related ICC-TRN. | ICC patients could be clustered to two groups based on the gene expression of nodes in CNV-ICC-TRN. | [71] |
| Genome and transcription data | Comparison of Genomic and transcriptional alterations from different populations | CCA with or without liver fluke infection are compared and their similarity and difference are identified | [72] |
For complex disease, information collected at different levels such as molecule level, organ/tissue level and individual patient level are all suggestive to the risk or genesis of disease. The information from different levels could be reasonably integrated for the improvement of prediction of the disease. Therefore, integrative studies are not only applied to the same omics level, such as genome, transcriptome, proteome etc. but also could be performed at cross-levels like molecular level and tissue/organ level by combining the gene expression information with the imaging features from magnetic resonance imaging (MRI), computed tomography (CT) and positron emission tomography (PET)/CT [66-69]. Although this kind of integrative study is not yet reported for the study of CCA, in the future the integration of gene expression or proteomics data with the clinical imaging features will provide a better way to the diagnosis of CCA and to the understanding the mechanism of CCA genesis and progression.
Bioinformatics for CCA biomarker and drug-target identification
Precision medicine needs to have novel methods to take use of the multiple dimensional big data which is often heterogeneous and unstructured. The synergic factors for CCA pathogenesis are needed to be precisely interconnected for the biomarker and risk factor screening [73-75]. Nowadays, with the accumulation of biomedical big data, many bioinformatics methods were developed for personalized and precision medicine of cancer, such as the precision classification and the immune treatment of lung cancer based on their gene expression profiles [76-78]. The bioinformatics models for CCA biomarker and drug-target identification are comparatively less than studies in other cancer types. Some of the examples are collected and listed in Table 5. The biomarkers identified could be grouped as diagnostic and therapeutic of CCA. It could be used to the classification of CCA. The biological data used here for biomarker discovery include deep sequencing data, proteomics data, copy number variations and the data from The Cancer Genome Atlas project (TCGA) [79] and Gene Expression Omnibus (GEO) [80]. Many putative biomarkers such as C-reactive protein (CRP), alpha-1-antitrypsin, circulating plasma miRNA-21, etc. were screened for the diagnosis of CCA. Moreover, AIFM3 was detected as a potential chemotherapeutic target for CCA treatment.
Clinical applications of the bioinformatics analyses
| Application | Method | Number of patients involved | Discovery | Ref. |
|---|---|---|---|---|
| Diagnostic Biomarker | Integrative analysis of data from TCGA and GEO database and identify the differentially expressed genes and validated experimentally. | 103 iCCA and 384 other adenocarcinoma patients | C-reactive protein (CRP) was identified as putative diagnostic biomarker better than N-cadherin for distinguish intra-hepatic cholangiocarcinoma (iCCA) from CRP expression indicates a better overall survival. | [17] |
| Comparative and quantitative proteomics study of the bile fluid of patients | Six CCA patients and two non-CCA patients | Alpha-1-antitrypsin is identified as a potential marker for early diagnosis of cholangiocarcinoma. | [81] | |
| Comparative deep sequencing miRNA expression between tumor and control samples | 25 ICC patients and 7 healthy controls | Circulating plasma miRNA-21 and miRNA-221 are identified as potential diagnostic markers for primary iCCA | [82] | |
| Chemotherapeutic Target | A shotgun proteomic approach Using SDS-PAGE coupled with LC-MS/MS to screen mitochondrial proteins overexerted in CCA. | 25 CCA patients with 11 non-papillary and 14 papillary types | AIFM3 was found as a potential CCA chemotherapeutic target. | [83] |
| Prognostic nomogram | Using data from ICCA patients to develop and evaluate the nomogram by concordance index and testing calibration. | Information from 185 iCCA patients was used for nomogram creation | A nomogram integrated ten clinicopathological variables was developed to predict prognostic overall survival (OS) for iCCA patients after hepatectomy. | [84] |
| Biomarkers to distinguish CCA from benign biliary tract diseases(BBTDs) | Comparative proteomic with SDS-PAGE and LC-MS/MS | 19 CCA and 17 BBTDs patients | FAM19A5, MAGED4B, KIAA0321, RBAK, and UPF3B are screened as putative biomarkers to differentiate BBTDs and CCA. | [85] |
| Identification of biomarker for diagnosis of eCCA | Mass spectrometry | 165 extrahepatic cholangiocarcinoma and 21 non-cancerous patients | S100P, CEAM5, MUC5A, OLFM4, OAT, CAD17, FABPL, AOFA, K1C20 and CPSM were identified associated with eCCA could be acted as biomarker for diagnosis of eCCA. | [86] |
| Classification of iCCA | Copy number alterations and classification | 53 iCCA patients | iCCA can be grouped and targeted based on their copy number alterations areas such as 1p, 3p, 7p, etc. | [87] |
Limitation of the existed bioinformatics applications
Although many bioinformatics techniques and models have been applied to the study of CCA, precise models for deep analyses of trans-omics data of cholangiocarcinoma are still lacking for the investigation of the CCA heterogeneity, the interaction between the tumor cells and their microenvironment, the pharmacological effects, etc. The paired genotype and phenotype data, the time series and dynamic data for the CCA developing will be needed to study these questions with translational bioinformatics models.
