Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. Methods and Results
3. Discussion
Acknowledgements
Conflicts of Interest
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2006; 2(2):54-60. doi:10.7150/ijbs.2.54 This issue Cite
Research Paper
Identification and Characterisation of five novel Miniature Inverted-repeat Transposable Elements (MITEs) in amphioxus (Branchiostoma floridae)
PW. Osborne1 *, GN. Luke2 *, PWH. Holland1, DEK. Ferrier1 ![]()
1. Department of Zoology, University of Oxford, Tinbergen Building, South Parks Road, Oxford, OX1 3PS, UK.
2. School of Biological Sciences, University of Reading Whiteknights, Reading, RG6 6AJ, UK.
* These authors contributed equally to this work.
Received 2006-1-25; Accepted 2006-2-13; Published 2006-4-10
Abstract
As the sister group to vertebrates, amphioxus is consistently used as a model of genome evolution for understanding the invertebrate/vertebrate transition. The amphioxus genome has not undergone massive duplications like those in the vertebrates or disruptive rearrangements like in the genome of Ciona, a urochordate, making it an ideal evolutionary model. Transposable elements have been linked to many genomic evolutionary changes including increased genome size, modified gene expression, massive gene rearrangements, and possibly intron evolution. Despite their importance in genome evolution, few previous examples of transposable elements have been identified in amphioxus. We report five novel Miniature Inverted-repeat Transposable Elements (MITEs) identified by an analysis of amphioxus DNA sequence, which we have named LanceleTn-1, LanceleTn-2, LanceleTn-3a, LanceleTn-3b and LanceleTn-4. Several of the LanceleTn elements were identified in the amphioxus ParaHox cluster, and we suggest these have had important implications for the evolution of this highly conserved gene cluster. The estimated high copy numbers of these elements implies that MITEs are probably the most abundant type of mobile element in amphioxus, and are thus likely to have been of fundamental importance in shaping the evolution of the amphioxus genome.
Keywords: Cephalochordate, Miniature Inverted-repeat Transposable Elements (MITEs), Genome evolution, ParaHox
1. Introduction
Genome sequencing projects have revealed that transposable elements (TEs) account for a startlingly large proportion of genetic material in eukaryotes. TEs for example encode up to 45% of the human genome [1] and even more in some plants (for example [2]). There are two classes of transposable elements which are grouped on the basis of their transposition mechanism. Class I elements move via an RNA intermediate, whereby the entire element is transcribed into RNA and subsequently reverse transcribed back into the genome at another location. In contrast, class II elements ‘cut and paste’ themselves around the genome using a protein called Transposase. Each class has both autonomous and non-autonomous elements. An element which encodes proteins for its own transposition is termed autonomous, whereas non-autonomous elements are thought to transpose by ‘hijacking’ the proteins coded by related autonomous elements.
One group of non-autonomous elements are called miniature inverted-repeat transposable elements (MITEs). These were originally believed to be part of the class I non-autonomous group of elements called SINEs (for example [3]). This classification is now thought to be erroneous and MITEs have been moved into class II. Some MITEs have even been classified into specific class II superfamilies [4]. MITEs are diverse elements which tend to be defined on a similar structural basis rather than sequence similarity. The common structural requirements include a small size (usually less than 600 bp), generally AT rich sequence, terminal inverted repeats (TIRs), target site duplications (TSDs) and non-coding internal sequence.
MITEs have been identified in many animal species including Caenorhabditis elegans, mosquitoes, Xenopus, fish, humans (reviewed in [4]), Drosophila [5], Ciona intestinalis [6] and beetles [7]. Many of the identified MITEs appear to be related to one of several DNA transposon superfamilies (Tc1/mariner, PIF/Harbinger, piggyBac or hAT) [4]. A few individual C. elegans MITEs are also classified into superfamilies such as Mutator, Mirage or Merlin on the basis of TSDs and TIRs. The remainder are simply unclassified.
