Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Results
Discussion
Materials and Methods
Abbreviations
Acknowledgements
Supplementary Material
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(9):1122-1132. doi:10.7150/ijbs.25881 This issue Cite
Research Paper
KDM5B demethylates H3K4 to recruit XRCC1 and promote chemoresistance
Wenxia Xu1, Bingluo Zhou2, Xiaoya Zhao2, Liyuan Zhu1, Jinye Xu2, Zhinong Jiang3, Dingwei Chen4, Qi Wei1, Mengjiao Han2, Lifeng Feng1, Shouyu Wang5, Xian Wang2, Jianwei Zhou5, Hongchuan Jin1 ![]()
1. Laboratory of Cancer Biology, Key Laboratory of Biotherapy in Zhejiang, Sir Run Run Shaw hospital, Medical School of Zhejiang University, China;
2. Department of Medical Oncology, Sir Run Run Shaw hospital, Medical School of Zhejiang University, China;
3. Department of Pathology, Sir Run Run Shaw hospital, Medical School of Zhejiang University, China;
4. Department of general surgery, Sir Run Run Shaw hospital, Medical School of Zhejiang University, China;
5. Department of Molecular Cell Biology and Toxicology, School of Public Health, Nanjing Medical University, Nanjing, China.
Received 2018-3-5; Accepted 2018-6-3; Published 2018-6-22
Abstract

Chemotherapy is the main treatment for human cancers including gastric cancer. However, in response to chemotherapeutic drugs, tumor cells can develop drug resistance by reprogramming intracellular metabolic and epigenetic networks to maintain their intrinsic homeostasis. Previously, we have established cisplatin-resistant gastric cancer cells as a drug resistant model, and elucidated the XRCC1 as the core DNA repair mechanism of drug resistance. This study investigated the regulation of XRCC1 by lysine demethylase 5B (KDM5B) in drug resistance. We found that the methylation level of H3K4 decreased significantly in drug-resistant cells. The chemical inhibitor of H3K4 demethylases, JIB-04, restored the methylation of H3K4 and blocked the co-localization of XRCC1 and γH2AX, eventually improved drug sensitivity. We further found that the expression level of KDM5B increased significantly in drug-resistant cells. Knockdown of KDM5B increased the methylation level of H3K4 and blocked the localization of XRCC1 to the DNA damage site, leads to increased drug sensitivity. In the sensitive cells, overexpression of KDM5B suppressed H3K4 methylation levels, which resulted to resistance to cisplatin. Moreover, we found that the posttranslational modification of KDM5B is responsible for its high expression in drug-resistant cells. Through mass spectrometry screening and co-immunoprecipitation validation, we found that the molecular chaperone HSP90 forms a complex with KDM5B in drug resistance cells. Interestingly, HSP90 inhibitor 17-AAG induced KDM5B degradation in a time-and-dose-dependent manner, indicating that HSP90 protected KDM5B from protein degradation. Targeting inhibition of HSP90 and KDM5B reversed drug resistance both in vitro and in vivo. Taken together, molecular chaperon HSP90 interacted with KDM5B to protect it from ubiquitin-dependent proteasomal degradation. Increased KDM5B demethylated H3K4 and facilitated the recruitment of XRCC1 to repair damaged DNA. Therefore, inhibition of HSP90 or KDM5B represented a novel approach to reverse chemoresistance in human cancers.
Keywords: KDM5B, XRCC1, H3K4 demethylation, Chemoresistance, gastric cancer
Introduction
Cancer is a systemic and complex disease that seriously affects human health[1]11. In East Asia, especially in China, gastric cancer is the cancer with the highest morbidity and mortality. Chemotherapy is one of the main treatments for gastric cancer. However, tumor cells have natural and acquired resistance to chemotherapy, leading to the failure of chemotherapy[2-3].
Cisplatin is a commonly used drug for chemotherapy, and plays a cytotoxic role mainly through the induction of DNA damages such as DNA adducts[4], double strand breaks[5]. Tumor cells generate cisplatin resistance by enhancing DNA damage repair capabilities[6]. Repair pathways including nucleotide excision, homologous recombination, and non-homologous end joining are involved in the repair of DNA damages caused by cisplatin. XRCC1 plays an important role in the repair of both single-strand DNA damage and double-strand DNA breaks[7-8]. By establishing cisplatin-resistant tumor cell model, we previously revealed that the increased XRCC1 contributes to drug resistance in human cancer cells. We found that XRCC1 may join the DSB repair process triggered by cisplatin through the NHEJ pathway, because XRCC1 was co-located with DNA-PK, which was the core repair complex in NHEJ[9-10]. However, it remains unknown how XRCC1 reaches the site of DNA damage during DNA repair.
The spatial structure of chromatin plays an important regulatory role in DNA damage response, and the remodeling of chromatin spatial structure mainly depends on the post-translational modification of lysine residues in histones[11]. Recently, the role of histone methylation modification in DNA damage response is receiving more and more attention[12-13]. Histone methylation occurs mainly in H3K4, H3K27, H3K36, and H4K20. Demethylation of H3K4 can adjust the spatial structure of chromatin in the event of DNA damage to fully accessible to large protein complex for DNA damage repair[14-16]. The methylation status of H3K4 is regulated by methyltransferase and demethylase. Methyltransferase is mainly MLL family proteins, while demethylase includes KDM5A, KDM5B, KDM5C and KDM5D. However, the role of H3K4 methylation in gastric cancer drug resistance is not well known.
Here, we found that H3K4 methylation levels were reduced and KDM5B was increased in gastric cancer resistant cells. Reverting H3K4 methylation levels by chemical or genetic inhibition of KDM5B reversed chemoresistance by impairing the recruitment of XRCC1 for efficient DNA damage repair. Molecular chaperone HSP90 protects KDM5B from biquitin-proteasomal degradation, thus increasing KDM5B expression to promote chemoresistance. Therefore, targeting KDM5B and HSP90 to attenuate DNA damage repair might be a valuable strategy for reversing chemoresistance and improving chemotherapeutic effects.