For the application of the findings of basic biological research to clinical diagnosis or treatment, further validation is needed since these findings based on the screening in small number of patients. When applied the findings to big population, we need to consider the genetic and phenotypic similarities between the patients. Big data and personalized information will be essential to build the personalized models.
The Future Perspectives on Precision Medicine of Cholangiocarcinoma
With the big data driven modeling and systems level thinking paradigm popularized, more and more bioinformatics applications will be developed and implemented to the cancer diagnosis and treatment. The cross-disciplinary communication and cooperation is still a barrier for the precision medicine development. The challenges and opportunities coexisted in the future for the application of bioinformatics to cholangiocarcinoma study. We propose to develop CCA specific databases, knowledge bases and systems medicine model considering the dynamic changes of the cancer evolution, the big data based monitoring the CCA genesis for the healthcare will become the reality.
Database building is the basic for the translational research
Bioinformatics is a discipline using database as the basic tool for annotation, analysis and modeling of biological systems. For the CCA studies, we are lacking of CCA specific databases for the gene alterations, for the genotype-phenotype relationship at genomic, imaging or clinical symptom level. For the clinical data collection, we also need to consider the ethical and privacy issues, the personalized information need to be de-identified and preserved. As presented in Figure 2, we need to first consider the privacy issue when we build a database including patients' clinical information. In this aspect, many efficient cryptographic models have been developed and proposed for the privacy preserving [88-90]. With the specific database built, data scientists and clinical researchers could cooperate together to mining the association between the risk factors and the sub-types of the studied diseases, to make precision classification and to explore the complex mechanisms for the special clinical phenotype and at last to screen actionable biomarkers, risk factors for the diagnosis and prevention of disease.
Data-driven discovery for cholangiocarcinoma study
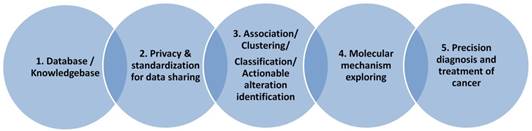
Knowledge based informatics models are the essential
Knowledge base is an advanced database, it stores the expert annotated information often with structural relationship and standardized terminology. It promotes the knowledge sharing and precise application. Since the complex disease like CCA often related to many factors like genetics, lifestyle and environmental factors (see Figure 3A), these factors may be associated with each other. So a knowledge base which can describe their relationship precisely will be very useful for the understanding of their interaction, then to identify the driver changes or events from these complex data.
Knowledge guided model for personalized medicine of cholangiocarcinoma
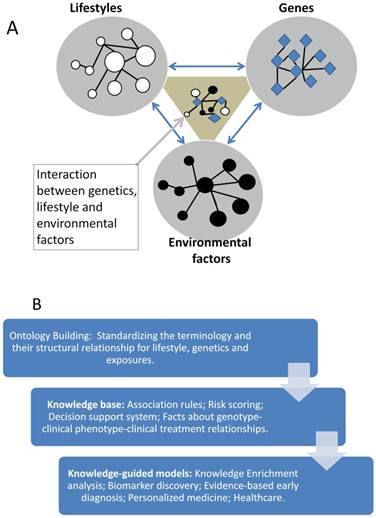
Ontology is successful in the annotation of gene products' function in the last several decades, in the future the clinical phenotypes for the CCA needs to be developed to standardize the CCA associated clinical symptom and phenotypes, then applied to the mapping of genotype-phenotype relationship for CCA study and bioinformatics model developing (see Figure 3B).