TEs are a major force of genomic change and evolution. In the Diptera, for example, TEs have been linked to hybrid dysgenesis, horizontal transfer, rearrangements, alternative gene splicing, modified gene expression, creation of heterochromatin and even construction of a new form of telomere (reviewed in [8]). When a TE transposes into a novel genomic location, there is a good chance it will land in or near the coding sequence of a gene. Indeed, there appears to be a high correlation between MITEs and their proximity to genes; they are often found in either introns or sequences flanking the coding sequence [6, 9, 10]. The most likely outcome of landing in coding sequence would be a disruption of the gene’s function, either through a frame-shift or the addition of premature stop codons. TEs landing in genes have also been observed to create novel introns [11] and even exons [12], providing clear examples of functional evolution. A TE does not need to land in a coding sequence to affect the gene’s function. Modified gene expression can also occur if TEs are incorporated into a cis-regulatory region; for example, Yang et al. [13] demonstrated that a MITE (Kiddo) was responsible for modifying rice rubq2 gene expression. As TEs increase in copy number, further biological effects can be observed. The most obvious is an increase in genome size, leading to a greater metabolic load on the organism. Recombination between repeats is also a result of genomes containing multiple TE copies. This can cause significant genome rearrangements such as deletions, duplications, inversions and translocations all of which can lead to developmental defects or cancers.
In contrast to these widespread effects around the rest of the genome, some Hox gene clusters appear to avoid the effects of TEs. Vertebrate Hox clusters for instance, seem to exclude, or be inaccessible to, TEs [14]. Such an absence of TEs may contribute to the maintenance of Hox cluster integrity by reducing the opportunity for disruptive recombinative events. Indeed, there may even be a causal link between TE invasion and disruption of the C. intestinalis Hox cluster [15]. However, the exclusion of TEs from Hox clusters does not extend to the mammalian ParaHox cluster, the evolutionary sister of the Hox cluster [16]. Despite the invasion of TEs into ParaHox clusters of mammals, one full gene cluster has still been conserved in humans for example, from the ancestral vertebrate condition of four ParaHox clusters.
Given this potential link between TEs and the organisation of homeobox gene clusters and the link of homeobox gene function with their organisation in a cluster (for example, [17]), we have performed an analysis of the repetitive transposable elements of amphioxus (Branchiostoma floridae) around the ParaHox cluster and NK homeobox genes. The amphioxus Hox cluster has recently been sequenced and this will be analysed elsewhere (Chris Amemiya, pers. communication.). Only two reports of amphioxus transposable elements have been published. The first is a class I non-LTR retrotransposon [18], while the second is an unusual class II element called ATE-1 [19]. Although the ATE-1 element had some characteristics of a MITE, no definitive amphioxus MITE sequences have been published.
Here we present five novel MITE sequences from B. floridae. We provide a consensus sequence for each element and tentatively classify three of the five elements into transposon superfamilies. This adds to our knowledge of amphioxus transposable elements and will form a starting point for a more thorough investigation of all TEs once the recently sequenced B. floridae genome is assembled (forecast for 2006). Some of the MITEs are present in the amphioxus ParaHox cluster, indicating that like vertebrates TEs are not excluded from this region of the genome. This analysis suggests MITEs are a common TE in the amphioxus genome and will be one of the major agents of genomic evolution in this animal.
2. Methods and Results
Five novel elements have been characterised from the amphioxus sequences around NK homeobox genes and the ParaHox cluster. Initial analyses of cosmid sequences containing the NK genes were performed on the NIX implementation of BLASTn at [
Characterisation of LanceleTn-1
An initial BLASTn analysis of the cosmid sequence Acc. AL671994 (which is part of a contig containing AmphiNK3 and AmphiNK4, [22]) revealed a match to a reported intron in the genomic sequence of AmphiHox-2 (Acc. AB028207.1). BLASTn analyses of this intron on the stand alone database revealed 39 highly significant matches, 23 of which were EST sequences. Alignments between the AmphiHox-2 intron and the 39 BLASTn matching sequences allowed us to assemble a consensus sequence and define its general characteristics.
The characteristics of LanceleTn-1 (Table 1 and Fig. 1) fulfil the criteria for a MITE. These include a 21 bp terminal inverted repeat, and 8 bp target site duplications. In addition, the internal sequence has no coding ability or similarity to any known proteins. MITEs are frequently located within or close to non-coding regions of genes [6, 9, 23-26]. Of the 18 genomic DNA copies of LanceleTn-1 found in this analysis, 10 were closely associated with genes. LanceleTn-1 is the longest of the five amphioxus MITEs at 433 bp (excluding the TSDs).
