Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(8):930-937. doi:10.7150/ijbs.24582 This issue Cite
Research Paper
Computational Design of Antiangiogenic Peptibody by Fusing Human IgG1 Fc Fragment and HRH Peptide: Structural Modeling, Energetic Analysis, and Dynamics Simulation of Its Binding Potency to VEGF Receptor
Lin Ning1,2, Zhongyan Li1,2, Zhengya Bai1,2, Shasha Hou1,2, Bifang He1,2, Jian Huang1,2, ![]() , Peng Zhou1,2,
, Peng Zhou1,2, ![]()
1. Center for Informational Biology, University of Electronic Science and Technology of China (UESTC), Chengdu 611731, China
2. School of Life Science and Technology, University of Electronic Science and Technology of China (UESTC), Chengdu 610054, China
Received 2017-12-26; Accepted 2018-1-24; Published 2018-5-22
Abstract

Peptibodies represent a new class of biological therapeutics with combination of peptide activity and antibody-like properties. Previously, we discovered a novel peptide HRH that exhibited a dose-dependent angiogenesis-suppressing effect by targeting vascular endothelial growth factor receptors (VEGFRs). Here, we computationally designed an antiangiogenic peptibody, termed as PbHRH, by fusing the HRH peptide to human IgG1 Fc fragment using the first approved peptibody drug Romiplostim as template. The biologically active peptide of Romiplostim is similar with HRH peptide; both of them have close sequence lengths and can fold into a α-helical conformation in free state. Molecular dynamics simulations revealed that the HRH functional domain is highly flexible, which is functionally independent of Fc fragment in the designed PbHRH peptibody. Subsequently, the intermolecular interactions between VEGFR-1 domain 2 (D2) and PbHRH were predicted, clustered and refined into three representatives. Conformational analysis and energetic evaluation unraveled that the PbHRH can adopt multiple binding modes to block the native VEGF-A binding site of VEGFR-1 D2 with its HRH functional domain, although the binding effectiveness of HRH segments in peptibody context seems to be moderately decreased relative to that of free HRH peptide. Overall, it is suggested that integrating HRH peptide into PbHRH peptibody does not promote the direct intermolecular interaction between VEGFR-1 D2 and HRH. Instead, the peptibody may indirectly help to improve the pharmacokinetic profile and bioavailability of HRH.
Keywords: peptibody, Romiplostim, PbHRH, HRH peptide, VEGF, VEGFR, antiangiogenesis
Introduction
Peptibodies, the hybrid of antibody IgG Fc fragment and biologically active peptide, have been raised as a flexible alternative format to monoclonal antibodies and represent a novel class of biological therapeutics [1]. By fusing a functional peptide to part or all of an antibody, a peptibody combines the peptide activity with some antibody-like properties such as long in vivo circulation half-life and easy expression and purification, making it an attractive platform for therapeutic drugs [2]. Because the peptibodies are smaller than full-length antibodies, they could penetrate more deeply into tissues. It is also possible to attach an antibody portion with multiple peptide segments to make polydrugs or to allow faster and tighter binding to the target cells [3]. In recent years, peptibodies have been recognized as a new and promising therapeutic strategy combating a variety of diseases; a number of peptibody drugs such as Romiplostim, Dulaglutide and AMG 386 have been approved or are in preclinical studies and clinical trials [4-6].
By screening against a Ph.D.-12 phage display library using VEGFR-Fc fusion protein as the bait, we recently identified a novel peptide HRH (HRHTKQRHTALH) that exhibited a dose-dependent anti-angiogenesis effect by inhibiting the proliferation of human umbilical vein endothelial cells through binding to vascular endothelial growth factor receptors (VEGFRs) [7]. Here, we attempted to computationally design angiogenesis-suppressing peptibodies by integrating the HRH peptide into IgG Fc fragment. We also systematically examined the effects of substitution position, valence state and linkage mode on the structural conformation and binding affinity of designed peptibodies to VEGFR-1. This work would help to further guide the rational development of peptibody drugs for antiangiogenic therapy.
Materials and Methods
Template
Romiplostim is the first FDA-approved peptibody medicine used to treat low blood platelet counts in adults with chronic immune thrombocytopenia (ITP) [4]. The protein is a homodimer consisting of two identical subunits; each contains an IgG1 Fc that is covalently fused to two 14-mer biologically active peptides via Gly-rich flexible linkers (Figure 1). The primary sequence of Romiplostim was retrieved from the DrugBank database [8] with accession number DB05332. Sequence comparison revealed that the Romiplostim shared a fully consistent Fc fragment with Conbercept, an engineered anti-VEGF protein agent and we have previously modeled its complex structure with VEGF-A [9]. Here, the Romiplostim scaffold will be used as a template to derive new peptibody drugs through fusion of HRH peptide to the template's IgG1 Fc region.
Sequence annotation of a Romiplostim subunit, consisting of an IgG1 Fc region (N-terminus, CH2 and CH3) and two 14-mer biologically active peptides 1 and 2 that are covalently connected to the Fc region via Gly-rich flexible linkers 1 and 2.

Molecular dynamics simulation
Molecular dynamics (MD) simulations of designed peptibodies and their complexes with VEGFR-1 were carried out using amber ff03 force field [10]. Here, the simulation protocol was modified from our previous works [11-16]. Briefly, the investigated systems were solvated in a periodic rectangular box full of explicit TIP3P water molecules [17]; each direction of the box was extended 10 Å away from any protein atom. Counterions were placed around the protein components based on Columbic potential to keep the whole systems electroneutral. First, all hydrogen atoms, counterions, water molecules and the whole system were in turn minimized with the combination of steepest descent and conjugate gradient methods. Second, the systems were heated to 300 K in 500 ps followed by constant temperature equilibration at 300 K for 1 ns. Third, MD production simulations with an integration step of 2 fs were run in an isothermal isobaric (NPT) ensemble with periodic boundary conditions. The particle mesh Ewald (PME) [18] and SHAKE [19] methods were employed to calculate the long-range electrostatic interactions and to restrain covalent bonds involving hydrogen atoms, respectively. A cut-off distance of 10 Å was applied for van der Waals interactions.
Energetic analysis of intermolecular interaction
The intermolecular interaction energy between the designed peptibodies and VEGFR-1 were calculated and decomposed using molecular mechanics/Poisson-Boltzmann surface area (MM/PBSA) [20] and normal mode analysis (NMA) [21] based on the dynamics conformational snapshots extracted evenly from the MD production simulation trajectories. The total binding energy of VEGFR-1 D2 to peptibody can be expressed as follows:
ΔGttl = ΔEint + ΔGslv - TΔS (1)
where the ΔEint is noncovalent interaction energy between peptibody and VEGFR-1 D2, which was computed via force field approach. The ΔGslv denotes solvent effect associated with peptibody-VEGFR-1 binding, which is composed of polar and nonpolar components -- the former was calculated by finite difference solution of nonlinearized Poisson-Boltzmann (PB) equation, while the latter was derived from an additive surface area (SA) model by weighting solvent accessible surface areas of the binding interface [22]. In addition, the -TΔS represents free energy change due to conformational entropy penalty upon the binding, which was estimated through a harmonic energy approximation with frequencies of the vibrational modes obtained at 300 K [23].
Results and Discussion
Structural modeling of HRH peptide and its analogs
According to the sequence analysis of Romiplostim subunit, two biologically active peptides AF12505 (IEGPTLRQWLAARA) [24] are covalently connected to the IgG Fc fragment via two flexible Gly-rich linkers, i.e. GKGGGGG (linker 1) and GGGGGGGG (linker 2). Here, we considered to separately replace the two AF12505 peptides in Romiplostim by two HRH peptides (HRHTKQRHTALH) to obtain a new peptibody entity with HRH as its functional domain. The AF12505 and HRH peptides have close sequence lengths of 14 and 12 amino acids long, respectively. Their atomic structures were computationally modeled using the PEP-FOLD, a de novo peptide structure prediction server [25]. As can be seen in Figure 2AB, the two peptides are highly structured and both of them can fold into a α-helical conformation in free state; the AF12505 peptide exhibits a slight intrinsic disorder at its N-terminus. We also analyzed the distribution of electrostatic and hydrophobic potentials on HRH helix surface and, evidently, the peptide N-terminus is positively charged and highly hydrophilic, while a moderately neutral region can be found in its C-terminus, thus exhibiting a dipole/hydrophobic moment along the helical orientation.
De novo structure prediction of AF12505 peptide (IEGPTLRQWLAARA) (A), HRH peptide (HRHTKQRHTALH) (B), linker 1 + HRH peptide (GKGGGGG-HRHTKQRHTALH) (C), and linker 2 + HRH peptide (GGGGGGGG-HRHTKQRHTALH) (D). The prediction was carried out using PEP-FOLD server [25].
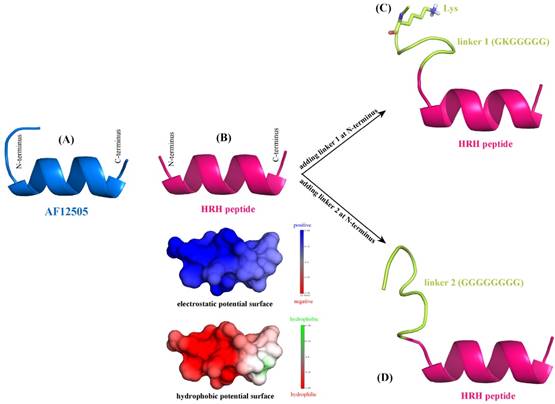
Next, linkers 1 and 2 were separately added at the N-terminus of HRH peptide to generate two extended versions of linker 1 + HRH (GKGGGGG-HRHTKQRHTALH) and linker 2 + HRH (GGGGGGGG-HRHTKQRHTALH). We rebuilt the atomic structures of the two extended peptides with PEP-FOLD server [25] to examine the effect of linkers on HRH peptide. As might be expected, the two linkers cannot influence the HRH structure substantially due to their large flexibility. It is evident in Figure 2CD that the linker regions are intrinsically disordering, which do not directly contact and then disrupt the helical conformation of HRH peptide. In particular, the positively charged Lys residue in linker 1 is far away from HRH peptide and thus can only exert a very modest effect on the peptide. All these suggest that fusing HRH peptides to IgG Fc fragment via the Gly-rich linkers would not alter helical conformation and biological activity of the peptides essentially.
Structural modeling of HRH-fused peptibody
The Romiplostim IgG Fc is taken from human IgG1 Fc domain, whose crystal structure can be retrieved from the protein data bank (PDB) database [26] with accession number 3DNK (Figure 3A). This structure has been successfully used in our previous modeling of the Fc region of Conbercept [8]. By sequence analysis it was indicated that the human IgG1 Fc crystal structure misses a N-terminus sequence MDKTHTCPPCPAPELL relative to Romiplostim, which can be directly grafted from the Conbercept structure [8], where the conformation of this N-terminus sequence was modeled using ModLoop server [27]. Consequently, we obtained the grafted structure of IgG1 Fc + N-terminus (Figure 3B).
Schematic representation of modeling HRH-fused peptibody PbHRH structure. (A) Crystal structure of human IgG1 Fc domain (PDB: 3DNK). (B) Adding N-terminus to the IgG1 Fc crystal structure, resulting in IgG1 Fc + N-terminus. (C) Adding HRH functional domain to IgG1 Fc + N-terminus, resulting in PbHRH peptibody.
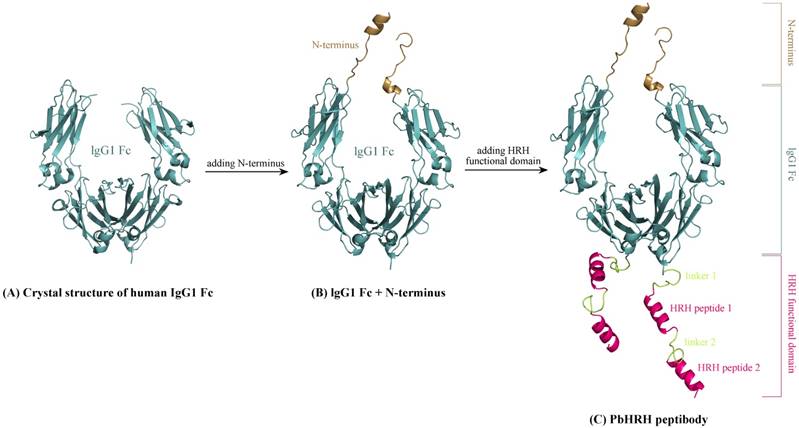
Next, above modeled peptides of linker 1 + HRH and linker 2 + HRH (HRH functional domain) were manually added to the C-terminus of IgG1 Fc + N-terminus to generate the structural architecture of full-length HRH-fused peptibody, termed as PbHRH, which was then subjected to 120-ns MD simulations for conformational equilibrium and relaxing.
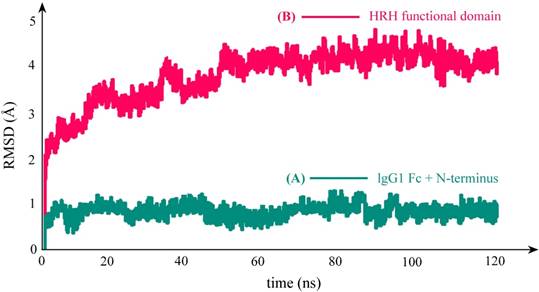
As can be seen in Figure 3C, the equilibrium structure of designed PbHRH peptibody contains three modules: a N-terminus, a IgG1 Fc fragment and a HRH functional domain, where the HRH functional domain is consisted of two repeat regions, i.e. linker 1 + HRH and linker 2 + HRH. It is revealed that, in the context of PbHRH, the HRH peptides can maintain in α-helix, while the Gly-rich linkers remain in intrinsic disorder. In addition, the HRH functional domain exhibits an extended conformation and has no intramolecular interactions with rest of the peptibody, indicating that the IgG1 Fc + N-terminus region would not influence either the conformational state of HRH functional domain or the intermolecular recognition between the domain and its target VEGFR. Instead, the antibody module primarily helps to improve the pharmacokinetic profile and bioavailability of PbHRH peptibody. It is revealed from the MD dynamics trajectory of PbHRH peptibody that the IgG1 Fc + N-terminus region and the HRH functional domain exhibit distinct RMSD fluctuation profiles (Figure 4). As expected, the former has no considerable structural change as compared to its initial state, indicating that the antibody scaffold is highly rigid that can be well structured in solution, whereas the latter possesses a large thermal motion, suggesting that the linear HRH functional domain can swing swiftly by the flexible linkers.
RMSD fluctuation profile of the backbone atoms in PbHRH peptibody protein during 120-ns MD simulations. (A) IgG1 Fc + N-terminus. (B) HRH functional domain.
Structural modeling and energetic analysis of PbHRH-VEGFR-1 complex
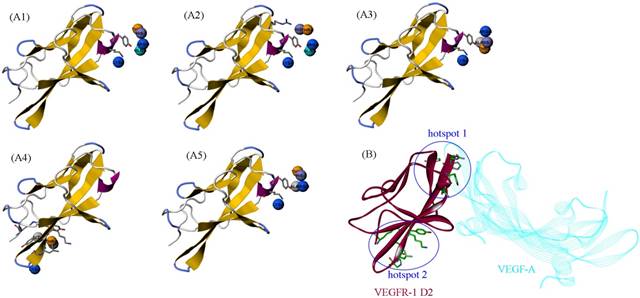
The HRH peptide was identified as VEGFR blocker with dose-dependent inhibition on the proliferation of human umbilical vein endothelial cells stimulated by VEGF [6]. Thus, VEGFR is the putative target of PbHRH peptibody. Previously, deletion studies showed that the second extracellular domain (D2) of VEGFR-1 is the primary binding site of VEGF, which has also been exploited as the effective target of many anti-VEGFR-1 drugs [28]. Here, the intermolecular interaction between PbHRH and VEGFR-1 D2 was investigated at structural and energetic levels. The crystal structure of VEGFR-1 D2 was retrieved from the PDB database [26] with accession number 1FLT, which is a co-crystallized complex with VEGF-A [29]. The VEGFR-1 D2 structure stripped from the complex was submitted to the PepSite server [30] to infer its potential binding sites of HRH peptide. Consequently, top-5 binding sites were predicted (Figure 5A1-A5) and then mapped onto the surface of VEGFR-1 D2 (Figure 5B), where the co-crystallized VEGF-A is also shown for comparison. As can be seen, the top-5 binding sites are roughly concentrated to two hotspots that separately locate at the head and tail of VEGFR-1 D2, in which the hotspot 1 contains four predicted sites (Figure 5A1, A2, A3 and A5), whereas only one site (Figure 5A4) is assigned to hotspot 2. A further visual comparison revealed that the hotspot 1 is well overlapped with the native binding site of VEGF-A, while the hotspot 2 is far away from the native site (Figure 5B). Overall, VEGFR-1 D2 hotspot 1, which consists of residues His147, Tyr199, Lys200, His223 and Arg224, is recognized as the direct targeting site of HRH peptide and, yet, PbHRH peptibody.
(A1-A5) The top-5 binding sites of HRH peptide to VEGFR-1 D2, predicted using PepSite server [30]. (B) Mapping the five sites onto the surface of VEGFR-1 D2 in complex with VEGF-A (PDB: 1FLT). The five sites can be clustered to two hotspots, in which the hotspot 1 is overlapped with the native binding site of VEGF-A.
Next, the intermolecular interaction between VEGFR-1 D2 and PbHRH peptibody was automatically predicted using the sophisticated protein docking method ZDOCK [31], where the hotspot 1 of VEGFR-1 D2 and the HRH functional domain of PbHRH were manually selected as contacting sections for the docking calculations. The docking can generate a large number of potential binding modes of VEGFR-1 D2 to PbHRH. It is seen in Figure 6A that the multiple VEGFR-1 D2 binding modes are either located between the two arms of HRH functional domain or surrounded one single arm, exhibiting a typical region distribution. These predicted binding modes were then clustered into three representatives by using MaxCluster algorithm [32] and separately shown in Figure 6BCD. In representative 1, the VEGFR-1 D2 interacts with both the second HRH segment of first arm and the first HRH segment of second arm, although the interaction is primarily dominated by nonspecific contacts such as hydrophobic and van der Waals forces. In addition, only the first HRH segment of second arm is able to touch the VEGF-A binding site of VEGFR-1 D2, indicating that this segment plays a major role in competitive disruption of VEGFR-1 D2-VEGF-A association --- this can be further supported by representatives 2 and 3, where the VEGFR-1 D2 only interacts with the first HRH segment of second arm using its VEGF-A binding site, albeit the interaction modes of representatives 2 and 3 are not fully consistent.
(A) Multiple binding modes of VEGFR-1 D2 to the HRH functional domain of PbHRH peptibody, predicted using ZDOCK server [31]. (BCD) Clustering of the multiple binding modes into three representatives 1, 2 and 3 by using MaxCluster algorithm [32]. (E) Schematic representation of the nonbonded interactions across the complex interface of VEGFR-1 D2 with HRH representative 3, generated using LigPlot program [34].
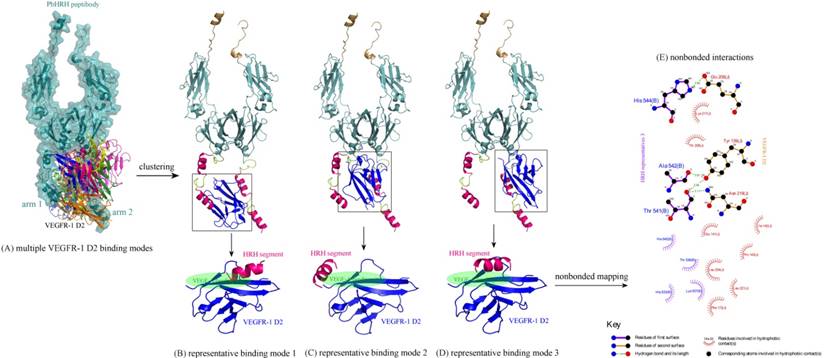
The binding energetics of representatives 1, 2 and 3 were calculated and decomposed with MD simulations and MM/PBSA + NMA analysis (Table 1). The binding is associated with a very favorable interaction energy between VEGFR-1 D2 and PbHRH (ΔEint << 0), which, however, would be largely counteracted by the unfavorable solvent effect (ΔGslv > 0) and entropy penalty (-TΔS > 0), finally resulting in a moderate or modest affinity for the binding. The unfavorable factors indicate that the formed complex interface is partially polar and hydrophilic, and the large flexibility of HRH function domain would cause indirect readout against the binding [33]. In addition, it is revealed a considerable difference between VEGFR-1 D2 binding to full-length PbHRH and the first HRH segment of second arm. The former possesses a generally higher affinity than the latter (ΔGttl = -25.4, -21.7 and -29.1 kcal/mol versus -12.6, -14.0 and -18.9 kcal/mol, respectively); this is in line with the fact that the protein context should play a crucial role in determining the interaction affinity and specificity of protein-protein and protein-peptide recognition [Stein A, et al. PLoS ONE 2008, 3, e2524]. In particular, the VEGFR-1 D2 exhibits a high binding potency to both the full-length PbHRH and HRH segment in representative 3 (ΔGttl = -29.1 and -18.9 kcal/mol, respectively), of which the nonbonded interactions across its complex interface were identified and mapped onto a 2D schematic plot in Figure 6E using LigPlot program [34], revealing an intense noncovalent network of hydrogen bonds, hydrophobic forces and van der Waals contacts that confer both specificity and stability to the complex system of representative 3.
The calculated binding energetics of VEGFR-1 D2 to its interacting partners (full-length PbHRH peptibody, HRH segment in the peptibody and free HRH peptide)
| Partner | Representative | Region | Energetics (kcal/mol) | |||
|---|---|---|---|---|---|---|
| ΔEint | ΔGslv | -TΔS | ΔGttl | |||
| PbHRH | representative 1 | full-length | -78.6 | 18.5 | 34.7 | -25.4 |
| HRH segment | -49.0 | 13.2 | 23.2 | -12.6 | ||
| PbHRH | representative 2 | full-length | -67.2 | 15.9 | 29.6 | -21.7 |
| HRH segment | -53.7 | 16.8 | 22.9 | -14.0 | ||
| PbHRH | representative 3 | full-length | -82.4 | 28.3 | 25.0 | -29.1 |
| HRH segment | -64.8 | 24.5 | 21.4 | -18.9 | ||
| HRH | representative 1 | HRH peptide | -71.5 | 26.0 | 24.8 | -20.7 |
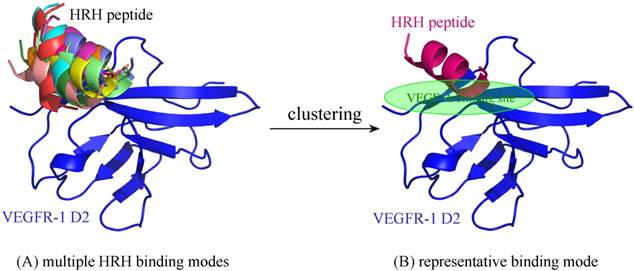
For comparison purpose, the free HRH peptide was redocked, clustered and refined to VEGFR-1 D2, resulting in a single representative (Figure 7). As can be seen, the representative binding mode of free HRH peptide lays on the core region of VEGF-A binding site. In contrast, the HRH segments in peptibody context can only bind nearby or partial overlapping with the cognate site. The energetic analysis also supported this finding, that is, free HRH peptide can bind tightly to VEGFR-1 D2 as compared with the HRH segments in three representatives (ΔGttl = -20.7 kcal/mol versus -12.6, -14.0 and -18.9 kcal/mol) (Table 1), where the HRH segment of representative 3 is most close to the free peptide in both binding conformation and interaction energy. Overall, integrating HRH peptide into PbHRH peptibody may not improve the direct intermolecular interaction between VEGFR-1 D2 and HRH.
(A) Multiple binding modes of free HRH peptide to VEGFR-1 D2, predicted using ZDOCK server [31]. (B) Clustering of the multiple binding modes into one representative. The interaction details of VEGFR-1 D2 with the HRH representative were refined using PepCrawler server [32].
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Nos. 61571095 [J. Huang] and 31671361 [P. Zhou]) and the Fundamental Research Funds for the Central Universities of China (Nos. ZYGX2015Z006 [J. Huang] and ZYGX2016J186 [P. Zhou]).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Shimamoto G, Gegg C, Boone T, Quéva C. Peptibodies: A flexible alternative format to antibodies. mAbs. 2012;4:586-591
2. Huang C. Receptor-Fc fusion therapeutics, traps, and MIMETIBODY technology. Curr Opin Biotechnol. 2009;20:692-699
3. Torchia J, Weiskopf K, Levy R. Targeting lymphoma with precision using semisynthetic anti-idiotype peptibodies. Proc Natl Acad Sci USA. 2016:113 5376-5381
4. Cines DB, Yasothan U, Kirkpatrick P. Romiplostim. Nat Rev Drug Discov. 2008:7 887-888
5. Amblee A. Dulaglutide for the treatment of type 2 diabetes. Drugs Today. 2014:50 277-289
6. Robson EJ, Ghatage P. AMG 386: profile of a novel angiopoietin antagonist in patients with ovarian cancer. Expert Opin Investig Drugs. 2011;20:297-304
7. Zhang Y, He B, Liu K, Ning L, Luo D, Xu K, Zhu W, Wu Z, Huang J, Xu X. A novel peptide specifically binding to VEGF receptor suppresses angiogenesis in vitro and in vivo. Sig Transd Target Ther. 2017;2:17010
8. Wishart DS, Knox C, Guo AC, Cheng D, Shrivastava S, Tzur D, Gautam B, Hassanali M. DrugBank: a knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 2008;36:D901-D906
9. Wu Z, Zhou P, Li X, Wang H, Luo D, Qiao H, Ke X, Huang J. Structural characterization of a recombinant fusion protein by instrumental analysis and molecular modeling. PLoS ONE. 2013;8:e57642
10. Duan Y, Wu C, Chowdhury S, Lee MC, Xiong G, Zhang W, Yang R, Cieplak P, Luo R, Lee T, Caldwell J, Wang J, Kollman P. A point-charge force field for molecular mechanics simulations of proteins. J Comput Chem. 2003;24:1999-2012
11. Yang C, Wang C, Zhang S, Huang J, Zhou P. Structural and energetic insights into the intermolecular interaction among human leukocyte antigens, clinical hypersensitive drugs and antigenic peptides. Mol Simul. 2015;41:741-751
12. Zhou P, Zhang S, Wang Y, Yang C, Huang J. Structural modeling of HLA-B*1502/peptide/carbamazepine/T-cell receptor complex architecture: implication for the molecular mechanism of carbamazepine-induced Stevens-Johnson syndrome/toxic epidermal necrolysis. J Biomol Struct Dyn. 2016;34:1806-1817
13. Yang C, Zhang S, He P, Wang C, Huang J, Zhou P. Self-binding peptides: folding or binding? J Chem Inf Model. 2015;55:329-342
14. Yang C, Zhang S, Bai Z, Hou S, Wu D, Huang J, Zhou P. A two-step binding mechanism for the self-binding peptide recognition of target domains. Mol Biosyst. 2016;12:1201-1213
15. Bai Z, Hou S, Zhang S, Li Z, Zhou P. Targeting self-binding peptides as a novel strategy to regulate protein activity and function: a case study on the proto-oncogene tyrosine protein kinase c-Src. J Chem Inf Model. 2017;57:835-845
16. Zhou P, Hou S, Bai Z, Li Z, Wang H, Chen Z, Meng Y. Disrupting the intramolecular interaction between proto-oncogene c-Src SH3 domain and its self-binding peptide PPII with rationally designed peptide ligands. Artif Cells Nanomed Biotechnol. 2017 [Epub ahead of print, doi: 10.1080/21691401.2017.1360327]
17. Jorgensen WL, Chandrasekha J, Madura JD, Impey RW, Klein ML. Comparison of simple potential function for simulating liquid water. J Chem Phys. 1983;79:926-935
18. Darden T, York D, Pedersen L. Particle mesh Ewald: an N·log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089-10092
19. Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys. 1977;23:327-341
20. Homeyer N, Gohlke H. Free energy calculations by the molecular mechanics Poisson-Boltzmann surface area method. Mol Inf. 2012;31:114-122
21. Case D. Normal mode analysis of protein dynamics. Curr Opin Struct Biol. 1994;4:285-290
22. Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong L, Lee M, Lee T, Duan Y, Wang W, Donini O, Cieplak P, Srinivasan J, Case DA, Cheatham TE. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc Chem Res. 2000;33:889-897
23. Hou T, Chen K, McLaughlin WA, Lu B, Wang W. Computational analysis and prediction of the binding motif and protein interacting partners of the Abl SH3 domain. PLoS Comput Biol. 2006;2:e1
24. Yang AS. Development of romiplostim: a novel engineered peptibody. Semin Hematol. 2015:52 12-15
25. Maupetit J, Derreumaux P, Tuffery P. PEP-FOLD: an online resource for de novo peptide structure prediction. Nucleic Acids Res. 2009;37:W498-W503
26. Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucleic Acids Res. 2000;28:235-242
27. Fiser A, Sali A. ModLoop: automated modeling of loops in protein structures. Bioinformatics. 2003:19 2500-2501
28. Di Stasi R, Diana D, Capasso D, Palumbo R, Romanelli A, Pedone C, Fattorusso R, D'Andrea LD. VEGFR1(D2) in drug discovery: Expression and molecular characterization. Biopolymers. 2010;94:800-809
29. Wiesmann C, Fuh G, Christinger HW, Eigenbrot C, Wells JA, de Vos AM. Crystal structure at 1.7 A resolution of VEGF in complex with domain 2 of the Flt-1 receptor. Cell. 1997;91:695-704
30. Trabuco LG, Lise S, Petsalaki E, Russell RB. PepSite: prediction of peptide-binding sites from protein surfaces. Nucleic Acids Res. 2012;40:W423-W427
31. Chen R, Li L, Weng Z. ZDOCK: an initial-stage protein-docking algorithm. Proteins. 2003;52:80-87
32. de Hoon MJ, Imoto S, Nolan J, Miyano S. Open source clustering software. Bioinformatics. 2004;20:1453-1454
33. Yu H, Zhou P, Deng M, Shang Z. Indirect readout in protein-peptide recognition: a different story from classical biomolecular recognition. J Chem Inf Model. 2014;54:2022-2032
34. Wallace AC, Laskowski RA, Thornton JM. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995;8:127-134
Author contact
![]() Corresponding authors: Jian Huang (hjedu.cn) and Peng Zhou (p_zhouedu.cn)
Corresponding authors: Jian Huang (hjedu.cn) and Peng Zhou (p_zhouedu.cn)