At present, we cannot diagnose and treat the CCA precisely since we do not have the precise relationship between the clinical symptoms and the genes, the lifestyles or the environmental factors. Without the knowledge, we cannot build the precision models, and we even cannot identify the risk factors and the pathogens at the systems level. The building of knowledge base for CCA can help us to connect the reported factors and to build the systems biological model for the investigation of heterogeneity of CCA and to identify the key players or the drivers in different steps of the CCA evolution [91].
Exploring the regulatory mechanism of CCA genesis and progress
The discovery of the specific molecular mechanism for CCA genesis and progress is the basic for the identification of efficient therapeutic targets or diagnostic biomarkers. It is reported that the long term inflammation is one of the causing factors for CCA [92], the autocrine and paracrine mechanisms will promote the chemo-resistance in CCA [93]. However, CCA is very heterogeneous and the effective druggable targets or biomarkers still missing, the modeling at network or systems level is required to understand the controllability of the CCA systems.
With the rapid developing of diverse omics techniques, it is easy to collect the trans-omics data such as the mutation/CNV/gene fusion profiles, to detect the methylation and the transcription landscapes, even the different clinical and physiological phenotyping data. The integration of the information can facilitate the building of systems biological model for exploring the molecular mechanism of CCA genesis and progress.
Understanding the evolutionary mechanisms is the necessary to precision medicine of cholangiocarcinoma
Cancer is also an evolutionary disease [94-97], the over-diagnosis and over-treatment is often happened since we cannot efficiently predict the tendency of the cancer evolution. The accumulation of gene alternations is an evolutionary way to destruct the biological system step by step. To consider the cancer as a chronic disease and try to change the negative styles or prevent to exposure to the harmful environments may be an alternative way to decrease the mortality of CCA. It could be expected that in the future, with more data and knowledge available for the modeling of CCA, the prevention of CCA could become possible.
Figure 4 displays the progress of CCA, from its cellular origins and then accumulate the gene alterations like mutations, gene abnormal expression, etc. The heterogeneity of CCA is increased and the complex of the CCA is also changed to the stage difficult to be controlled and managed. Therefore, it is always needed to develop a way to screen CCA as early as possible, but it is not easy to collect the data or information in the early stage of CCA, since no clear signs or symptoms presented in the early stage of CCA. The only way is to find an evolutionary rule or model to infer the early changes at the molecule level. Then we could have a way to predict and act early for the prevention of the progress of CCA.
Evolutionary study of cholangiocarcinoma and preventative medicine
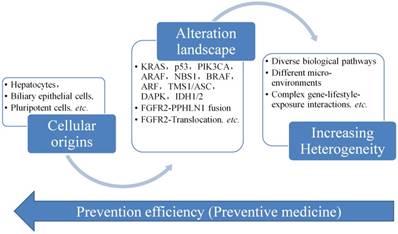
Conclusions
Precision medicine of CCA needs not only the data, we also need precise information or systems level models. Bioinformatics is the key discipline for translational study of cancers like CCA. At present, the models applied to translational CCA study are simple and not so precise or CCA specific, since we are lacking of the CCA specific databases, knowledge bases and therefore no in-depth applications of bioinformatics in CCA study. To construct the CCA specific database and knowledge base will be the basic for the model developing in CCA study. This offers us great opportunities and challenges in the next step bioinformatics-guided precision medicine for CCA biomarker and risk factor discovery.