Characteristics of five novel miniature inverted-repeat transposable elements.
| Name | TSD | TIR* | Total length+ | Estimated Copy No.† | Superfamily classification | A+T content % |
|---|---|---|---|---|---|---|
| LanceleTn-1 | 8 bp§ | 21 bp TAGGGCTGGGTATCGGTACAG | 433 bp | 2500 | hAT | 56 |
| LanceleTn-2 | ? | ≤59 bp TWAGGCTAAGGTCACATTTCCA | 207 bp | 2200 | ? | 55 |
| LanceleTn-3a | 9 bp | 21 bp TAATCTCCAAGCAGATCCTAC | 173 bp | 3600 | Mutator | 55 |
| LanceleTn-3b | 9 bp | 19 bp TACTCTCCAAGCAGAGGTT | 205 bp | 2900 | Mutator | 63 |
| LanceleTn-4 | ? | ≤53 bp TNAAGGTGGTATCTCACTGCAC | 253 bp | 4800 | ? | 56 |
? Not enough information is present to provide a definitive answer (see text for details)
* For LanceleTn-2, 3a, 3b and 4 only the most highly conserved terminal sequence of the TIR is presented. In all four cases loose inverted repeats can be extended further. In addition, the possible TSDs are also included in the TIR sequence for LanceleTn-2 and 4 as the first base of the TIR cannot be assigned without knowing the TSD.
+ Total length is based on the length of the consensus sequence and includes the TIR. The TSDs of LanceleTn-2 and LanceleTn-4 (but not the other three) are also included because it is not clear where the elements end. The putative TSDs in these sequences are underlined.
† The copy number of each element is estimated based on the number of copies found in the known amount of sequence on the NCBI database (see text)
§ As deduced from multiple alignments and confirmed by the AmphiHox-2 sequence within Acc. AB028207.1.
Consensus sequences of the 5 LanceleTn MITEs from amphioxus. Putative TSD sequences are in bold and TIR sequences (as in Table 1) are underlined. The numbers in brackets refer to the number of full length sequences used to create the consensus sequence.

As introns are spliced out of mRNAs after transcription, their splice sites can be identified through a comparison of cDNA and genomic DNA. After comparing a genomic phage clone of AmphiHox-2 (Acc. AB028207.1) and a cDNA of AmphiHox-2, Wada et al. [27] concluded there is an intron in the 3’ UTR of this gene. This putative intron is composed of the LanceleTn-1 sequence described above. In contrast however, the AmphiHox-2 gene in two different PAC clones (Accs. AC124805 and AC129949) lacks LanceleTn-1, indicating the presence or absence of this MITE at this locus is an allelic condition. Indeed, a PCR screen of genomic DNA from 49 further individuals of B. floridae (using primers flanking LanceleTn-1 in AmphiHox-2) failed to detect its presence [28], indicating that the allelic variant of AmphiHox-2 with LanceleTn-1 is present at low frequency in the amphioxus population of Tampa Bay, Florida. It has not yet proven possible to test if the LanceleTn-1 sequence in the AmphiHox-2 phage clone represents a true intron. It is interesting however, that a GT/AG spliceosomal consensus sequence exists in the 5’ TSD and at the border of the 3’ TIR/TSD of the element in this clone (Fig. 2), suggesting splicing may be possible.
Conserved Terminal Inverted Repeat (RED =TIR) and Target Site Duplication (BLUE =TSD) motifs in the flanking regions of the AmphiHox2 LanceleTn-1 insertion. The underlining represents conserved residues in the 21 bp TIR. The arrows represent the suggested, intron/exon boundary following the GT/AG major spliceosome rule as deduced by Wada et al., [27] from cDNA/genomic DNA sequence comparison

Characterisation of LanceleTn-2
LanceleTn-2 was first identified from BLASTn analysis of the cosmid sequence Acc. AL671994 as a significant match within an intron of one of the B. floridae EF1-alpha genes (Acc. AB070234). BLASTn analysis of this intron on the stand-alone database provided 45 sequences (10 of which were ESTs) from which an initial consensus was obtained. This was subsequently refined by the addition of nine further sequences. LanceleTn-2 (Table 1 and Fig. 1) has a probable target site duplication of the dinucleotide TA (although it may actually be TWA, where W represents T or A) and has TIRs of up to 59 bp. The total length of the element is 207 bp. One unusual copy of LanceleTn-2 was identified in AmphiPRHOXNB, a gene flanking the ParaHox cluster. This gene is conserved in vertebrates and Strongylocentrotus purpuratus having only one intron (data not shown). In amphioxus, however a novel intron has been created, breaking the second exon. This intron is made of a LanceleTn-2 element and some unrelated flanking sequence. It is probable that a LanceleTn-2 element jumped into this new intron, in contrast to the other possibility of a LanceleTn-2 element with flanking sequences creating the intron. Subsequently, the 5’ end of this novel intron (which includes half of the LanceleTn-2 element) has undergone multiple tandem duplications to result in a larger intron.
