Impact Factor ISSN: 1449-2288
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Newly developed methods for APA...
Bioinformatics pipelines for APA...
Wet and dry laboratory...
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(12):1709-1714. doi:10.7150/ijbs.27168 This issue Cite
Review
Alternative polyadenylation analysis in animals and plants: newly developed strategies for profiling, processing and validation
Yunqi Zhang, Shane A. Carrion, Yangzi Zhang, Xiaohui Zhang, Amy L. Zinski, Jennifer J. Michal, Zhihua Jiang ![]()
Department of Animal Sciences and Center for Reproductive Biology, Washington State University, Pullman, WA 99164-7620
Received 2018-5-8; Accepted 2018-8-5; Published 2018-9-7
Abstract

Alternative polyadenylation is an essential RNA processing event that contributes significantly to regulation of transcriptome diversity and functional dynamics in both animals and plants. Here we review newly developed next generation sequencing methods for genome-wide profiling of alternative polyadenylation (APA) sites, bioinformatics pipelines for data processing and both wet and dry laboratory approaches for APA validation. The library construction methods LITE-Seq (Low-Input 3'-Terminal sequencing) and PAC-seq (PolyA Click sequencing) tag polyA+ cDNA, while BAT-seq (BArcoded, three-prime specific sequencing) and PAPERCLIP (Poly(A) binding Protein-mediated mRNA 3′End Retrieval by CrossLinking ImmunoPrecipitation) enrich polyA+ RNA. Interestingly, only WTTS-seq (Whole Transcriptome Termini Site sequencing) targets both polyA+ RNA and polyA+ cDNA. Varieties of bioinformatics pipelines are well established to pursue read quality control, mapping, clustering, characterization and pathway analysis. The RHAPA (RNase H alternative polyadenylation assay) and 3'RACE-seq (3' rapid amplification of cDNA end sequencing) methods directly validate APA sites, while WTSS-seq (whole transcriptome start site sequencing), RNA-seq (RNA sequencing) and public APA databases can serve as indirect validation methods. We hope that these tools, pipelines and resources trigger huge waves of interest in the research community to investigate APA events underlying physiological, pathological and psychological changes and thus understand the information transfer events from genome to phenome relevant to economically important traits in both animals and plants.
Keywords: alternative polyadenylation, profiling tools, processing pipelines, validation approaches, genome function.
Introduction
Alternative polyadenylation, which causes the same gene to produce multiple RNA transcripts, is an evolutionarily conserved phenomenon in both animals and plants [1-4]. This process prepares the 3' untranslated regions (3'UTRs) of RNAs with either variable sequence composition or different nucleotide lengths due to use of alternative polyadenylation (APA) sites [5]. In fact, 3'UTRs often harbor variable cis- and trans-acting regulatory elements so that alternative polyadenylation plays essential roles in regulation of RNA stability, localization, translation and degradation [6]. Consequently, the same gene can function quantitatively, qualitatively or epigenetically, depending on APA position, 3'UTR features and regulatory modes.
Two alternative transcripts can encode for the same protein. For example, the transcript with a short 3'UTR would avoid the negative regulation targeted by microRNA, but potentially lose the stability provided by RNA-binding proteins in comparison to another with a long 3'UTR. This regulatory mode contributes quantitatively to gene function [7]. Qualitative modes occur when APA sites from the same gene yield transcript isoforms that encode distinct proteins, potentially with distinct properties [8]. When a protein-coding transcript is converted into a non-coding, truncated or unstable protein due to a switch in APA usage, this regulatory mode is called an epigenetic effect, which ultimately silences the target gene [9].
These findings clearly indicate that alternative polyadenylation plays essential roles in coordination of genetic information transfer from genome to phenome. This has triggered a great wave of interest in the research community to develop methods and techniques that can comprehensively capture the 3'-ends of transcripts and thus thoroughly characterize how APA sites influence various physiological, pathological and psychological processes [1, 10-11]. In 2015, Jiang and colleagues [5] reviewed 15 next generation sequencing methods and technologies specifically designed to profile the 3' termini of RNAs with or without restriction enzyme digestion. In the present review, we evaluate methods advanced during the last 2 - 3 years, summarize data processing strategies and discuss ways to validate the functional significance of APA sites.