Abbreviations
CCA: Cholangiocarcinoma, OS: overall survival, iCCA: intrahepatic cholangiocarcinoma, eCCA: extra-hepatic cholangiocarcinoma, SBML: systems biology markup language, MAGE-ML: microarray gene expression markup language, CabosML: carbohydrate sequence markup language, CMap: Connectivity Map, EMT: Epithelial-mesenchymal transition, MRI: magnetic resonance imaging, CT: computed tomography, PET: positron emission tomography, TCGA: The Cancer Genome Atlas project, PPI: Protein-protein interaction, GEO: Gene Expression Omnibus, KEGG: Kyoto Encyclopedia of Genes and Genomes, GO: Gene ontology.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (NSFC) (grant nos. 31670851, 31470821 and 91530320) and National Key R&D programs of China (2016YFC1306605).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Erice O, Merino-Azpitarte M, Arbelaiz A, Gutierrez-Larranaga M, Jimenez-Aguero R, Perugorria MJ. et al. Molecular Mechanisms of Cholangiocarcinogenesis: New Potential Targets for Therapy. Current drug targets. 2017;18:932-49
2. Jusakul A, Cutcutache I, Yong CH, Lim JQ, Huang MN, Padmanabhan N. et al. Whole-Genome and Epigenomic Landscapes of Etiologically Distinct Subtypes of Cholangiocarcinoma. Cancer Discov. 2017;7:1116-35
3. Lin Y, Chen J, Shen B. Interactions Between Genetics, Lifestyle, and Environmental Factors for Healthcare. Advances in experimental medicine and biology. 2017;1005:167-91
4. Kongpetch S, Jusakul A, Ong CK, Lim WK, Rozen SG, Tan P. et al. Pathogenesis of cholangiocarcinoma: From genetics to signalling pathways. Best Pract Res Clin Gastroenterol. 2015;29:233-44
5. Andersen JB, Spee B, Blechacz BR, Avital I, Komuta M, Barbour A. et al. Genomic and genetic characterization of cholangiocarcinoma identifies therapeutic targets for tyrosine kinase inhibitors. Gastroenterology. 2012;142:1021-31.e15
6. Jusakul A, Kongpetch S, Teh BT. Genetics of Opisthorchis viverrini-related cholangiocarcinoma. Current opinion in gastroenterology. 2015;31:258-63
7. Wadsworth CA, Dixon PH, Wong JH, Chapman MH, McKay SC, Sharif A. et al. Genetic factors in the pathogenesis of cholangiocarcinoma. Digestive diseases (Basel, Switzerland). 2011;29:93-7
8. Wiwanitkit V. Pesticides, fresh water fish, liver flukes and nitrosamines: A story of cholangiocarcinoma development in Thailand. Asian Pac J Cancer Prev. 2009;10:961-2
9. Gatto M, Alvaro D. Cholangiocarcinoma: risk factors and clinical presentation. European review for medical and pharmacological sciences. 2010;14:363-7
10. Kirstein MM, Vogel A. Epidemiology and Risk Factors of Cholangiocarcinoma. Visceral medicine. 2016;32:395-400
11. Gupta A, Dixon E. Epidemiology and risk factors: intrahepatic cholangiocarcinoma. Hepatobiliary surgery and nutrition. 2017;6:101-4
12. Palmer WC, Patel T. Are common factors involved in the pathogenesis of primary liver cancers? A meta-analysis of risk factors for intrahepatic cholangiocarcinoma. J Hepatol. 2012;57:69-76
13. Songserm N, Promthet S, Sithithaworn P, Pientong C, Ekalaksananan T, Chopjitt P. et al. Risk factors for cholangiocarcinoma in high-risk area of Thailand: role of lifestyle, diet and methylenetetrahydrofolate reductase polymorphisms. Cancer epidemiology. 2012;36:e89-94
14. Massarweh NN, El-Serag HB. Epidemiology of Hepatocellular Carcinoma and Intrahepatic Cholangiocarcinoma. Cancer control: journal of the Moffitt Cancer Center. 2017;24:1073274817729245
15. Walker NJ, Crockett PW, Nyska A, Brix AE, Jokinen MP, Sells DM. et al. Dose-additive carcinogenicity of a defined mixture of "dioxin-like compounds". Environmental health perspectives. 2005;113:43-8
16. Bond GG, McLaren EA, Sabel FL, Bodner KM, Lipps TE, Cook RR. Liver and biliary tract cancer among chemical workers. American journal of industrial medicine. 1990;18:19-24