Characterisation of LanceleTn-3a and LanceleTn-3b
LanceleTn-3a was initially characterised from 65 BLASTn matches in the stand-alone database to a region of the third intron of an NK-like gene AmphiLcx [22, 28]. Twenty-eight of these matches were to ESTs. Subsequent analysis of the amphioxus ParaHox cluster revealed an additional closely related MITE, LanceleTn-3b. These two elements have been grouped together because 14 of their terminal 15 bp are identical (see Table 1 and Fig. 1). This 15 bp is possibly the size of the functional TIR but the most conserved sequence of each element’s TIR extends to 21 bp in LanceleTn-3a and 19 bp in LanceleTn-3b (and in both cases a loose inverted repeat is extended even further). In addition, both LanceleTn-3 elements share a 9 bp TSD (although in some of the LanceleTn-3a copies only 8 bp TSDs can be confidently assigned). The length of LanceleTn-3a is 175 bp which is 30 bp shorter than LanceleTn-3b (205 bp). The internal sequence has no similarity between the elements or to any proteins but is conserved within each element.
Characterisation of LanceleTn-4
LanceleTn-4 (Table 1 and Fig. 1) was also identified from BLAST analyses of the ParaHox cluster. It is 253 bp long with up to 53 bp TIRs. Unlike LanceleTn-1, 3a and 3b, identifying the exact sequence of the TSD was difficult. The second and second to last nucleotides varied mostly between T and A, but there were also several G nucleotides. The most likely TSD sequence is TA but other TSDs such as TTAA, TTA or TAA cannot be excluded. Again the internal sequence does not match any other known sequences.
Estimation of the number of LanceleTn elements
The LanceleTn consensus sequences were used as a BLASTn query without low-complexity filtering and limiting the search to Branchiostoma sequences on the Genbank nr database. The number of hits to the CHORI-302 B. floridae BAC contigs on the Genbank nr database (as of Jan 2006) was then counted. As of 5th January 2006 there were 31 sequenced B. floridae BAC contigs with an average length of 170 kb on the NCBI database, making a total of 5.3 Mb of amphioxus DNA sequence. The full genome size of B. floridae is 500-600 Mb [29, 30] which is approximately 100 times greater than this figure. Using this multiplier, we estimated the copy number of each LanceleTn MITE element to range from 2,200 (LanceleTn-2) to 4,800 (LanceleTn-5) (Table 1).
Classification of LanceleTn elements
MITE classification is often not possible due to the lack of sequence similarity between the central regions of MITEs and known transposons. It is therefore usually necessary to base classifications on the size/sequence of the TSD and the sequence of the TIR. The most systematic classification of MITEs is based on a four level designation [4], where levels 1 and 2 have strong evidence for a MITE originating from a larger element. Level 3 provides strong evidence that a MITE is transposed by a particular autonomous element but supplies no information as to the MITE’s origin. A level 4 classification suggests only tentative evidence is available for a relationship between a MITE and a larger element.
As the MITE sequences presented here have no sequence similarity to known transposases from other animals and no amphioxus autonomous DNA transposons are known, the only classification we can use is at level 4. LanceleTn-1 is proposed to fall into the hAT (hobo, Ac, Tam3) superfamily. hAT transposons often have 8 bp TSDs [4] but more conclusively, transposons of the Ac group have a conserved sequence (TAGGGATG) at the ends of the TIR [3]. These are the same characteristics of LanceleTn-1 having only 1 bp different in the conserved sequence (Table 1). LanceleTn-3a and LanceleTn-3b should be classified into the same superfamily as they share the same TSD size and the beginning of their TIR. The sequence of the TIR is not similar to any other elements, but the only group of transposons to have a 9 bp TSD is the Mutator-like superfamily [4]. Thus we group LanceleTn-1, LanceleTn-3a and 3b into the two different superfamilies. LanceleTn-2 and LanceleTn-4 however, do not have TIR sequence similarity to any transposons and their TSDs are less informative than the other MITEs and will therefore have to remain unclassified.