Results
Demethylation of H3K4 resulted in chemoresistance
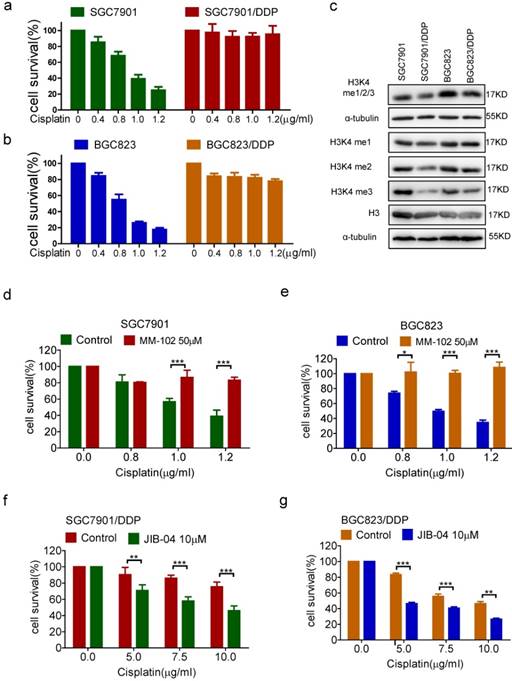
In previous studies, we have established chemoresistant cancer cells by chronic low-dose cisplatin exposure of primarily sensitive gastric cancer cell lines SGC7901 and BGC823, respectively[9-10, 17]. As shown in Figure 1a and b, the survival rates of sensitive cells SGC7901 and BGC823 showed a dose-dependent decline in response to cisplatin. However, the resistant cells SGC7901/DDP and BGC823/DDP still maintained a high survival rate under the effect of cisplatin. We first examined the methylation level of H3K4 in drug-resistant cells and -sensitive cells, and found that the overall methylation level of H3K4 especially di-methylation and tri-methylation levels were decreased in both drug-resistant cells, and the most significantly decreased was tri-methylation (Figure 1c). To elucidate the relevance of H3K4 methylation to drug resistance, we used the methyltransferase inhibitor MM-102 and the demethylase inhibitor JIB-04 to alter intracellular H3K4 methylation (Supplementary Figure 1a and b). The results showed that the inhibition of H3K4 methylation by MM-102 significantly increased the survival rates of sensitive cells SGC7901 and BGC823 under the treatment of cisplatin (Figure 1d and e). Conversely, the use of JIB-04 to promote methylation of H3K4 significantly reduced the survival rates of drug-resistant cells under cisplatin treatment (Figure 1f and g). In summary, these results suggested that demethylation of H3K4 could promote the drug resistance of cancer cells.
(a-b) The viability of cells treated with cisplatin 24h in concentrations as indicated was determined CCK-8 assay. (c) H3K4 mono/di/tri-me (me1/2/3), H3K4 mono-me(me1), H3K4 di-me(me2), H3K4 tri-me(me3), H3 in the cells as indicated were determined by Western blotting. (d-e) The viability of cells treated with cisplatin and MM-102 for 24h in concentrations as indicated was determined CCK-8 assay. (f-g) The viability of cells treated with cisplatin and JIB-04 for 24h in concentrations as indicated was determined by CCK-8 assay.
Restored H3K4 methylation repressed DNA repair and promoted cell apoptosis
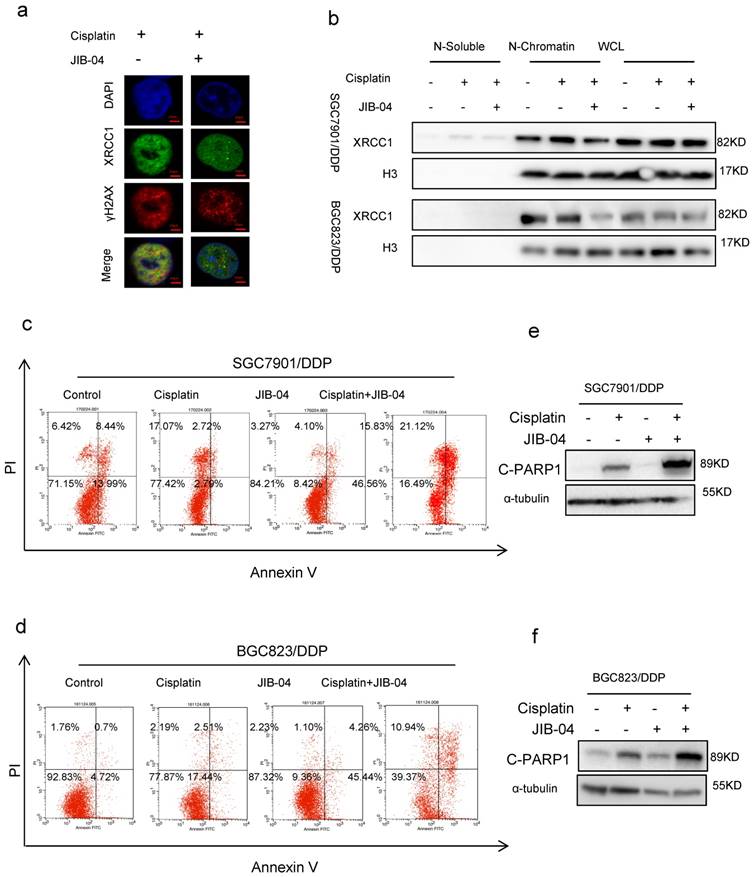
Our previous study has demonstrated that the DNA repair protein XRCC1 restrained cisplatin-induced cell death through DNA damage-induced apoptosis.[9] To investigate the relevance of XRCC1 to H3K4 demethylation-promoted chemoresistance, we determined the co-localization of cisplatin-triggered XRCC1 and γH2AX in cancer cells with or without the treatment of JIB-04. Immunofluorescence assay data showed that JIB-04 weakened the co-localization of XRCC1 with γH2AX (Figure 2a). Moreover, JIB-04 treatment reduced XRCC1's binding to chromatin under cisplatin treatment while the total amount of XRCC1 did not change (Figure 2b), suggesting that methylation of H3K4 may hinder the recruitment of XRCC1 for efficient DNA damage repair. As a consequence, cisplatin combined with JIB-04 resulted in more apoptosis of drug-resistant cells SGC7901/ DDP and BGC823/DDP than cisplatin alone (Figure 2c and d, Flow-cytometry analysis; 2e and f, Western blotting). On the other hand, cisplatin in combination with MM-102 reduced apoptosis in sensitive cells when compared with cisplatin alone (Supplementary Figure 2a and b). The levels of cleaved-PARP1 in MM-102 and cisplatin treated cells were significantly lower than that in cells treated with cisplatin alone (Supplementary Figure 2c and d). These results suggested that H3K4 hypermethylation can inhibit XRCC1's recruitment for effective DNA repair to promote chemotherapy-induced apoptosis.
(a) Immunofluorescence staining determined XRCC1and γH2AX foci by exposure of SGC7901/DDP cells to 5μg/ml cisplatin and 10μM JIB-04 for 24h (×1000). (b) Western blotting determined the expression of XRCC1 and H3 in each component as indicated. Cells were treated with 5μg/ml cisplatin and 10μM JIB-04 for 24h. (c-d) Annexin-PI staining determined the apoptotic cells. Cells were treated with 5μg/ml cisplatin and 10μM JIB-04 for 24h. (e-f) Western blotting determined the expression of cleaved-PARP1(c-PARP1) as indicated. Cells were treated with 5μg/ml cisplatin and 10μM JIB-04 for 24h.
Demethylation of H3K4 promoted chemoresistance due to the increased KDM5B expression
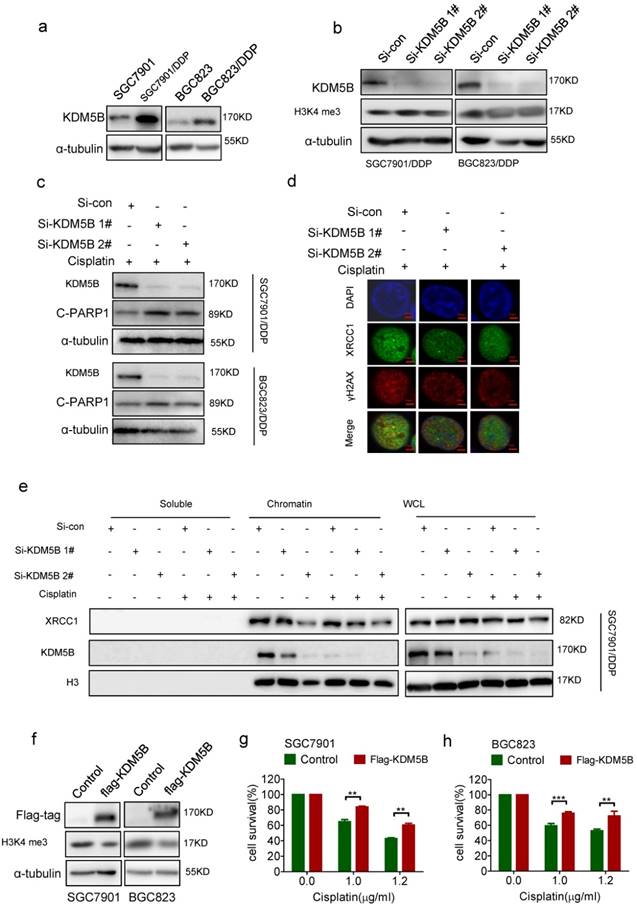
To explore the mechanism for decreased methylation level of H3K4 in chemoresistance cells, we screened the expression of the hitherto known H3K4 tri-methylated demethylases including KDM5A, KDM5B, KDM5C, and KDM5D. As a result, only KDM5B was highly expressed in both drug-resistant cells respectively compared with their parental cells (Figure 3a, and Supplementary Figure 3a-c). Knockdown of KDM5B succeeded to increase the tri-methylation level of H3K4 in drug-resistant cells (Figure 3b). Moreover, knockdown of KDM5B induced more cleaved-PARP1 (Figure 3c), together with reduced co-localization of XRCC1 with γH2AX (Figure 3d), in drug resistant cancer cells treated with cisplatin. In consistance with this, knockdown of KDM5B resulted in a decrease of XRCC1's binding to the chromatin upon cisplatin treatment, while the total amount of XRCC1 in the cells did not changed (Figure 3e, and Supplementary Figure 3d). On the other hand, enforced overexpression of KDM5B in SGC7901 and BGC823 cells by transfection of flag-KDM5B plasmid resulted in reduced H3K4 tri-methylation (Figure 3f) and cell death with cisplatin treatment (Figure 3g and h). Taken together, these results indicated that increased expression of KDM5B demethylated H3K4 to facilitate XRCC1's recruitment for efficient DNA repair in drug resistant cancer cells.
(a) Western blotting determined the expression of KDM5B in each cell as indicated. (b) Western blotting determined the expression of KDM5B and H3K4 tri-me (me3) in KDM5B knockdown cells. (c) Western blotting determined the expression of KDM5B and cleaved-PARP (c-PARP1) in KDM5B knockdown cells treated with 5μg/ml cisplatin for 24h. (d) Immunofluorescence staining determined XRCC1and γH2AX foci by exposure of KDM5B knockdown SGC7901/DDP cells to 5μg/ml cisplatin for 24h (×1000). (e) Western blotting determined the expression of XRCC1 and KDM5B in each component as indicated. KDM5B knockdown SGC7901/ DDP Cells were treated with 5μg/ml cisplatin for 24h. (f) Western blotting determined the expression of flag-KDM5B and H3K4 tri-me (me3) in KDM5B overexpressed cells. (g-h) The viability of KDM5B overexpressed cells treated with cisplatin 24h in concentrations as indicated was determined CCK-8 assay.
HSP90 protected KDM5B from ubiquitin-dependent proteasomal degradation
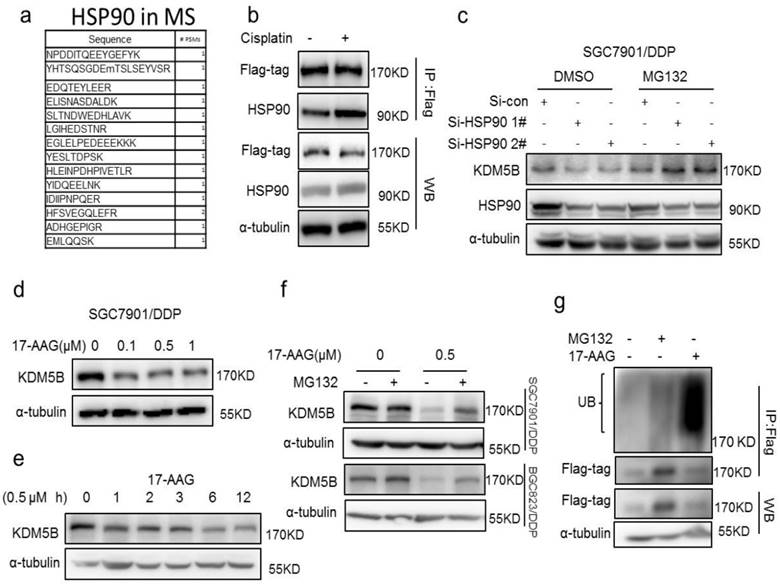
To investigate the mechanism of KDM5B overexpression in drug resistant cells, we first examined the mRNA levels of KDM5B in the two pairs of cells, and found no significant differences in KDM5B mRNA level between drug sensitive and resistant cells (Supplementary figure 4a). In contrast, the proteasome inhibitor MG132 increased KDM5B expression significantly (Supplementary figure 4b), suggesting that high expression of KDM5B may be due to the blockage of ubiquitination-proteasomal degradation.
Interestingly, in an effort to understand the underlying mechanism of increased KDM5B expression in drug resistant cancer cells, we identified the molecular chaperone HSP90 as a novel interaction partner of KDM5B by mass spectrometry screening and co-immunoprecipitation validation (Figure 4a, Supplementary Figure 4c). However, we found no significant changes in the expression of KDM5B and HSP90 in cisplatin-treated cells, while the interaction of flag-tagged KDM5B with HSP90 was actually increased (Figure 4b), indicated this interaction is a dynamic process. In addition, there was no significant difference in HSP90 expression between drug-resistant cells and sensitive cells (Supplementary figure 4d), suggesting an increase of the binding of KDM5B to HSP90 during the development of drug resistance. Indeed, knockdown of HSP90 resulted in down-regulation of KDM5B expression, which could be blocked by MG132 (Figure 4c). Moreover, HSP90 inhibitor 17-AAG also down-regulated KDM5B expression in both time- and dose-dependent manners (Figure 4d and e, Supplementary Figure 4e and f). Likewise, MG132 could also block 17-AAG-induced KDM5B down-regulation. Similar to genetic knockdown of HSP90, 17-AAG also induced significant poly-ubiquitination of KDM5B (Figure 4g). Therefore, these results suggested that HSP90 protected KDM5B from ubiquitinion-dependent proteasomal degradation in chemoresistance.
(a) The peptide of HSP90 was detected by Mass spectrometry. (b) immunoprecipitation determined the interaction of flag-KDM5B (flag-tag) and HSP90 in SGC7901 cells treated with 1μg/ml cisplatin for 24h. (c) Western blotting determined the expression of KDM5B and HSP90 in HSP90 knockdown cells with or without 50μM MG132 treatment for 6h. (d-e) Western blotting determined the expression of KDM5B in SGC7901/DDP cells treated with 17-AAG as indicated. (f) Western blotting determined the expression of KDM5B in cells treated with 17-AAG and MG132 (50μM, 6h) as indicated. (g) Immunofluorescence staining determined the ubiquitination of KDM5B (flag-tag) in the SGC7901/DDP cells treated with 17-AAG and MG132 (50μM, 6h).
Reversing in vivo chemoresistance by inhibiting KDM5B and HSP90
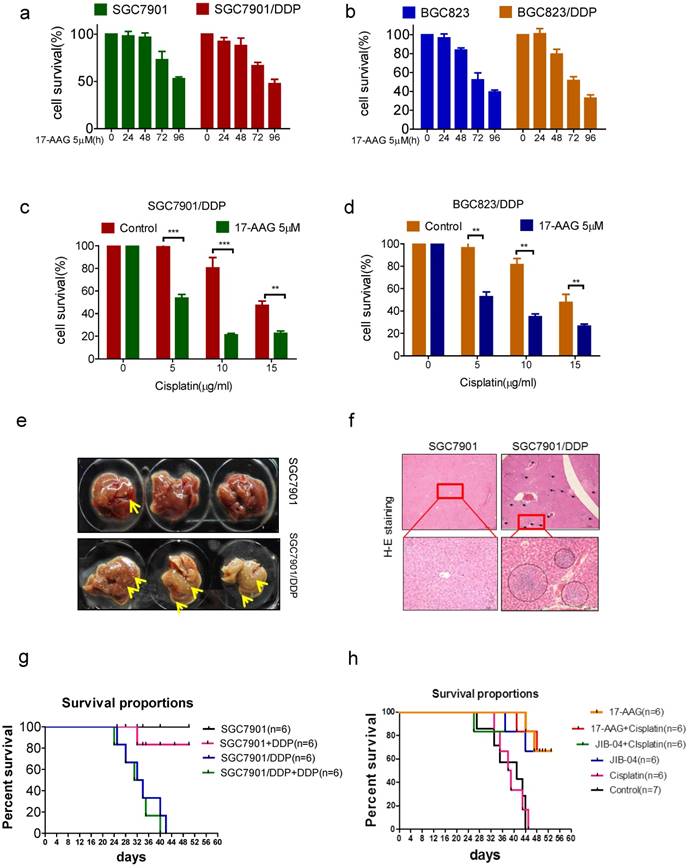
To further confirm the relevance of HSP90 to chemoresistance, we tried to reverse drug resistance with 17-AAG in vitro. Like that of sensitive cells, the survival rate of drug-resistant cell was decreased in a time-dependent manner with 5uM 17-AAG treatment (Figure 5a and b). Furthermore, 17-AAG combined with cisplatin resulted in a significant decline of cell viability compared with using cisplatin alone (Figure 5c and d). We further tried to reverse drug resistance with 17-AAG in vivo. We injected drug-sensitive and resistant cells into mice subcutaneously, we found that in situ tumors of both cells did not grow, but liver metastases occurred, and the liver metastasis of drug-resistant cells was significantly more than that of sensitive cells (figure 5e and f). Moreover, cisplatin treatment does not prolong the survival of mice in the resistant cell group (figure 5g). Then, we injected drug-resistant cells SGC7901/DDP into mice subcutaneously and treated with 17-AAG or JIB-04, with or without cisplatin. The results showed that 17-AAG alone or in combination with cisplatin significantly increased the survival of mice (Figure 5h). Similarly, JIB-04 alone or combined with cisplatin significantly increased the survival of mice (Figure 5h). Therefore, targeting KDM5B and HSP90 could reverse drug resistance both in vitro and in vivo.
(a-b) The viability of cells treated with 5μM 17-AAG for various times as indicated was determined CCK-8 assay. (c-d) The viability of cells treated with cisplatin and 5μM 17-AAG for 24h in concentrations as indicated was determined CCK-8 assay. (e) The liver metastasis of tumor. (f) HE stain of the liver (40× and 200×). (h) The survival of tumor-bearing mice treated with cispaltin. (g) The survival of cisplatin resistant tumor-bearing mice treated with 17-AAG, JIB-04, cisplatin alone or in combination.
Discussion
Drug resistance is the leading cause of chemotherapy failure. Increased DNA damage repair is an important mechanism leading to resistance to DNA targeting drugs such as cisplatin[18-19]. DNA repair process depends on the activation of DNA repair molecules on one hand, and remodeling of chromatin spatial structure on the other hand[20-21]. The covalent modification of histones plays an important role in regulating chromosome structure, controlling gene transcription and participating in DNA damage repair[22-24]. Histone H3K4 methylation is an important type of chromatin covalent modifications, which is closely related to tumorigenesis[25-27]. There are three modes of methylation of H3K4, monomethylation, dimethylation, and trimethylation. H3K4 methylation can promote the transcriptional activation of many oncogenes to promote tumorigenesis. In addition, H3K4 methylation maintains tumor cell survival by regulating DNA damage response. We found that the level of H3K4 methylation in chemoresistant cancer cells was decreased. Reverting H3K4 methylation levels can restore the sensitivity of resistant cells to chemotherapeutic drugs, whereas inhibition of H3K4 methylation can confer drug resistance to sensitive cancer cells. H3K4 demethylation promotes drug resistance by recruiting XRCC1 for efficient DNA repair and thereby inhibiting apoptosis. How H3K4 methylation modulates the chromatin spatial structure to affect the localization of XRCC1 at the damage site, and the dynamic regulation of XRCC1 deserves further studies.
Histone methylation is catalyzed by methyltransferase and demethylase. To date, two major classes of demethylases have been discovered. One is the class of lysine-specific demethylases 1, members of the amino acid oxidase family, such as LSD1[28]. The other is a family of proteins that contain the Jumonji domain. This species has diverse catalytic substrates but generally require the involvement of Fe2 + and α-ketoglutarate for the demethylation [29-30]. Although all of these proteins can catalyze the demethylation of histones, they have some differences in substrate selection. Enzymes that catalyze H3K4 demethylation include KDM5A[31], KDM5B[32], KDM5C[33], and KDM5D[34]. We tested the expression of these proteins in resistant cells and found that only KDM5B is highly expressed in chemoresistant cancer cells. Knockdown of KDM5B expression could increase the methylation level of H3K4 in drug-resistant cells and blocked the localization of XRCC1 to DNA damage, thereby enhancing the drug sensitivity. While overexpression of KDM5B in sensitive cells decreased the methylation level of H3K4 and led to the resistance to cisplatin, indicating that KDM5B promotes the formation of drug resistance in cancer cells. It has been reported that KDM5B played oncogenic roles in a variety of cancers, such as breast cancer, esophageal cancer, and lung cancer[35]. After the treatment of melanoma cells with chemotherapeutic drugs such as cisplatin or molecular targeting drugs as vemurafenib, a group of subpopulations with high KDM5B expression were uniformly enriched as resistant clones[36]. Targeting these subpopulations significantly sensitized melanoma cells to therapy independent of their genotypes, further pinpointing the relevance of KDM5B to drug resistance. By demethylates H3K4, KDM5B not only regulates gene transcription, but also correlates with DNA damage repair. For example, KDM5B is crucial for accurate location of DNA repair molecules such as Ku70, Ku80 and BRCA1 at the damage site[15]. Certainly, some important questions remain open, such as the dynamic regulation of KDM5B in the short process of DNA repair.
Post-translational modifications play an important regulatory role in the structure and function of proteins. A variety of post-translational modifications of KDM5B such as ubiquitination, sumoylation have been reported to regulate its expression and function36.[37] For instance, E3 ligase TRAF6 catalyzes the mono-ubiquitination of KDM5B K242 to promote KDM5M enzymatic activity, while K242 can also be recognized by E3 sumo enzyme RNF4 for sumolyation[38-39]. In this study, we found that HSP90 was crucial for the protection of KDM5B from ubiquitination-dependent proteasomal degradation. Further studies are needed to identify the enzyme as well as the lysine sites for the ubiquitnation of KDM5B.
Importantly, both HSP90 inhibitor 17-AAG and the KDM5B inhibitor JIB-04 reversed chemoresistance in vitro and in vivo[40-43]. As 17-AAG and other chemical inhibitors of HSP90 has been evaluated in clinical trials for their potential clinical application in cancer treatment, this study might provide added values in the design of combination treatment strategies for various human cancers.
Materials and Methods
Chemicals and Cell lines
Cisplatin, MM-102, JIB-04, 17-AAG, MG132 were bought from Selleck Chemicals (Shanghai, China). Human gastric cancer cell lines BGC823 and SGC7901 were purchased from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). The cells were cultured in RPMI 1640 medium supplemented with 10% of fetal bovine serum (FBS), 100 U/ml of penicillin, and 100 μg/ml of streptomycin (Life Technologies/Gibco, Grand Island, NY, USA). The cells were grown at 37℃ in a humidified incubator with 5% CO2. The cisplatin-resistant cells were developed from the parental cells subjected to persistent gradient exposure to cisplatin for about 12 month, through increasing cisplatin concentration from 0.05 μg/ml until the cells acquired resistance to 1 μg/ml.
Cytotoxicity assay
One day before treatment, BGC823, SGC7901, BGC823/DDP, SGC7901/DDP cells were plated at a density of 5000 cells per well in 96-well plates. The cells were treated with various concentrations of drugs. After 24 h treatment, the cell viability was determined using Cell Counting Kit-8 (CCK-8) according to the manufacturer's instructions (Dojindo, Kumamoto, Japan). The cell survival rates were expressed as mean±S.D. from at least three independent experiments.
Flow cytometry analysis
Apoptotic cell death was determined by using the FITC Annexin V Apoptosis Detection Kit I (BD Bioscience, Bedford, MA, USA) according to the manufacturer's instruction. Cells were washed twice with cold PBS and then suspended at a concentration of 1×106 cells/ml in 1×Binding Buffer. Then 100 μl cellular suspensions were added with 5 μl of FITC Annexin V and 5 μl PI, and then incubated for 15 min at room temperature in darkness before analyzed by flow cytometry.
Plasmid, siRNA and transfection
The flag-KDM5B plasmid was constructed by GeneChem Company (Shanghai, China). The plasmid was transfected into cells with X-tremeGENE HP DNA Transfection Reagent (Roche, Basel, Switzerland). In brief, cells were seeded overnight in 6-well plates (3-5×105/well). DNA was diluted to a final concentration of 1 µg plasmid DNA /100 µl medium before adding 2µl DNA Transfection Reagent. The transfection reagent:DNA complex was added for to the cells in a dropwise manner after incubating for 15 min. The transfected cells can be harvested for further analysis after incubation for 1-3 days at 37°C.
The siRNA was transfected into cells with Lipofectamine RNAiMAX Reagent (Life Technologies, CA, USA) according to manufacturer's instructions. In brief, cells were seeded overnight in 6-well plates(3-5×105/well). 9μl Lipofectamine RNAiMAX Reagent or 30pmol siRNA were diluted separately in 150μl Opti-MEM Medium and mixed them for 5min before adding to cells. The transfected cells were incubated for 1-3 days before harvesting for further analysis. Small interfering RNA (siRNA) specific for KDM5B (1# 5'-CACTGGAGCTATTCAATTA-3', 2# 5'-GCGTATCCGTTTGGAACAA-3'), HSP90(1# 5'-GGAGAAGGAACGTGATAAATT -3', 2# 5'-GCGGAAGATAAAGAGAACTTT -3') and control siRNA was synthesized in GenePharma (Shanghai, China).
Western blotting
In brief, cells were lysed in whole-cell lysate buffer containing 1% phosphatase inhibitor cocktail and NPC tumor tissues were lysed in RIPA buffer (Beyotime, Jiangsu, China) with 1% phosphatase inhibitor cocktail. Lysates containing 20-30 µg protein were loaded onto 8% or 12% sodium dodecyl sulfate-polyacrylamide gels for electrophoresis (SDS-PAGE) and the separated proteins transferred to poly vinylidene fluoride (PVDF) membranes (Pall, NY, USA). After blocking with 5% fat free milk for 1 h in Tris-buffered saline (TBS), the membranes were incubated with the primary antibody overnight at 4°C and then with the peroxidase labelled secondary antibody (agilent, CA, USA) for 1 h on the next day. WB bands were visualized with an enhanced chemiluminescence kit (EMD Millipore, MA, USA). The primary antibodies used were γH2AX, XRCC1, KDM5B, UB(1:2000 dilution, abcam, Cambridge, UK), HSP90, KDM5A, KDM5C, KDM5D(1:2000 dilution, proteintech, IL, USA), flag (1:2000 dilution, sigma, Louis, MO, USA ) and monoclonal anti-cleaved PARP-1, α-tubulin, H3K4 mono/di/tri-me, H3K4 mono-me, H3K4 di-me, H3K4 tri-me, H3(1:2000 dilution, Cell SignalingTechnology, Danvers, MA, USA).
Indirect immunofluorescence microscopy
Indirect immunofluorescence stains were performed as previously reported. [9] The cells were incubated with anti-γH2AX mouse monoclonal antibody, anti-XRCC1 rabbit monoclonal antibody at a 1:200 dilution, respectively. The confocal images of cells were sequentially acquired with Zeiss AIM software on a Zeiss LSM 710 confocal microscope system.
Quantitative real-time RT-PCR
In brief, Total RNA was extracted using Trizol reagent (Life Technologies, CA, USA) according to the manufacturer's instructions. RNAs were quantified using a NanoDrop 2000c instrument (Thermo Scientific, MA, USA). Reverse transcription reaction was performed using 1μg of total RNA with Quantscript RT kit (Tiagen Biotechnology, Beijing, China). The mRNA expression level was determined by quantitative real-time PCR using SYBR Green Master Mix (Applied Biosystens, CA, USA) and ABI 7500 Real-Time PCR System (Applied Biosystems, CA, USA). Human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal control of RNA integrity. The Primers: (KDM5B-F 5'-GAATTCGGGAATCTTAAATTTG-3', KDM5B-R 5'-TATCTCGAGTTCCTGTTCGGAATAGG-3'), (GAPDH-F 5'-GGAGTCAACGGATTTGGT-3', GAPDH-R 5'-GTGATGGGATTTCCATTGAT-3').
Immunoprecipitation
Briefly, the cells were harvested and lyzed in cold lysis buffer [50 mM Tris (pH 7.4), 150 mM NaCl, 1 mM EDTA, 0.5% (v/v) NP-40, 10% (v/v) glycerol, 1 mM PMSF]. The cell extracts were centrifuged at 12 000 g at 4℃ for 15 min, and the supernatant was then incubated with protein A/G agarose beads (Santa Cruz) as a pretreatment. Precleared lysates were then incubated with anti-flag antibody or control IgG for 1 h, then incubated overnight with protein A/G agarose beads. The beads were collected by centrifugation, washed three times with the lysis buffer and resuspended in 1×SDS loading buffer. The immunoprecipitates were eluted from the beads by incubation at 95℃for 5 min. The eluted proteins were separated by SDS-PAGE and western blotting or Mass spectrometry (Quanchuan biotechnology, Hangzhou, China) analysis were subsequently performed.
In vivo nude mice assay
All animal procedures were approved by the Committee on Animal Experimentation of Zhejiang University, and the procedures complied with the NIH Guide for the Care and Use of Laboratory Animals. SGC7901/DDP cells were injected into the flanks of BALB/c nude mice (Nu/Nu, female, 4-6 weeks old) purchased from the Center of Experimental Animals of zhejiang University and maintained under pathogen-free conditions (n=6/ group). When the tumors were 100-150 mm3 in size, the mice were treated with cisplatin (4 mg/kg body weight; i.p., every 4 days), 17-AAG(50 mg/kg body weight; i.p., every 4 days), or JIB-04 (55 mg/kg body weight; i.p., every 4 days). Tumor development was monitored by mortality.
Statistical analysis
Data are expressed as the mean ± SD. The statistical significance of the differences between the cell lines was determined by the parametric unpaired Student's t test. Differences were considered significant when P < 0.05.
Abbreviations
HSP90, heat shock protein 90; H3K4, histone 3 lysine 4; KDM5B, lysine demethylase 5B; PARP1, poly (ADP-ribose) polymerase 1; Ub, Ubiquitin; XRCC1, X-ray repair cross complementing group1; γH2AX, phosphorylated histone H2AX.
Acknowledgements
This work was supported by Natural Science foundation of China (81773189, 81502609, 8171101166, 91740106), Natural Science Foundation of Zhejiang province (LY18H160003), High Level Innovative Talents Program and 151 Talents program in Zhejiang, and Department of Health in Zhejiang Province (WKJ-ZJ-1520; 2016143571).
Supplementary Material
Supplementary figures.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Lochhead P, El-Omar E M. Molecular Predictors of Gastric Neoplastic Progression. Cancer Cell. 2018;33:9-11
2. Shi W J, Gao J B. Molecular mechanisms of chemoresistance in gastric cancer. World J Gastrointest Oncol. 2016;8:673-81
3. Lee S Y, Oh S C. Changing strategies for target therapy in gastric cancer. World J Gastroenterol. 2016;22:1179-89
4. Ziehe M, Esteban-Fernandez D, Hochkirch U, Thomale J, Linscheid M W. On the complexity and dynamics of in vivo Cisplatin-DNA adduct formation using HPLC/ICP-MS. Metallomics. 2012;4:1098-104
5. Rancoule C, Guy J B, Vallard A, Ben M M, Rehailia A, Magne N. [50th anniversary of cisplatin]. Bull Cancer. 2017;104:167-176
6. Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O. et al. Molecular mechanisms of cisplatin resistance. Oncogene. 2012;31:1869-83
7. Horton J K, Stefanick D F, Zhao M L, Janoshazi A K, Gassman N R, Seddon H J. et al. XRCC1-mediated repair of strand breaks independent of PNKP binding. DNA Repair (Amst). 2017;60:52-63
8. Moser J, Kool H, Giakzidis I, Caldecott K, Mullenders L H, Fousteri M I. Sealing of chromosomal DNA nicks during nucleotide excision repair requires XRCC1 and DNA ligase III alpha in a cell-cycle-specific manner. Mol Cell. 2007;27:311-23
9. Xu W, Wang S, Chen Q, Zhang Y, Ni P, Wu X. et al. TXNL1-XRCC1 pathway regulates cisplatin-induced cell death and contributes to resistance in human gastric cancer. Cell Death Dis. 2014;5:e1055
10. Xu W, Chen Q, Wang Q, Sun Y, Wang S, Li A. et al. JWA reverses cisplatin resistance via the CK2-XRCC1 pathway in human gastric cancer cells. Cell Death Dis. 2014;5:e1551
11. Chatterjee S, Senapati P, Kundu T K. Post-translational modifications of lysine in DNA-damage repair. Essays Biochem. 2012;52:93-111
12. Khoury-Haddad H, Nadar-Ponniah P T, Awwad S, Ayoub N. The emerging role of lysine demethylases in DNA damage response: dissecting the recruitment mode of KDM4D/JMJD2D to DNA damage sites. Cell Cycle. 2015;14:950-8
13. Lu F, Wu X, Yin F, Chia-Fang L C, Yu M, Mihaylov I S. et al. Regulation of DNA replication and chromosomal polyploidy by the MLL-WDR5-RBBP5 methyltransferases. Biol Open. 2016;5:1449-1460
14. Mosammaparast N, Kim H, Laurent B, Zhao Y, Lim H J, Majid M C. et al. The histone demethylase LSD1/KDM1A promotes the DNA damage response. J Cell Biol. 2013;203:457-70
15. Li X, Liu L, Yang S, Song N, Zhou X, Gao J. et al. Histone demethylase KDM5B is a key regulator of genome stability. Proc Natl Acad Sci U S A. 2014;111:7096-101
16. Gong F, Clouaire T, Aguirrebengoa M, Legube G, Miller K M. Histone demethylase KDM5A regulates the ZMYND8-NuRD chromatin remodeler to promote DNA repair. J Cell Biol. 2017;216:1959-1974
17. Zhang Y, Xu W, Ni P, Li A, Zhou J, Xu S. MiR-99a and MiR-491 Regulate Cisplatin Resistance in Human Gastric Cancer Cells by Targeting CAPNS1. Int J Biol Sci. 2016;12:1437-1447
18. Blasiak J. DNA-Damaging Anticancer Drugs - A Perspective for DNA Repair- Oriented Therapy. Curr Med Chem. 2017;24:1488-1503
19. Cree I A, Charlton P. Molecular chess? Hallmarks of anti-cancer drug resistance. BMC Cancer. 2017;17:10
20. Hauer M H, Gasser S M. Chromatin and nucleosome dynamics in DNA damage and repair. Genes Dev. 2017;31:2204-2221
21. Diaz M, Pecinka A. Scaffolding for Repair: Understanding Molecular Functions of the SMC5/6 Complex. Genes (Basel). 2018:9
22. Fyodorov D V, Zhou B R, Skoultchi A I, Bai Y. Emerging roles of linker histones in regulating chromatin structure and function. Nat Rev Mol Cell Biol. 2017
23. Lai W, Pugh B F. Understanding nucleosome dynamics and their links to gene expression and DNA replication. Nat Rev Mol Cell Biol. 2017;18:548-562
24. Dabin J, Fortuny A, Polo S E. Epigenome Maintenance in Response to DNA Damage. Mol Cell. 2016;62:712-27
25. Zhou A, Lin K, Zhang S, Chen Y, Zhang N, Xue J. et al. Nuclear GSK3beta promotes tumorigenesis by phosphorylating KDM1A and inducing its deubiquitylation by USP22. Nat Cell Biol. 2016;18:954-966
26. Wong S H, Goode D L, Iwasaki M, Wei M C, Kuo H P, Zhu L. et al. The H3K4-Methyl Epigenome Regulates Leukemia Stem Cell Oncogenic Potential. Cancer Cell. 2015;28:198-209
27. Benard A, Goossens-Beumer I J, van Hoesel A Q, de Graaf W, Horati H, Putter H. et al. Histone trimethylation at H3K4, H3K9 and H4K20 correlates with patient survival and tumor recurrence in early-stage colon cancer. BMC Cancer. 2014;14:531
28. Gamble M J, Kraus W L. Visualizing the histone code on LSD1. Cell. 2007;128:433-4
29. Park S Y, Park J W, Chun Y S. Jumonji histone demethylases as emerging therapeutic targets. Pharmacol Res. 2016;105:146-51
30. Accari S L, Fisher P R. Emerging Roles of JmjC Domain-Containing Proteins. Int Rev Cell Mol Biol. 2015;319:165-220
31. Klose R J, Yan Q, Tothova Z, Yamane K, Erdjument-Bromage H, Tempst P. et al. The retinoblastoma binding protein RBP2 is an H3K4 demethylase. Cell. 2007;128:889-900
32. Xiang Y, Zhu Z, Han G, Ye X, Xu B, Peng Z. et al. JARID1B is a histone H3 lysine 4 demethylase up-regulated in prostate cancer. Proc Natl Acad Sci U S A. 2007;104:19226-31
33. Iwase S, Lan F, Bayliss P, de la Torre-Ubieta L, Huarte M, Qi H H. et al. The X-linked mental retardation gene SMCX/JARID1C defines a family of histone H3 lysine 4 demethylases. Cell. 2007;128:1077-88
34. Lee M G, Norman J, Shilatifard A, Shiekhattar R. Physical and functional association of a trimethyl H3K4 demethylase and Ring6a/MBLR, a polycomb-like protein. Cell. 2007;128:877-87
35. Han M, Xu W, Cheng P, Jin H, Wang X. Histone demethylase lysine demethylase 5B in development and cancer. Oncotarget. 2017;8:8980-8991
36. Roesch A, Vultur A, Bogeski I, Wang H, Zimmermann K M, Speicher D. et al. Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1B(high) cells. Cancer Cell. 2013;23:811-25
37. Hendriks I A, Treffers L W, Verlaan-de V M, Olsen J V, Vertegaal A C. SUMO-2 Orchestrates Chromatin Modifiers in Response to DNA Damage. Cell Rep. 2015
38. Bueno M T, Richard S. SUMOylation negatively modulates target gene occupancy of the KDM5B, a histone lysine demethylase. Epigenetics. 2013;8:1162-75
39. Lu W, Liu S, Li B, Xie Y, Adhiambo C, Yang Q. et al. SKP2 inactivation suppresses prostate tumorigenesis by mediating JARID1B ubiquitination. Oncotarget. 2015;6:771-88
40. Calero R, Morchon E, Martinez-Argudo I, Serrano R. Synergistic anti-tumor effect of 17AAG with the PI3K/mTOR inhibitor NVP-BEZ235 on human melanoma. Cancer Lett. 2017;406:1-11
41. Siegelin M D. Inhibition of the mitochondrial Hsp90 chaperone network: a novel, efficient treatment strategy for cancer? Cancer Lett. 2013;333:133-46
42. Wang L, Chang J, Varghese D, Dellinger M, Kumar S, Best A M. et al. A small molecule modulates Jumonji histone demethylase activity and selectively inhibits cancer growth. Nat Commun. 2013;4:2035
43. Dalvi M P, Wang L, Zhong R, Kollipara R K, Park H, Bayo J. et al. Taxane-Platin-Resistant Lung Cancers Co-develop Hypersensitivity to JumonjiC Demethylase Inhibitors. Cell Rep. 2017;19:1669-1684
Author contact
![]() Corresponding author: Dr. Hongchuan Jin, email: jinhcedu.cn
Corresponding author: Dr. Hongchuan Jin, email: jinhcedu.cn