Newly developed methods for APA profiling
Here we review five recently developed methods for capturing the 3'-ends of transcripts associated with polyA+ tails: 1) LITE-Seq (Low-Input 3'-Terminal sequencing method) [4]; 2) PAC-seq (PolyA Click sequencing method) [12]; 3) BAT-seq (BArcoded, Three-Prime specific sequencing method) [13]; 4) WTTS-seq (Whole Transcriptome Termini Site sequencing method) [14] and 5) PAPERCLIP (Poly(A) binding Protein-mediated mRNA 3′End Retrieval by CrossLinking ImmunoPrecipitation) [15]. Although all of these methods use total RNA as their starting materials, their strategies to enrich the polyA+ ends of transcripts are different. Broadly, the first two methods enrich polyA+ cDNA (complementary DNA), while the last three methods enrich polyA+ RNA to complete construction of the next generation sequencing libraries (Figures 1A and 1B).
Outline of library construction procedures involved in five newly developed methods. (A) LITE-seq and PAC-seq enrich polyA+ cDNA. (B) BAT-seq, WTTS-seq and PAPERCLIP-seq target polyA+ RNAs.
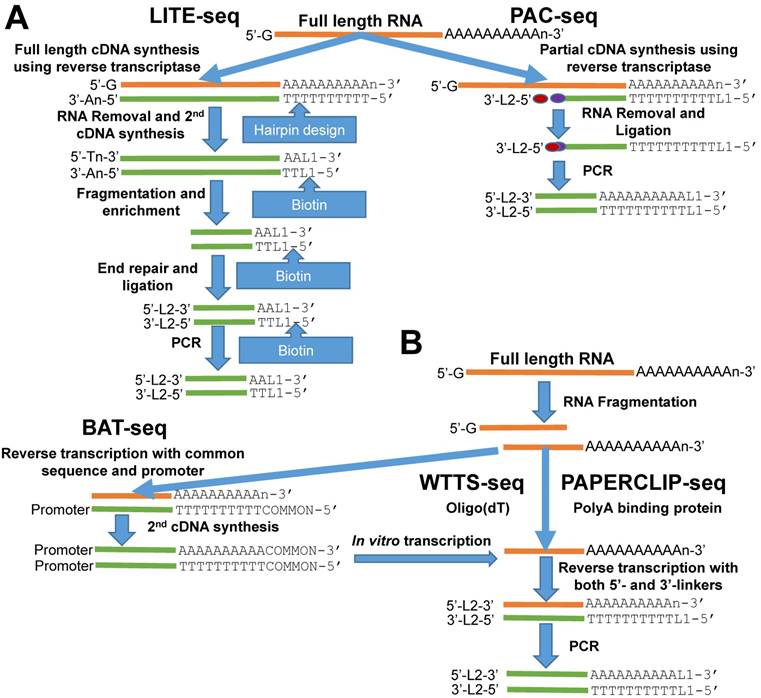
The LITE-Seq library preparation method does not deplete rRNA molecules and begins with synthesis of full-length cDNA, targeting polyA+ RNAs by reverse transcription using oligo (dT) primers containing a hairpin structure. Next, a polyA tail is added to the first-strand cDNA and second-strand synthesis completed using PCR, which integrates biotinylated half-hairpin primers at the 3' ends (Figure 1A). These full-length, double-stranded cDNA molecules are then fragmented and polyA+ cDNAs enriched using streptavidin beads. Like conventional RNA-seq, the remaining steps include end repair, dA tailing, adaptor ligation and PCR amplification using primers that fit with the sequencing platforms. In contrast, PAC-seq synthesizes a partial cDNA because the reverse transcription reaction utilizes azido-nucleotides, which induce termination of cDNA synthesis once incorporated (Figure 1A). Oligo (dT) primers containing 3' partial adaptors dictate first-strand cDNA synthesis so that only fragments associated with polyA products are enriched. The click ligation reaction, which is catalyzed by vitamin C and Cu-TBTA at room temperature, is then performed to join the azido-terminated cDNA and the 5 hexynyl-functionalized DNA oligos. The chemically ligated products are purified, amplified by PCR and size-selected for sequencing. While the PAC-seq method involves relatively few steps, the efficiency in the chemical ligation step can be very low [12].