3. Discussion
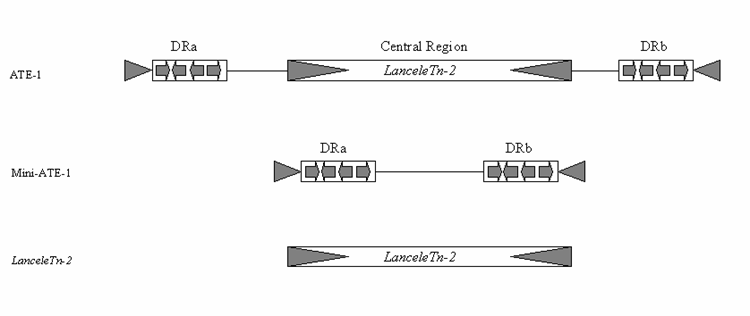
Transposons are major factors in the evolution of animal genomes and amphioxus is a key organism for understanding this evolution. This is due to its proposed sister group relationship to the vertebrates, its derivation from the unduplicated genome state prior to vertebrate origins and the trend for amphioxus to have retained prototypical gene cluster organisation; lost in other invertebrate deuterostome models. Prior to the present report of five amphioxus MITE elements, only two transposable elements have been previously reported in this animal. These are a class I element named BfCR1 [18] and an unusual class II element called ATE-1 [19]. Although all five MITE elements reported here are novel, there is a relationship between LanceleTn-2 and ATE-1. Comparison of their DNA sequences revealed that LanceleTn-2 is almost identical to the central part of ATE-1. In addition to the usual TSD and TIRs, ATE-1 also contained sub-terminal direct repeats and a central region. Cañestro et al. [19] were not able to classify this element, although they did suggest the direct repeats could have originally been two smaller MITEs. It is now clear however, that the central region of this ATE-1 element is actually LanceleTn-2. As there are multiple copies of LanceleTn-2 on its own, the two repetitive elements should be considered separately, and it is valid to assign them different names. Indeed, a BLASTn search of the ATE-1 element identified only the one published copy, which is located in the EF1-alpha gene. There are also up to 10 loosely related copies of a miniature ATE-1 element (without LanceleTn-2) in the CH302 BAC sequences (corresponding to approximately 1000 copies in the genome). This indicates that a ‘mini’ or precursor ATE-1 is an independent mobile DNA element, and distinct from LanceleTn-2 (Fig. 3). It therefore seems likely that a LanceleTn-2 element inserted into a ‘mini’ ATE-1 element which was subsequently identified as the composite ATE-1 element found by Cañestro et al. [19].
Comparison between Cañestro et al.’s [19] composite ATE-1, the ‘Mini’ ATE-1 and LanceleTn-2. The ATE1 element was published as having terminal inverted repeats (grey arrow heads), sub-terminal direct repeats made up of smaller repeated modules (DRa & DRb) and a Central Region. Sequence comparison to the LanceleTn-2 element presented here demonstrates the central region of ATE-1 is actually an independent MITE (LanceleTn-2) and a few copies of a ‘Mini’ ATE-1 without the LanceleTn-2 element were also found in the Branchiostoma sequences on Genbank. This demonstrates the ability for new TEs to form through a fusion of two independent TEs.
One aspect of the MITE analysis reported here that proved difficult was the classification into larger superfamilies. This was because none of the MITEs had significant sequence similarity to other TEs. It will become important in subsequent studies, therefore, to identify autonomous transposons from amphioxus that may be involved in mobilising the MITE elements. If MITEs are simply derivatives of larger elements, a comparison of DNA sequences from autonomous elements would hopefully provide clearer evidence for the evolutionary origin of these LanceleTn elements. However, it has been suggested by Feschotte et al. that “new MITEs may arise de novo from the fortuitous association of TIRs flanking unrelated segments of DNA” [4]. In cases where this has occurred (such as the composite ATE-1), MITEs could only be classified on the basis of which transposon mobilises them rather than where they originated from.