17. Yeh YC, Lei HJ, Chen MH, Ho HL, Chiu LY, Li CP. et al. C-Reactive Protein (CRP) is a Promising Diagnostic Immunohistochemical Marker for Intrahepatic Cholangiocarcinoma and is Associated With Better Prognosis. Am J Surg Pathol. 2017
18. Wei M, Lu L, Lin P, Chen Z, Quan Z, Tang Z. Multiple cellular origins and molecular evolution of intrahepatic cholangiocarcinoma. Cancer letters. 2016;379:253-61
19. Terada M, Horisawa K, Miura S, Takashima Y, Ohkawa Y, Sekiya S. et al. Kupffer cells induce Notch-mediated hepatocyte conversion in a common mouse model of intrahepatic cholangiocarcinoma. Sci Rep. 2016;6:34691
20. Sirica AE. Notching up on the cellular origins of intrahepatic cholangiocarcinoma. Hepatology (Baltimore, Md). 2013;57:1668-71
21. Sekiya S, Suzuki A. Intrahepatic cholangiocarcinoma can arise from Notch-mediated conversion of hepatocytes. The Journal of clinical investigation. 2012;122:3914-8
22. Mu X, Pradere JP, Affo S, Dapito DH, Friedman R, Lefkovitch JH. et al. Epithelial Transforming Growth Factor-beta Signaling Does Not Contribute to Liver Fibrosis but Protects Mice From Cholangiocarcinoma. Gastroenterology. 2016;150:720-33
23. Moeini A, Sia D, Bardeesy N, Mazzaferro V, Llovet JM. Molecular Pathogenesis and Targeted Therapies for Intrahepatic Cholangiocarcinoma. Clin Cancer Res. 2016;22:291-300
24. Medeiros JD, Leite LR, Pylro VS, Oliveira FS, Almeida VM, Fernandes GR. et al. Single-cell sequencing unveils the lifestyle and CRISPR-based population history of Hydrotalea sp. in acid mine drainage. Molecular ecology. 2017
25. Jaitin DA, Weiner A, Yofe I, Lara-Astiaso D, Keren-Shaul H, David E. et al. Dissecting Immune Circuits by Linking CRISPR-Pooled Screens with Single-Cell RNA-Seq. Cell. 2016;167:1883-96.e15
26. Datlinger P, Rendeiro AF, Schmidl C, Krausgruber T, Traxler P, Klughammer J. et al. Pooled CRISPR screening with single-cell transcriptome readout. Nature methods. 2017;14:297-301
27. Lussier YA, Li H. The rise of translational bioinformatics. Genome biology. 2012;13:319
28. Shen B, Shen HB, Tian T, Lu Q, Hu G. Translational bioinformatics and computational systems medicine. Computational and mathematical methods in medicine. 2013;2013:375641
29. Shameer K, Badgeley MA, Miotto R, Glicksberg BS, Morgan JW, Dudley JT. Translational bioinformatics in the era of real-time biomedical, health care and wellness data streams. Briefings in bioinformatics. 2017;18:105-24
30. Shen B, Teschendorff AE, Zhi D, Xia J. Biomedical data integration, modeling, and simulation in the era of big data and translational medicine. BioMed research international. 2014;2014:731546
31. Centenera MM, Raj GV, Knudsen KE, Tilley WD, Butler LM. Ex vivo culture of human prostate tissue and drug development. Nature reviews Urology. 2013;10:483-7
32. Doroshow JH, Kummar S. Translational research in oncology-10 years of progress and future prospects. Nature reviews Clinical oncology. 2014;11:649-62
33. Kikuchi N, Kameyama A, Nakaya S, Ito H, Sato T, Shikanai T. et al. The carbohydrate sequence markup language (CabosML): an XML description of carbohydrate structures. Bioinformatics (Oxford, England). 2005;21:1717-8
34. Spellman PT, Miller M, Stewart J, Troup C, Sarkans U, Chervitz S. et al. Design and implementation of microarray gene expression markup language (MAGE-ML). Genome biology. 2002;3:Research0046
35. Hucka M, Finney A, Sauro HM, Bolouri H, Doyle JC, Kitano H. et al. The systems biology markup language (SBML): a medium for representation and exchange of biochemical network models. Bioinformatics (Oxford, England). 2003;19:524-31
36. Yu C, Shen B. XML, Ontologies, and Their Clinical Applications. Advances in experimental medicine and biology. 2016;939:259-87
37. Zhang S, Xiao J, Chai Y, Du YY, Liu Z, Huang K. et al. LncRNA-CCAT1 Promotes Migration, Invasion, and EMT in Intrahepatic Cholangiocarcinoma Through Suppressing miR-152. Dig Dis Sci. 2017
38. Zhang F, Wan M, Xu Y, Li Z, Leng K, Kang P. et al. Long noncoding RNA PCAT1 regulates extrahepatic cholangiocarcinoma progression via the Wnt/beta-catenin-signaling pathway. Biomed Pharmacother. 2017;94:55-62
39. Wang P, Lv L. miR-26a induced the suppression of tumor growth of cholangiocarcinoma via KRT19 approach. Oncotarget. 2016;7:81367-76
40. Palumbo T, Poultsides GA, Kouraklis G, Liakakos T, Drakaki A, Peros G. et al. A functional microRNA library screen reveals miR-410 as a novel anti-apoptotic regulator of cholangiocarcinoma. BMC Cancer. 2016;16:353
41. Zhang J, Han C, Zhu H, Song K, Wu T. miR-101 inhibits cholangiocarcinoma angiogenesis through targeting vascular endothelial growth factor (VEGF). Am J Pathol. 2013;182:1629-39
42. Ma SL, Li AJ, Hu ZY, Shang FS, Wu MC. Coexpression of the carbamoylphosphate synthase 1 gene and its long noncoding RNA correlates with poor prognosis of patients with intrahepatic cholangiocarcinoma. Mol Med Rep. 2015;12:7915-26
43. Khoontawad J, Hongsrichan N, Chamgramol Y, Pinlaor P, Wongkham C, Yongvanit P. et al. Increase of exostosin 1 in plasma as a potential biomarker for opisthorchiasis-associated cholangiocarcinoma. Tumour Biol. 2014;35:1029-39
44. Qu ZL, Cui NQ, Xi ZH, Du Z. [Investigation of hepatitis B virus integration sites in hilar cholangiocarcinoma tissues]. Zhonghua Wai Ke Za Zhi. 2011;49:741-5
45. Mustafa MZ, Nguyen VH, Le Naour F, De Martin E, Beleoken E, Guettier C. et al. Autoantibody signatures defined by serological proteome analysis in sera from patients with cholangiocarcinoma. J Transl Med. 2016;14:17
46. Loffler MW, Chandran PA, Laske K, Schroeder C, Bonzheim I, Walzer M. et al. Personalized peptide vaccine-induced immune response associated with long-term survival of a metastatic cholangiocarcinoma patient. J Hepatol. 2016;65:849-55
47. Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic acids research. 1999;27:29-34
48. Yeh I, Karp PD, Noy NF, Altman RB. Knowledge acquisition, consistency checking and concurrency control for Gene Ontology (GO). Bioinformatics (Oxford, England). 2003;19:241-8
49. Hu Y, Li J, Yan W, Chen J, Li Y, Hu G. et al. Identifying novel glioma associated pathways based on systems biology level meta-analysis. BMC systems biology. 2013;7(Suppl 2):S9
50. Tang Y, Yan W, Chen J, Luo C, Kaipia A, Shen B. Identification of novel microRNA regulatory pathways associated with heterogeneous prostate cancer. BMC systems biology. 2013;7(Suppl 3):S6
51. Wang Y, Chen J, Li Q, Wang H, Liu G, Jing Q. et al. Identifying novel prostate cancer associated pathways based on integrative microarray data analysis. Comput Biol Chem. 2011;35:151-8
52. Chen MH, Lin KJ, Yang WL, Kao YW, Chen TW, Chao SC. et al. Gene expression-based chemical genomics identifies heat-shock protein 90 inhibitors as potential therapeutic drugs in cholangiocarcinoma. Cancer. 2013;119:293-303
53. Wu X, Xia M, Chen D, Wu F, Lv Z, Zhan Q. et al. Profiling of downregulated blood-circulating miR-150-5p as a novel tumor marker for cholangiocarcinoma. Tumour Biol. 2016;37:15019-29
54. Zou S, Li J, Zhou H, Frech C, Jiang X, Chu JS. et al. Mutational landscape of intrahepatic cholangiocarcinoma. Nat Commun. 2014;5:5696
55. Braconi C, Swenson E, Kogure T, Huang N, Patel T. Targeting the IL-6 dependent phenotype can identify novel therapies for cholangiocarcinoma. PLoS One. 2010;5:e15195
56. Wei GH, Liu DP, Liang CC. Charting gene regulatory networks: strategies, challenges and perspectives. The Biochemical journal. 2004;381:1-12
57. Wang Z, Wei GH, Liu DP, Liang CC. Unravelling the world of cis-regulatory elements. Medical & biological engineering & computing. 2007;45:709-18
58. Hristov BH, Singh M. Network-Based Coverage of Mutational Profiles Reveals Cancer Genes. Cell systems. 2017;5:221-9.e4
59. Zhang W, Zang J, Jing X, Sun Z, Yan W, Yang D. et al. Identification of candidate miRNA biomarkers from miRNA regulatory network with application to prostate cancer. J Transl Med. 2014;12:66
60. Zhu F, Liu Q, Zhang X, Shen B. Protein interaction network constructing based on text mining and reinforcement learning with application to prostate cancer. IET systems biology. 2015;9:106-12
61. Zhu J, Wang S, Zhang W, Qiu J, Shan Y, Yang D. et al. Screening key microRNAs for castration-resistant prostate cancer based on miRNA/mRNA functional synergistic network. Oncotarget. 2015;6:43819-30
62. Yang W, Li Y, Song X, Xu J, Xie J. Genome-wide analysis of long noncoding RNA and mRNA co-expression profile in intrahepatic cholangiocarcinoma tissue by RNA sequencing. Oncotarget. 2017;8:26591-9
63. Chen Y, Liu D, Liu P, Yu H, Zhang Q. Identification of biomarkers of intrahepatic cholangiocarcinoma via integrated analysis of mRNA and miRNA microarray data. Mol Med Rep. 2017;15:1051-6
64. Li C, Shen W, Shen S, Ai Z. Gene expression patterns combined with bioinformatics analysis identify genes associated with cholangiocarcinoma. Comput Biol Chem. 2013;47:192-7
65. Vaquero J, Guedj N, Claperon A, Nguyen Ho-Bouldoires TH, Paradis V, Fouassier L. Epithelial-mesenchymal transition in cholangiocarcinoma: From clinical evidence to regulatory networks. J Hepatol. 2017;66:424-41
66. Gevaert O, Xu J, Hoang CD, Leung AN, Xu Y, Quon A. et al. Non-small cell lung cancer: identifying prognostic imaging biomarkers by leveraging public gene expression microarray data-methods and preliminary results. Radiology. 2012;264:387-96
67. Kim GR, Ku YJ, Cho SG, Kim SJ, Min BS. Associations between gene expression profiles of invasive breast cancer and Breast Imaging Reporting and Data System MRI lexicon. Annals of surgical treatment and research. 2017;93:18-26
68. Moonen CT. Spatio-temporal control of gene expression and cancer treatment using magnetic resonance imaging-guided focused ultrasound. Clin Cancer Res. 2007;13:3482-9
69. Popovici V, Budinska E, Capkova L, Schwarz D, Dusek L, Feit J. et al. Joint analysis of histopathology image features and gene expression in breast cancer. BMC bioinformatics. 2016;17:209
70. Farshidfar F, Zheng S, Gingras MC, Newton Y, Shih J, Robertson AG. et al. Integrative Genomic Analysis of Cholangiocarcinoma Identifies Distinct IDH-Mutant Molecular Profiles. Cell Rep. 2017;18:2780-94
71. Li L, Lian B, Li C, Li W, Li J, Zhang Y. et al. Integrative analysis of transcriptional regulatory network and copy number variation in intrahepatic cholangiocarcinoma. PLoS One. 2014;9:e98653
72. Ito T, Sakurai-Yageta M, Goto A, Pairojkul C, Yongvanit P, Murakami Y. Genomic and transcriptional alterations of cholangiocarcinoma. J Hepatobiliary Pancreat Sci. 2014;21:380-7
73. Beckmann JS, Lew D. Reconciling evidence-based medicine and precision medicine in the era of big data: challenges and opportunities. Genome medicine. 2016;8:134
74. Capobianco E. Systems and precision medicine approaches to diabetes heterogeneity: a Big Data perspective. Clinical and translational medicine. 2017;6:23
75. Doherty M, Metcalfe T, Guardino E, Peters E, Ramage L. Precision medicine and oncology: an overview of the opportunities presented by next-generation sequencing and big data and the challenges posed to conventional drug development and regulatory approval pathways. Annals of oncology: official journal of the European Society for Medical Oncology. 2016;27:1644-6
76. Li B, Cui Y, Diehn M, Li R. Development and Validation of an Individualized Immune Prognostic Signature in Early-Stage Nonsquamous Non-Small Cell Lung Cancer. JAMA oncology. 2017;3:1529-37
77. Karasaki T, Nagayama K, Kuwano H, Nitadori JI, Sato M, Anraku M. et al. An Immunogram for the Cancer-Immunity Cycle: Towards Personalized Immunotherapy of Lung Cancer. Journal of thoracic oncology: official publication of the International Association for the Study of Lung Cancer. 2017;12:791-803
78. Liu X, Cho WC. Precision medicine in immune checkpoint blockade therapy for non-small cell lung cancer. Clinical and translational medicine. 2017;6:7
79. Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, Ellrott K. et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nature genetics. 2013;45:1113-20
80. Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic acids research. 2002;30:207-10
81. Laohaviroj M, Potriquet J, Jia X, Suttiprapa S, Chamgramol Y, Pairojkul C. et al. A comparative proteomic analysis of bile for biomarkers of cholangiocarcinoma. Tumour Biol. 2017;39:1010428317705764
82. Correa-Gallego C, Maddalo D, Doussot A, Kemeny N, Kingham TP, Allen PJ. et al. Circulating Plasma Levels of MicroRNA-21 and MicroRNA-221 Are Potential Diagnostic Markers for Primary Intrahepatic Cholangiocarcinoma. PLoS One. 2016;11:e0163699
83. Chua-On D, Proungvitaya T, Techasen A, Limpaiboon T, Roytrakul S, Wongkham S. et al. High expression of apoptosis-inducing factor, mitochondrion-associated 3 (AIFM3) in human cholangiocarcinoma. Tumour Biol. 2016;37:13659-67
84. Yeh CN, Wang SY, Chen YY, Chen MH, Chiang KC, Cheng CT. et al. A Prognostic Nomogram for Overall Survival of Patients After Hepatectomy for Intrahepatic Cholangiocarcinoma. Anticancer Res. 2016;36:4249-58
85. Janvilisri T, Leelawat K, Roytrakul S, Paemanee A, Tohtong R. Novel Serum Biomarkers to Differentiate Cholangiocarcinoma from Benign Biliary Tract Diseases Using a Proteomic Approach. Dis Markers. 2015;2015:105358
86. Maeda S, Morikawa T, Takadate T, Suzuki T, Minowa T, Hanagata N. et al. Mass spectrometry-based proteomic analysis of formalin-fixed paraffin-embedded extrahepatic cholangiocarcinoma. J Hepatobiliary Pancreat Sci. 2015;22:683-91
87. Dalmasso C, Carpentier W, Guettier C, Camilleri-Broet S, Borelli WV, Campos Dos Santos CR. et al. Patterns of chromosomal copy-number alterations in intrahepatic cholangiocarcinoma. BMC Cancer. 2015;15:126
88. Jiang X, Zhao Y, Wang X, Malin B, Wang S, Ohno-Machado L. et al. A community assessment of privacy preserving techniques for human genomes. BMC medical informatics and decision making. 2014;14(Suppl 1):S1
89. Wang S, Zhang Y, Dai W, Lauter K, Kim M, Tang Y. et al. HEALER: homomorphic computation of ExAct Logistic rEgRession for secure rare disease variants analysis in GWAS. Bioinformatics (Oxford, England). 2016;32:211-8
90. Chen F, Wang C, Dai W, Jiang X, Mohammed N, Al Aziz MM. et al. PRESAGE: PRivacy-preserving gEnetic testing via SoftwAre Guard Extension. BMC medical genomics. 2017;10:48
91. Shen L, Lin Y, Sun Z, Yuan X, Chen L, Shen B. Knowledge-Guided Bioinformatics Model for Identifying Autism Spectrum Disorder Diagnostic MicroRNA Biomarkers. Sci Rep. 2016;6:39663
92. Maemura K, Natsugoe S, Takao S. Molecular mechanism of cholangiocarcinoma carcinogenesis. J Hepatobiliary Pancreat Sci. 2014;21:754-60
93. Cadamuro M, Brivio S, Spirli C, Joplin RE, Strazzabosco M, Fabris L. Autocrine and Paracrine Mechanisms Promoting Chemoresistance in Cholangiocarcinoma. International journal of molecular sciences. 2017:18
94. Hornberg JJ, Bruggeman FJ, Westerhoff HV, Lankelma J. Cancer: a Systems Biology disease. Bio Systems. 2006;83:81-90
95. Thomas F, Rome S, Mery F, Dawson E, Montagne J, Biro PA. et al. Changes in diet associated with cancer: An evolutionary perspective. Evolutionary applications. 2017;10:651-7
96. Matsui Y, Niida A, Uchi R, Mimori K, Miyano S, Shimamura T. phyC: Clustering cancer evolutionary trees. PLoS computational biology. 2017;13:e1005509
97. Alberghina L, Chiaradonna F, Vanoni M. Systems biology and the molecular circuits of cancer. Chembiochem: a European journal of chemical biology. 2004;5:1322-33
Author contact
![]() Corresponding author: Professor Bairong Shen, email: bairong.shenedu.cn
Corresponding author: Professor Bairong Shen, email: bairong.shenedu.cn