Fragmentation of total RNAs is the first step involved in the BAT-seq, WTTS-seq and PAPERCLIP-seq library preparation methods [13-15] (Figure 1B). However, the subsequent procedures for each are quite different, especially between the first and the last two methods (Figure 1B). By targeting polyA+ RNAs using oligo(dT) primers, BAT-seq builds constructs with promoters, which leads to an in vitro transcription to make polyA+ RNA fragments expressed one more time for enrichment. These multiple amplification steps are required because BAT-seq is designed to capture the 3'ends of transcripts in a single cell. In contract, WTTS-seq and PAPERCLIP-seq capture polyA+ RNAs immediately with either oligo(dT) beads or polyA binding protein after fragmentation. Adaptors that fit with the sequencing platforms are then added to the polyA+ RNA fragments by reverse transcription. Regardless of method, size selection is required to select appropriate-sized products for sequencing.
Among the five methods described above, only our WTTS-seq method was designed to fit with the Ion Torrent sequencing platform [14]. WTTS-seq utilizes a strand-specific sequencing approach so that each read begins with a polyT stretch, complementary to the polyA tail. Our assay can be redesigned to fit with the Illumina sequencing platform, however, a “low-diversity library” issue may occur if it runs alone on the Illumina sequencing platform, which requires equal proportions of the four nucleotides at each reading position. Furthermore, we use RNases H and I to destroy all RNA molecules after reverse transcription so that our WTTS-seq enriches both polyA+ RNA and polyA+ cDNA [14]. Oligo (dT) primers are used in all five of these methods to synthesize first-strand cDNA. Two-base anchors are included in the oligo (dT) primer for WTTS-seq and PAPERCLIP-seq, but only a one-base anchor is included in the primer for LITE-seq, and noanchor is used for BAT-seq and PAC-seq. The amount of total RNA required for successful library preparation are variable among these five methods, ranging from 125 ng (PAC-seq) to 5 µg (WTTS-seq). Time required for library construction among methods is also variable. For example, it takes a technician at least 40 hours to create a library using PAPERCLIP-seq protocol [15].
Bioinformatics pipelines for APA characterization
In general, data processing includes raw read quality control, 5' or 3' end trimming, genome mapping, APA clustering and characterization, assessment of differential expression and pathway enrichment. Once libraries are well separated with individual barcodes, raw reads are processed for quality control and sequencing scores using CIMS (crosslinking induced mutation sites) (http://zhanglab.c2b2.columbia.edu/index.php/CIMS), FastQC (a quality control tool for high throughput sequence data) (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) or FASTX Toolkit version 0.0.13.1 (http://hannonlab.cshl.edu/fastx_toolkit/), for example.
Depending on the library construction protocol and sequencing platform, the ideal reads that contain APA sites should have either poly(T) at their 5'-ends or poly(A) at their 3'-ends. To improve successful rate of read mapping, it works best to trim off these poly(T) or poly(A) because they do not exist at the DNA level. Software, such as Cutadapt [16] or Perl script [14] can be used to complete the trimming process.
For read mapping, selection of a well-assembled genome is essential, which can be downloaded from either UCSC Genome Browser (https://genome.ucsc.edu/) or the NCBI ftp site (https://ftp.ncbi.nih.gov/genomes/). NovoAlign (http://www.novocraft.com/products/novoalign/), GSNAP (genome short-read nucleotide alignment program) [17], Bowtie (http://bowtie-bio.sourceforge.net/index.shtml) [18] and TopHat2 (https://ccb.jhu.edu/software/tophat/index.shtml) [19] are frequently used to map reads to different genome/gene regions. CIMS (http://zhanglab.c2b2.columbia.edu/index.php/CIMS), F-seq [20], PAcluster [21] and PlantAPA [22] can be used to call APA clusters, usually within a 20 - 30 bp window.
There are several ways to characterize APA sites by type or category. 3'UTR and CDS-APA sites can be separated using closestBed with the BEDtools suite [23]. This simple classification allows examination of APA switching events within 3'UTRs or between coding regions and 3'UTRs [24]. Genomic features, such as 5'UTR, intron, exon, 3'UTR and intergenic regions can be used to classify APA sites [25]. Differentially expressed APA (DE-APA) sites can be determined using DEXSeq, DESeq2, edgeR, HTSeq and XBSeq2 [26-30]. Differentially expressed genes (DEGs) associated with DE-APA sites can be used to enrich GO terms using PantherDB [31], KEGG (Kyoto Encyclopedia of Genes and Genomes, http://www.genome.jp/kegg/), and DAVID (the database for annotation, visualization and integrated discovery, https://david.ncifcrf.gov/), for example.
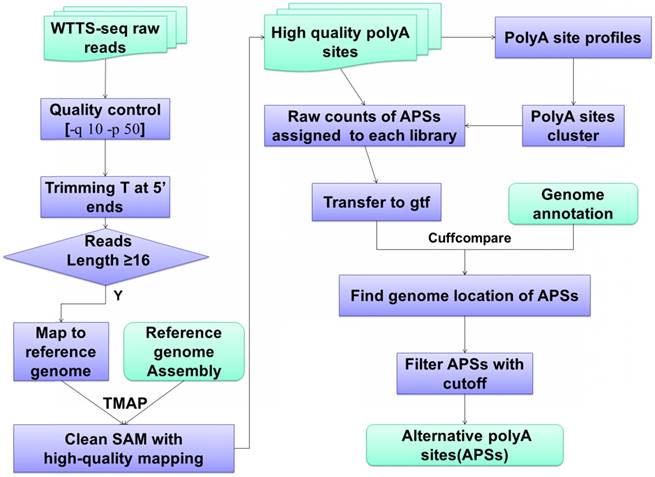
The bioinformatics pipelines for APA processing described above have been well tested by the scientific community [24, 32-35]. Figure 2 demonstrates the bioinformatics pipelines we have developed to analyze our WTTS-seq datasets [1, 14, 36-37]. First, we use TMAP (version 3.4.1, https://github.com/iontorrent/TMAP) to map reads to genomes because the package fits well with libraries sequenced on an Ion PGM™ Sequencer. Second, we explore gene biotypes, such as protein-coding genes, long non-coding genes, microRNAs, pseudogenes and small RNAs, which can be downloaded from NCBI databases for the species of interest. APA usage is significantly different in gene biotypes. Average APA usage per gene is extremely high in protein coding genes, moderate in lncRNAs and pseudogenes and low in small RNAs and miRNAs [1, 36-37]. Third, we use the Cuffcompare (v2.2.1) program [38] to classify APA sites into 1) genic regions with class codes c (or cAPA sites, confined in exonic regions), e (or eAPA sites, extended from exonic regions to intronic regions with at least 10 bp), i (or iAPA sites, completed in the intronic regions), o (or oAPA sites, exonic regions with extension), p (or pAPA sites, located within 2 kb downstream of reference transcripts) and x (or xAPA sites, exonic regions, but with opposite direction); and 2) intergenic regions with u (or uAPA sites, remain unknown). Different gene biotypes tend to utilize certain class codes. Lastly, we employ the Metascape program [39] for pathway enrichment. The unique feature about this program is that it can take multiple lists of genes to pursue pathway analyses simultaneously.
WTTS-seq raw data processing and bioinformatics pipeline. Data analysis usually involves quality control, reads mapping, APA clustering, location assignment and classification for characterization.
Wet and dry laboratory approaches for APA validation
It is preferable to use multiple methods to validate APA sites and their expression abundances. Here we focus on two wet laboratory approaches: RHAPA (RNase H alternative polyadenylation assay) [40] and 3'RACE-seq (3' rapid amplification of cDNA end sequencing) [41] that were designed to directly validate APA sites. For the former method, gene-specific oligonucleotides are synthesized and hybridized to all alternative polyadenylation transcripts for validation, followed by RNase H digestion. The digested products are then used to synthesize the first cDNA strand, just for RNA fragments containing polyA tails. Such procedures avoid any overlapping cDNAs synthesized among alternative transcripts. Finally, qRT-PCR is carried out using alternative transcript specific primers to directly measure and quantify each transcript. This method does not effectively identify APA sites that are less than 100 bp apart. The latter method is actually a combination between conventional RACE and high-throughput sequencing [41]. The authors used the commercial 3'RACE adaptor: 5'- GCGAGCACAGAATTAATACGACTCACTATAGGT12VN-3' (Promega, Madison, WI, USA) for cDNA synthesis, followed by the RACE amplification using oligonucleotides adjacent to the initiation codon as the forward primer. The amplified products are then sequenced using high-throughput platforms and reads are mapped to genes for APA counts. The potential drawback of this method is that some APA sites can be missed if they use different alternative transcriptional start sites. As such, we recently decided to use WTSS-seq (whole transcriptome start site sequencing) to indirectly validate WTTS-seq results. Interestingly, both methods match effectively in terms of functional pathways [36-37].
Dry laboratory approaches can be used to generate additional evidence for indirect validation of APA sites. For example, QAPA (Quantification of alternative polyadenylation) [42] and APAtrap [43] are recently released software packages that can be systematically used to retrieve and collect APA sites from RNA-seq data. In addition, at least three APA databases: PolyA_DB (http://www.polya-db.org/v3), APASdb (http://mosas.sysu.edu.cn/utr) and APADB (http://tools.genxpro.net/apadb/) [44-46] have been established to provide information on APA variants, location, usage and signals. PolyA_DB involves four species: human, mouse, rat and chicken, APASdb holds APA information on humans, mice and zebrafish, and APADB includes APA sites for humans, chickens and mice. We plan to establish our own APA resources for cattle, chicken, mouse, rat and Xenopus tropicalis in the near future.
In summary, tools, pipelines and resources to characterize alternative polyadenylation events in cells, tissues and even whole organisms derived from both animals and plants are well developed. Our recent studies clearly indicated that APA sites are sensitive and powerful biomarkers that illustrate information flows from genome to phenome under unique internal and external environments. As such, we believe that characterization of alternative polyadenylation events will provide novel insights into genome function related to genetic complexity of economically important traits in animals and plants.
Acknowledgements
This work was supported by the Eunice Kennedy Shriver National Institute of Child Health & Human Development of the National Institutes of Health under Award Number R21HD076845 and the National Institute of Food and Agriculture, United States Department of Agriculture under Award Number 2016-67015-24470 to ZJ.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Brutman JN, Zhou X, Zhang Y, Michal J, Stark B, Jiang Z. et al. Mapping diet-induced alternative polyadenylation of hypothalamic transcripts in the obese rat. Physiol Behav. 2018;188:173-80
2. Srivastava AK, Lu Y, Zinta G, Lang Z, Zhu JK. UTR-Dependent Control of Gene Expression in Plants. Trends Plant Sci. 2018;23:248-59
3. Wanke KA, Devanna P, Vernes SC. Understanding Neurodevelopmental Disorders: The Promise of Regulatory Variation in the 3'UTRome. Biol Psychiatry. 2018;83:548-57
4. West SM, Mecenas D, Gutwein M, Aristizábal-Corrales D, Piano F, Gunsalus KC. Developmental dynamics of gene expression and alternative polyadenylation in the Caenorhabditis elegans germline. Genome Biol. 2018;19:8
5. Jiang Z, Zhou X, Li R, Michal JJ, Zhang S, Dodson MV. et al. Whole transcriptome analysis with sequencing: methods, challenges and potential solutions. Cell Mol Life Sci. 2015;72:3425-39
6. Matoulkova E, Michalova E, Vojtesek B, Hrstka R. The role of the 3' untranslated region in post-transcriptional regulation of protein expression in mammalian cells. RNA Biol. 2012;9:563-76
7. Tian B, Manley JL. Alternative cleavage and polyadenylation: the long and short of it. Trends Biochem Sci. 2013;38:312-20
8. Shi Y. Alternative polyadenylation: new insights from global analyses. RNA. 2012;18:2105-17
9. Ma L, Pati PK, Liu M, Li QQ, Hunt AG. High throughput characterizations of poly(A) site choice in plants. Methods. 2014;67:74-83
10. Graham SV, Faizo AAA. Control of human papillomavirus gene expression by alternative splicing. Virus Res. 2017;231:83-95
11. Therrien M, Heiman M. Resolving CNS mRNA Heterogeneity: Examining mRNA Alternative Polyadenylation at a Cell-Type-Specific Level. Neuron. 2017;95:1232-3
12. Routh A, Ji P, Jaworski E, Xia Z, Li W, Wagner EJ. Poly(A)-ClickSeq: click-chemistry for next-generation 3΄-end sequencing without RNA enrichment or fragmentation. Nucleic Acids Res. 2017;45:e112
13. Velten L, Anders S, Pekowska A, Järvelin AI, Huber W, Pelechano V, Steinmetz LM. Single-cell polyadenylation site mapping reveals 3' isoform choice variability. Mol Syst Biol. 2015;11:812
14. Zhou X, Li R, Michal JJ, Wu XL, Liu Z, Zhao H. et al. Accurate Profiling of Gene Expression and Alternative Polyadenylation with Whole Transcriptome Termini Site Sequencing (WTTS-Seq). Genetics. 2016;203:683-97
15. Hwang HW, Darnell RB. Comprehensive Identification of mRNA Polyadenylation Sites by PAPERCLIP. Methods Mol Biol. 2017;1648:79-93
16. Martin M, 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17, pp. 10-12. doi:10.14806/ej.17.1.200.
17. Wu TD, Nacu S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics. 2010;26:873-81
18. Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25
19. Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36
20. Boyle AP, Guinney J, Crawford GE, Furey TS. F-Seq: a feature density estimator for high-throughput sequence tags. Bioinformatics. 2008;24:2537-8
21. Ji G, Lin Q, Long Y, Ye C, Ye W, Wu X. PAcluster: Clustering polyadenylation site data using canonical correlation analysis. J Bioinform Comput Biol. 2017;15:1750018
22. Wu X, Zhang Y, Li QQ. PlantAPA: A Portal for Visualization and Analysis of Alternative Polyadenylation in Plants. Front Plant Sci. 2016;7:889
23. Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841-2
24. Li J, He L, Zhang Y, Xue C, Cao Y. A novel method for genome-wide profiling of dynamic host-pathogen interactions using 3' end enriched RNA-seq. Sci Rep. 2017;7:8681
25. Hong L, Ye C, Lin J, Fu H, Wu X, Li QQ. Alternative polyadenylation is involved in auxin-based plant growth and development. Plant J. 2018;93:246-58
26. Anders S, Reyes A, Huber W. Detecting differential usage of exons from RNA-seq data. Genome Res. 2012;22:2008-17
27. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550
28. Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139-40
29. Anders S, Pyl PT, Huber W. HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166-9
30. Liu Y, Wu P, Zhou J, Johnson-Pais TL, Lai Z, Chowdhury WH, Rodriguez R, Chen Y. XBSeq2: a fast and accurate quantification of differential expression and differential polyadenylation. BMC Bioinformatics. 2017;18(Suppl 11):384
31. Mi H, Poudel S, Muruganujan A, Casagrande JT, Thomas PD. PANTHER version 10: expanded protein families and functions, and analysis tools. Nucleic Acids Res. 2016;44:D336-42
32. Fontes MM, Guvenek A, Kawaguchi R, Zheng D, Huang A, Ho VM. et al. Activity-Dependent Regulation of Alternative Cleavage and Polyadenylation During Hippocampal Long-Term Potentiation. Sci Rep. 2017;7:17377
33. Sanfilippo P, Miura P, Lai EC. Genome-wide profiling of the 3' ends of polyadenylated RNAs. Methods. 2017;126:86-94
34. Chen M, Lyu G, Han M, Nie H, Shen T, Chen W. et al. 3' UTR lengthening as a novel mechanism in regulating cellular senescence. Genome Res. 2018 Feb 12. doi: 10.1101/gr.224451.117. [Epub ahead of print]
35. Jereb S, Hwang HW, Van Otterloo E, Govek EE, Fak JJ, Yuan Y. et al. Differential 3' processing of specific transcripts expands regulatory and protein diversity across neuronal cell types. Elife. 2018 Mar 26;7. pii: e34042. doi: 10.7554/eLife.34042. [Epub ahead of print]
36. Zhang S, Zhang Y, Zhou X, Fu X, Michal JJ, Ji G, Du M, Davis JF, Jiang Z. Alternative polyadenylation drives genome-to-phenome information detours in the AMPKα1 and AMPKα2 knockout mice. Sci Rep. 2018;8:6462
37. Zhou X, Zhang Y, Michal JJ, Qu LJ, Zhang SW, Wildung MR. et al. Alternative Polyadenylation Coordinates Embryonic Development, Sexual Dimorphism and Longitudinal Growth in Xenopus tropicalis. Cell Mol Life Sci. 2018 (Resubmitted)
38. Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR. et al. 2012. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2002;7:562-78
39. Tripathi S, Pohl MO, Zhou YY, Rodriguez-Frandsen A, Wang GJ, Stein DA. et al. Meta- and Orthogonal Integration of Influenza "OMICs'' Data Defines a Role for UBR4 in Virus Budding. Cell Host Microbe. 2015;18:723-35
40. Cornett AL, Lutz CS. RHAPA: a new method to quantify alternative polyadenylation. Methods Mol Biol. 2014;1125:157-67
41. Wang H, Wang M, Cheng Q. Capturing the Alternative Cleavage and Polyadenylation Sites of 14 NAC Genes in Populus Using a Combination of 3'-RACE and High-Throughput Sequencing. Molecules. 2018:23 pii: E608
42. Ha KCH, Blencowe BJ, Morris Q. QAPA: a new method for the systematic analysis of alternative polyadenylation from RNA-seq data. Genome Biol. 2018;19:45
43. Ye C, Long Y, Ji G, Li QQ, Wu X. APAtrap: identification and quantification of alternative polyadenylation sites from RNA-seq data. Bioinformatics. 2018 Jan 18. doi: 10.1093/bioinformatics/bty029. [Epub ahead of print]
44. Wang R, Nambiar R, Zheng D, Tian B. PolyA_DB 3 catalogs cleavage and polyadenylation sites identified by deep sequencing in multiple genomes. Nucleic Acids Res. 2018;46:D315-D319
45. You L, Wu J, Feng Y, Fu Y, Guo Y, Long L. et al. APASdb: a database describing alternative poly(A) sites and selection of heterogeneous cleavage sites downstream of poly(A) signals. Nucleic Acids Res. 2015;43:D59-67
46. Müller S, Rycak L, Afonso-Grunz F, Winter P, Zawada AM, Damrath E. et al. APADB: a database for alternative polyadenylation and microRNA regulation events. Database (Oxford). 2014 pii: bau076
Author contact
![]() Corresponding author: Dr. Zhihua Jiang, Professor of Comparative Genomics. Phone: 509-335 8761; Email: jiangzedu
Corresponding author: Dr. Zhihua Jiang, Professor of Comparative Genomics. Phone: 509-335 8761; Email: jiangzedu