There has been considerable interest in the relationship between transposable elements and conserved gene clusters; for example, it has been noted that vertebrate Hox gene clusters are relatively devoid of TEs [14]. While a detailed analysis of the amphioxus Hox cluster is still underway, the presence of five MITEs lying between the three ParaHox genes (and another four immediately outside) suggests the amphioxus ParaHox gene cluster has not excluded TEs. This is similar to the situation in vertebrates, and marks a significant difference between Hox and ParaHox gene clusters [16]. In all tetrapods examined, there is one intact copy of the ParaHox cluster in addition to three degenerate clusters. Despite the presence of TEs in the intact ParaHox clusters, and the consequent scope for rearrangement they have remained whole, presumably due to a counteracting selective pressure. This selective pressure, for example through shared regulatory elements, may explain why the amphioxus ParaHox cluster has not been broken despite containing TEs. MITEs or other TEs in the ParaHox cluster could provide a basis for the degeneration of three out of four of the tetrapod ParaHox clusters.
In both previous studies of amphioxus transposable elements (BfCR1 and ATE-1; [18, 19]), the copy number of the element was estimated to be less than 20. In contrast, the estimated number for each the five LanceleTn MITEs reported here is over 2000 (Table 1), in the same order of magnitude as some human and Xenopus MITEs [4]. It will be interesting to see if this large copy number holds true once the entire genome is analysed for other elements; we do not anticipate that the five elements described here represent all amphioxus MITE elements. The current evidence suggests MITEs are the predominant form of transposable element in the amphioxus genome and are likely to have been important during the evolution of the amphioxus genome.
Acknowledgements
The authors wish to thank the reviewers for their critical comments and also Tom Butts and Rebecca Furlong for helpful discussions. Work in the authors’ laboratories is funded by the BBSRC. PWO is funded by a Clarendon scholarship and an Overseas Research Student award.
Conflicts of Interest
The authors have declared that no conflict of interest exists.
References
1. Lander ES, Linton LM, Birren B. et al. Initial sequencing and analysis of the human genome. Nature. 2001 ;409(6822):860-921
2. SanMiguel P, Tikhonov A, Jin Y-K. et al. Nested Retrotransposons in the Intergenic Regions of the Maize Genome. Science. 1996 ;274(5288):765-768
3. Unsal K, Morgan GT. A Novel Group of Families of Short Interspersed Repetitive Elements (Sines) in Xenopus - Evidence of a Specific Target Site for DNA-Mediated Transposition of Inverted-Repeat Sines. Journal of Molecular Biology. 1995 ;248(4):812-823
4. Feschotte C, Zhang X, Wessler S. Miniature Inverted-Repeat Transposable Elements and their Relationship to Established DNA Transposons. In: (ed.) Craig NL, Craigie R, Gellert M. et al. Mobile DNA II. Washington DC: ASM Press. 2002:1147-1158
5. Holyoake AJ, Kidwell MG. Vege and Mar: Two novel hAT MITE families from Drosophila willistoni. Molecular Biology and Evolution. 2003 ;20(2):163-167
6. Simmen MW, Bird A. Sequence analysis of transposable elements in the sea squirt, Ciona intestinalis. Molecular Biology and Evolution. 2000 ;17(11):1685-1694
7. Braquart C, Royer V, Bouhin H. DEC: a new miniature inverted-repeat transposable element from the genome of the beetle Tenebrio molitor. Insect Molecular Biology. 1999 ;8(4):571-574
8. Kidwell MG. Transposable Elements and the Evolution of Dipteran Genomes. In: (ed.) Yeates D.K, Wiegmann B.M. The Evolutionary Biology of Flies. New York: Columbia University Press. 2005:145-172
9. Tu ZJ. Three novel families of miniature inverted-repeat transposable elements are associated with genes of the yellow fever mosquito, Aedes aegypti. Proceedings of the National Academy of Sciences of the United States of America. 1997 ;94(14):7475-7480
10. Wessler SR, Bureau TE, White SE. Ltr-Retrotransposons and Mites - Important Players in the Evolution of Plant Genomes. Current Opinion in Genetics & Development. 1995 ;5(6):814-821
11. Giroux MJ, Clancy M, Baier J. et al. De-Novo Synthesis of an Intron by the Maize Transposable Element Dissociation. Proceedings of the National Academy of Sciences of the United States of America. 1994 ;91(25):12150-12154
12. Krull M, Brosius J, Schmitz J. Alu-SINE exonization: En route to protein-coding function. Molecular Biology and Evolution. 2005 ;22(8):1702-1711
13. Yang GJ, Lee YH, Jiang YM. et al. A two-edged role for the transposable element Kiddo in the rice ubiquitin2 promoter. Plant Cell. 2005 ;17(5):1559-1568
14. Fried C, Prohaska SJ, Stadler PF. Exclusion of repetitive DNA elements from gnathostome Hox clusters. Journal of Experimental Zoology Part B-Molecular and Developmental Evolution. 2004 ;302B(2):165-173
15. Ferrier DEK, Holland PWH. Ciona intestinalis ParaHox genes: evolution of Hox/ParaHox cluster integrity, developmental mode, and temporal colinearity. Molecular Phylogenetics and Evolution. 2002 ;24(3):412-417
16. Ferrier DE, Dewar K, Cook A. et al. The chordate ParaHox, cluster. Current Biology. 2005 ;15(20):R820-R822
17. Duboule D, Morata G. Colinearity and Functional Hierarchy among Genes of the Homeotic Complexes. Trends in Genetics. 1994 ;10(10):358-364
18. Albalat R, Permanyer J, Martinez-Mir C. et al. The first non-LTR retrotransposon characterised in the cephalochordate amphioxus, BfCR1, shows similarities to CR1-like elements. Cellular and Molecular Life Sciences. 2003 ;60(4):803-809
19. Cañestro C, Albalat R, Gonzalez-Duarte R. Isolation and characterization of the first non-autonomous transposable element in amphioxus, ATE-1. Gene. 2003 ;318:69-73
20. Altschul SF, Gish W, Miller W. et al. Basic Local Alignment Search Tool. Journal of Molecular Biology. 1990 ;215(3):403-410
21. Jurka J, Kapitonov VV, Pavlicek A. et al. Repbase update, a database of eukaryotic repetitive elements. Cytogenetic and Genome Research. 2005 ;110(1-4):462-467
22. Luke GN, Castro LFC, McLay K. et al. Dispersal of NK homeobox gene clusters in amphioxus and humans. Proceedings of the National Academy of Sciences of the United States of America. 2003 ;100(9):5292-5295
23. Bureau TE, Ronald PC, Wessler SR. A computer-based systematic survey reveals the predominance of small inverted-repeat elements in wild-type rice genes. Proceedings of the National Academy of Sciences of the United States of America. 1996 ;93(16):8524-8529
24. Bureau TE, Wessler SR. Tourist - a Large Family of Small Inverted Repeat Elements Frequently Associated with Maize Genes. Plant Cell. 1992 ;4(10):1283-1294
25. Bureau TE, Wessler SR. Mobile Inverted-Repeat Elements of the Tourist Family Are Associated with the Genes of Many Cereal Grasses. Proceedings of the National Academy of Sciences of the United States of America. 1994 ;91(4):1411-1415
26. Bureau TE, Wessler SR. Stowaway - a New Family of Inverted Repeat Elements Associated with the Genes of Both Monocotyledonous and Dicotyledonous Plants. Plant Cell. 1994 ;6(6):907-916
27. Wada H, Garcia-Fernandez J, Holland PWH. Colinear and segmental expression of amphioxus Hox genes. Developmental Biology. 1999 ;213(1):131-141
28. Luke GN. The NK Homeobox gene cluster of Branchiostoma floridae. Reading: The University of Reading. 2004
29. Atkin NB, Ohno S. DNA values of four primitive chordates. Chromosoma. 1967 ;23(1):10-13
30. Gibson-Brown JJ, Osoegawa K, McPherson JD. et al. A proposal to sequence the amphioxus genome submitted to the joint genome institute of the US department of energy. Journal of Experimental Zoology Part B-Molecular and Developmental Evolution. 2003 ;300B(1):5-22
Author contact
![]() Corresponding address:
Corresponding address: