Impact Factor ISSN: 1449-2288
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. Material and methods
3. Results
4. Discussion
Acknowledgements
Abbreviations
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2007; 3(2):71-76. doi:10.7150/ijbs.3.71 This issue Cite
Short Research Communication
Role of TAB1 in nitric oxide-induced p38 activation in insulin-producing cells
Natalia Makeeva, Godfried M. Roomans, Nils Welsh ![]()
Department of Medical Cell Biology, Uppsala University, Sweden
Received 2006-9-5; Accepted 2006-11-23; Published 2006-11-25
Abstract
The aim of present study was to elucidate the role of TAB1 in nitric oxide-induced activation of p38 MAPK. For this purpose we over-expressed TAB1 in insulin-producing β-TC6 cells. We observed in cells transiently over-expressing TAB1 that p38 activation was enhanced in response to DETA/NONOate. A lowering of TAB1 levels, using the siRNA technique, resulted in the opposite effect. The DETA/NONOate-induced cell death rate was increased in cells transiently overexpressing TAB1. In stable β-TC6 cell clones with very high TAB1 levels p38 phosphorylation was enhanced also at basal conditions. DETA/NONOate increased also the phosphorylation of JNK and ERK in β-TC6 cells, but these events were not affected by TAB1. Interestingly, the inhibitory effect of SB203580 on p38 phosphorylation was paralleled by a stimulatory effect on JNK phosphorylation and an inhibitory effect on ERK phosphorylation. In summary, we propose that TAB1 promotes nitric oxide-induced p38 autophosphorylation. In addition, nitric oxide-induced p38 activation seems to promote JNK inhibition and ERK activation, but this effect appears to not require TAB1. A better understanding of how the TAB1/p38 pathway promotes β-cell death in response to nitric oxide might help in the development of novel pharmacological approaches in the treatment of diabetes.
Keywords: apoptosis, nitric oxide, insulin producing cell, TAB1, p38 MAPK
1. Introduction
Type 1 diabetes is an autoimmune disease leading to extensive destruction of the pancreatic β-cells. Cell dysfunction and damage may result from direct contact with islet-infiltrating macrophages and T cells and/or exposure to soluble products of these cells, such as cytokines and free radicals. The radical nitric oxide (NO) is a possible mediator of pancreatic β-cell damage in insulin-dependent diabetes mellitus [1]. Increased production of NO, mediated by the inducible isoform of NO synthase (iNOS), in response to pro-inflammatory cytokines occurs not only in insulin producing β-cells [2], but also activated duct cells [3], macrophages [4] and endothelial cells [5] that are present in the islet micro-environment. NO participates in the regulation of the physiological activities of cells as well as in cytotoxic events. It possesses a biphasic effect on cell viability by both protecting against pro-apoptotic stimuli at moderate concentrations, and by inducing apoptosis when produced at high concentrations [6]. NO-induced cell death may involve multiple signaling pathways [7]. For example, NO has been shown to activate caspases and the tumor supressor p53, and down-regulate Bcl-2 [8,9]. NO-production inhibits the mitochondrial enzyme aconitase in rodent islet cells, leading to a suppressed mitochondrial activity and a defective insulin release [2,10]. We have also observed that NO-production results in a transient increase in p53 levels in RINm5F cells [11]. In addition, recent investigations indicate that NO promotes ER stress in insulin-producing cells [12].
The MAPKs, which include extracellular signal-regulated kinase 1/2 (ERK1/2), c-Jun N-terminal protein kinase (JNK), ERK/big MAP kinase 1 (BMK1) and p38 kinase play various roles in cellular signal pathways induced by numerous extracellular signals. These kinases have been implicated in the control of several diverse biological processes, such as cell proliferation, differentiation and apoptosis. The β-cell MAP kinases are rapidly activated in response to the cytokines IL-1β and TNF-α [2,13,14]. According to a recent report, the MAPK pathway is also stimulated by NO [15]. Activation of the MAP kinases may promote β-cell death as inhibition of JNK protects β-cell lines against IL-1β induced apoptosis [16,17] and human islets against the destruction mediated by IL-1β, TNF-α and IFN-γ [18,19]. In addition, inhibition of p38 protected against cytokine-induced rat islet [14] and human islet cell death [20].
It has been shown that p38 activation can be carried out not only by its upstream MAPK kinase (MKK3/6) but also by p38 autophosphorylation [21]. P38 autophosphorylation requires interaction of p38 with TAB1 [22]. TAB1 is a protein that was initially described as an activator of a member of MAPKK kinase TAK1 in response to stimulation of ΤGF-β [23]. The C-terminal 68-amino acid portion of TAB1 is sufficient for binding to and activation of TAK1 [24]. However, the portion of the TAB1 protein that is responsible for p38 interaction and activation is located N-terminal to the TAK1 binding site [21].
We have recently observed that p38 is autophosphorylated in response to cytokines in insulin producing cells [20]. The aim of the present investigation was to study whether also NO promotes p38 autophosphorylation and whether this occurs via the TAB1-dependent mechanism. We report that p38 phosphorylation is stimulated by TAB1 over-expression and that this is paralleled by increased rates of cell death.
2. Material and methods
Materials
The chemicals were obtained from the following sources: [4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-5-(4-pyridyl) imidazole] (SB203580) was from Calbiochem (San Diego, CA, U.S.A.). Recombinant human IL-1β, and recombinant mouse IFN-γ were from PeproTech EC Ltd (London, UK). Polyclonal antibodies against p38 MAP Kinase, SAPK/JNK, phospho- (Thr180/Tyr182) p38, phospho- (Thr183/Tyr185) SAPK/JNK and phospho- (Thr202/Tyr204) p42/p44, were all from Cell Signaling Technology (Beverly, MA, USA). Horseradish peroxidase-linked goat anti-rabbit Ig was from Amersham International (Amersham, UK). Polyclonal ERK-1(C-16), TAB1 antibodies were from Santa Cruz Biotechnology (Santa Cruz, Ca, USA). Streptozotocin was from Sigma (Sigma, St.Louis, MO) and DETA/NO was from Alexis (CH, Lausen, Switzerland).
Cell culture
Β-TC6 cells at the passage number 20-30 (American Tissue Culture Collection, Manassas, VA, USA) were maintained in D-MEM + 10% (vol/vol) fetal calf serum (FCS), benzylpenicilin (100 U/ml) and streptomycin (0.1 mg/ml) at 37°C and 5% CO2.
Overexpression of TAB1 in β-TC6 cells
For transient expression 2-3 x 106 β-TC6 cells, which had been seeded the day before, were transfected with 0.5 μg pEGFP-C1 (Clonetech) + 2 μg pcDNA3-TAB1 using the combination of 9 μl Lipofectamine (Invitrogen) and 9 μl Lipofectamine Plus (Invitrogen) as described by the manufacturer. Control cells were transfected with 2.5 μg of pEGFP-C1 only. The transfection mixture (200 μl) was dropped onto the cells which were in 1.5 ml serum-free D-MEM. The cells were centrifuged for 10 min at 600 g in a swing-out rotor. After 60 min at 37°C, the transfection medium was replaced with full culture medium. The day after transfection GFP-positive cells were sorted using a FACSCalibur flow cytometer (Becton-Dickinson). GFP-positive cells were identified by gating a region with high FL-1 intensity and normal size (FSC). In typical experiments 25-30 % of the cells were GFP-positive and 500000-550000 cells per group were collected. The sorted cells were then re-plated on cover slips in 24 well plates and cultured for another 24 hours.
For stable expression of ΤΑΒ1, β-TC6 cells were transfected with pcDNA3-ΤΑΒ1 using Lipofectamine and Lipofectamine Plus reagents as described above. One day after the transfection G418 (300 μg/ml) was added. At day 6 the G418 dose was reduced to 150 μg/ml. Resistant clones were picked after two weeks and analyzed for expression of ΤΑΒ1 by immunoblot analysis.
Immunoblot analysis
Β-TC6 cells (1x105) were washed in cold PBS and lysed in SDS-β-mercaptoethanol sample buffer containing 1 mM phenylmethyl sulfonyl-fluoride. Samples were then sonicated, boiled, separated on 9% SDS-polyacrylamide gels and electroblotted onto nitrocellulose filters. The filters were incubated with total p38 MAPK, total SAPK/JNK or total p42/p44 ERK antibodies diluted 1:1000 in PBS supplemented with 2.5% bovine serum albumin. The filters were also incubated with phospho-specific p38, phospho-specific SAPK/JNK and phospho-specific p42/44 antibodies diluted 1:1000 in TBS supplemented with 2.5% bovine serum albumin. In some experiments filters were probed for TAB1 (Santa Cruz). Stripping between the antibodies was performed by incubating for 40 min at 55°C in 2% SDS and 0.1 mM β-mercaptoethanol. Horseradish peroxidase-linked goat anti-rabbit Ig was used as a second layer. The immunodetection was performed as described for the ECL immunoblotting detection system (Amersham International). The intensities of the bands were quantified by densitometric scanning using Kodak Digital Science ID software (Eastman Kodak, Rochester, NY, USA).
Cell viability
β-TC6 cells transiently expressing GFP or TAB1+GFP were stained with propidium iodide (20 µg/ml) for 15 min at 37°C. After careful washing to remove the fluorescent dye and to exclude trypsinisation artefacts, cells were trypsinzed and analyzed analyzed for red fluorescence (FL-3) by flow cytometry using the FACSCalibur instrument (Becton-Dickinson). Cells with intermediate fluorescence were gated as apoptotic and cells with high fluorescence were gated as necrotic [25]. The two forms of cell death were combined to total cell death.
siRNA treatment of βTC-6 cells
βTC-6 cells were transfected with siRNA directed against TAB1 or GL3 lucifierase as previously described [20]. TAB1 and GL3 luciferase siRNA was kindly provided by Dr. Jason W. Myers at the and Department of Biochemistry, Stanford University School of Medicine, Stanford, CA, U.S.A. Two days after the transfection cells were incubated with DETA/NONOate (2 mM) for 30 min, which was followed by harvest of cells for immunoblot analysis as described above.
3. Results
Effect of transient TAB1 overexpression on NO-induced p38 phosphorylation and cell death
We have recently demonstrated that cytokine-induced β-cell death involves p38 autophosphorylation [20]. To evaluate whether NO acts via the same mechanism, and whether TAB1 promotes such an event, we presently analyzed the putative role of TAB1 in the NO donor DETA/NONOate-induced p38 autophosphorylation. For this purpose we transiently transfected the β-TC6 cells with GFP and TAB1 constructs. The FACS purified β-TC6 cells were then exposed to the proinflammatory cytokine IL-1β, NO donor DETA/NONOate and sorbitol for 20 minutes. All these treatments increased p38 phosphorylation (Figure 1, Panel A). Autophosphorylation of p38 at amino acids 180 and 182 was inhibited by the addition of SB203580. SB203580 is a pyridinyl imidazole compound that, by binding to the ATP-binding site, selectively inhibits the α- and β-isoforms of p38. In TAB1 overexpressing cells we observed an enhanced phosphorylation of p38 in response to IL-1β and DETA/NONOate (Figure 1, Panel A). In addition, both the IL-1β and the DETA/NONOate–induced p38 phosphorylation was partially counteracted by 10 μM SB203580. Hyperphosphorylation of p38 in response to sorbitol-induced osmotic stress was not affected by SB203580 in control or TAB1 expressing cells, indicating that the sorbitol pathway does not involve TAB1-induced p38 autophosphorylation.
(A) Transient overexpession of TAB1 affects cytokine- and DETA/NO–induced p38 phosphorylation. Two days after transfection and 1 day after FACS sorting, the GFP positive cells were pretreated with SB203580 (10 μM) for 30 min and then stimulated with IL-1β (50 U/ml), DETA/NONOate (2.5 mM) and sorbitol (0.4 M) for 30 min. Cells were lysed, separated by SDS gel electrophoresis and analysed by immunoblotting with p38 and phospho-specific antibodies. The immunoblot is representative for two separate experiments. (B) Effect of TAB1 overexpression on DETA/NO-induced cell death in transiently transfected cells. Two days after transfection and 1 day after cell sorting, GFP-positive β-TC6 cells were exposed to DETA/NONOate (2.5 mM) for 24 h. Cells were then stained with propidium iodide and cell viability was assessed using the FACSCalibur flow cytometer. Results are presented as percentage of dead cells of the total cell count. Bars are means±S.E.M for three independent experiments. * denotes P<0.05 using Student´s paired t-test. (C) Treatment with TAB1 siRNA decreases DETA/NON-induced p38 phosphorylation. Cells were transfected with GL3 lucifierase siRNA (control) or TAB1 siRNA. Two days later cells were incubated with DETA/NONOate (2.5 mM) for 30 min. Cells were lysed, separated by SDS gel electrophoresis and analysed by immunoblotting. The immunoblot is representative for two separate experiments.
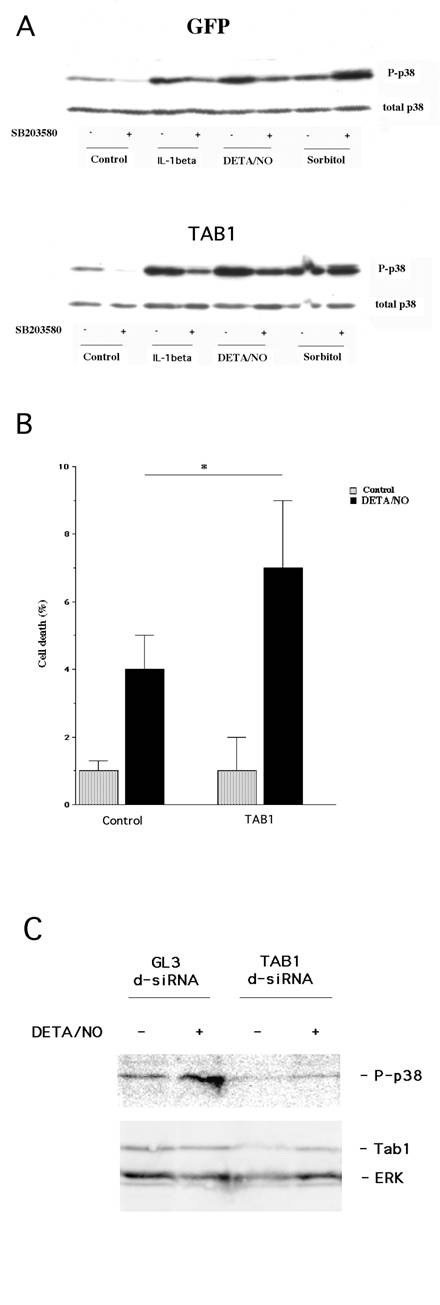
To establish whether TAB1 affects NO-induced cell death, the transiently transfected β-TC6 cells were exposed to DETA/NONOate for 24 hours and propidium iodide uptake was assessed by flow cytometry. We observed that TAB1 overexpression did not affect the basal cell death rate. However, the NO–induced cell death rate was increased in TAB1 overexpressing cells (Figure 1, Panel B).
Effect of TAB1 down-regulation on DETA/NONOate-induced p38 phosphorylation
A genetic loss of function approach was also employed to investigate whether TAB1 participates in nitric oxide-induced p38 activation. TAB1 levels were lowered by treatment with siRNA targeting TAB1 mRNA (Fig. 1C) and this was paralleled by a weaker nitric oxide-induced p38 phosphorylation as compared to control cells treated with GL3 luciferase siRNA (Fig. 1C).
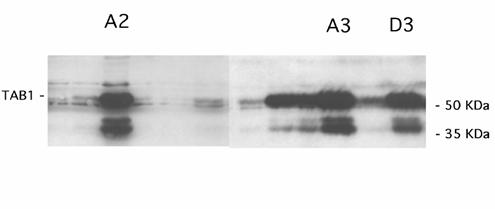
Effect of long-term TAB1 overexpression on p38, JNK, ERK phosphorylation
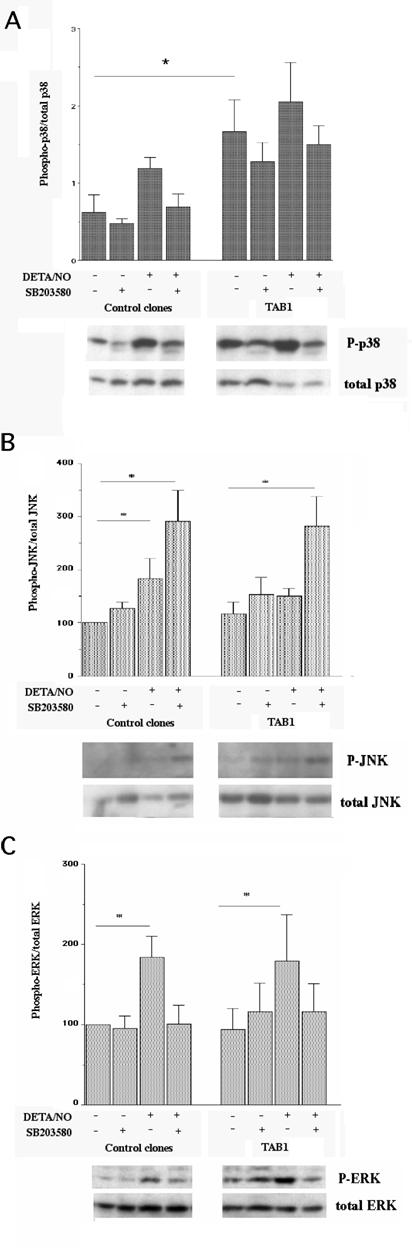
To study the details of TAB1-induced MAPK activation in closer detail, we analyzed three separate β-TC6 cell clones (A2, A3 and D3) with the highest levels of TAB1 expression (Figure 2A). Phosphorylation of p38 was more pronounced in the TAB1 clones than in control cells, particularly at basal conditions (Figure 3, Panel A). As observed with the transiently transfected β-TC6 cells, the p38 inhibitor SB203580 partially counteracted NO induced p38 phosphorylation, both in control and TAB1 clones. The DETA/NONOate-induced phosphorylation of JNK was not enhanced in the TAB1 cells as compared to control cells (Figure 3, Panel B). Instead, the inhibitory effect of SB203580 on p38 phosphorylation was paralleled by a stimulatory effect on the phosphorylation of JNK. Also the ERK phosphorylation was not affected by TAB1 overexpression (Figure 3, Panel C). In this case, however, SB203580 decreased NO-induced ERK phosphorylation.
TAB1 overexpression in stable cell clones. The β-TC6 cells were transfected for expression of TAB1 and neomycin resistance and selected by treatment with G418. After 10-14 days cells were lysed and analysed by immunoblot. The three clones A2, A3 and D3 were selected for further experimentation.
Effect of stable TAB1 overexpression on DETA/NO-stimulated p38, JNK and ERK phosphorylation. (A) The stable clones were pretreated with SB203580 (10 μM) and then stimulated with DETA/NO (2.5 mM) for 30 min. The cells were lysed, separated by SDS gel electrophoresis and analysed by immunoblotting with phospho-specific antibody recognizing phosphorylated p38 and total p38 (Panel A), phosphorylated JNK and total JNK (Panel B) or phosphorylated ERK and total ERK (Panel C). Values of phospho-protein bands were related to those of non-phospho-specific protein bands. Results shown are means±S.E.M. for 3-6 observations. * denotes P<0.05 using two-way ANOVA and Student´s t-test.
4. Discussion
It has been shown that NO plays a role for the β-cell demise in the multi-low dose streptozotocin mouse, which is a model for Type 1 diabetes [26]. NO, produced following cytokine treatment, or supplied exogenously by donor molecules, induces DNA damage, inhibits islet oxidative metabolism, mediates islet morphological degeneration, and induces islet cell death [27,28]. Although the type of islet cell death – apoptosis or necrosis - has been debated [27-29], it is clear that NO participates in the development of diabetes in certain mouse models [26,30]. On the other hand, experiments with human islets have shown that despite an increase in NO production, cytokine-induced human islet functional impartment and cell death cannot be prevented by blockers of NO production [31,32]. Thus, the exact role and biological significance of NO for human β-cell destruction remains uncertain.
We have recently reported that p38 activation is a necessary step in cytokine-induced rodent β-cell death [14,20]. As cytokines induce production of NO, we presently investigated the importance of and the mechanisms by which p38 is activated in response to NO. In the present paper we report (I) that high levels of NO activate p38, (II) that TAB1 augmented basal and NO-activated p38 phosphorylation, (III) that a lowering of TAB1 levels resulted in a weaker p38 activation and (IV) that increased TAB1 levels were associated with increased NO-induced cell death. These findings are compatible with a view that TAB1 plays a role in NO-induced p38 activation. TAB1 is a scaffold protein known to promote p38 autophosphorylation without involvement of TAK1 and MKK3/6 [21]. p38 autophosphorylation can be probed by using SB203580, an inhibitor that blocks p38 autophosphorylation but not MKK3/6-induced p38 phosphorylation. In the present study we consistently observed that SB203580 inhibits cytokine- and NO-induced p38 phosphorylation, but not sorbitol-induced p38 phosphorylation. This indicates that TAB1-stimulated p38 autophosphorylation represents the main p38-activating mechanism in response to cytokines and NO, but not to sorbitol.
Using the insulin producing cell line INS-1, it was recently reported that the NO-donor SNAP activated both p38 and JNK, whereas ERK activity was suppressed or unaffected [15]. In our experimental setup the NO donor DETA/NONOate induced p38, JNK and ERK phosphorylation in β-TC6 cells. This is in line with other studies using RINm5F cells, in which NO induced phosphorylation of ERK [33,34]. However, only the phosphorylation of p38, and not that of JNK or ERK, was presently enhanced in TAB1-overexpressing cells, indicating that NO-induced JNK and ERK phosphorylation does not involve TAB1. Interestingly, inhibition of p38 with SB203580 resulted in enhanced JNK phosphorylation. This finding concurs with a previous demonstration of a negative feed back mechanism in which p38 phosphorylation of TAB1 results in suppression of TAK1-mediated JNK phosphorylation [35]. More surprising was that SB203580 inhibited not only p38, but also NO-induced ERK phosphorylation. At a concentration of 10 μM, SB203580 does not inhibit ERK [36]. Thus, it is likely that SB203580-induced inhibition of ERK results from p38 inhibition, indicating that NO-induced p38 activation promotes a secondary ERK activation.
Activation of MAPK pathways in response to NO seems to occur in most cells, but the consequences of the different events probably differ from cell type to cell type. For example, ERK1/2 and p38 are activated by NO in colon cancer cells, but NO-induced apoptosis was mediated by p38 and not through ERK1/2 activation [37]. On the other hand, NO-mediated apoptosis in osteoblasts required JNK-activity and not p38 [38]. Moreover, in RINm5F cells NO stimulated the phosphorylation of JNK and p38 and selective inhibition of JNK, but not p38 or ERK, completely prevented β-cell recovery from NO-mediated damage [33]. In the present investigation enhanced p38 activity was paralleled by increased NO-induced cell death in cells transiently overexpressing TAB1.
It has been suggested that specific inhibitors of MAP kinases could be used for the treatment of inflammatory disorders, by inhibition of iNOS and NO production [39]. It may be a useful strategy also in the context of diabetes, considering the possible role of cytokines, NO and MAP kinases in the pathogenesis of Type 1 diabetes. However, due to the complex counter-regulatory mechanisms that are activated in response to MAP kinase inhibitors, such a pharmacological approach must be chosen with great care so that the activity of one MAP kinase is not replaced by another.
Acknowledgements
We thank Dr. Jiahuai Han at the Scripps Institute, CA, USA, for providing us with the TAB1 constructs.This work was supported in part by Swedish Medical Research Council grants (72P-12995, 12X-11564 and 12X-109), the Swedish Diabetes Association, the family Ernfors Fund, the Novo-Nordisk Fund and the European Foundation for the Study of Diabetes.
Abbreviations
DETA/NONOate: 2, 2´-(hydroxynitrosohydrazono)bis-ethanamine; ERK: extracellular –signal regulated kinase; GFP: green fluorescent protein; TAB1: transforming growth factor-β-activated protein kinase 1-binding protein 1; IL-1β: interleukin-1β; JNK: c-Jun terminal kinase; MAPK: mitogen activated protein kinase; NO: nitric oxide; TGF-β: transforming growth factor-β
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. Eizirik DL, Pavlovic D. Is there role for NO in β-cell dysfunction and damage in IDDM? Diabetes/metab Rev. 1997 ;13:293-307
2. Welsh N, Eizirik DL, Bendtzen K. et al. Interleukin-1β-induced NO production in isolated rat pancreatic islets requires gene transcription and may lead to inhibition of the Krebs cycle enzyme aconitase. Endocrinology. 1991 ;129:3167-73
3. Pavlovic D, Chen MC, Bouwens L. et al. Contribution of ductal cells to cytokine responses by human pancreatic islets. Diabetes. 1999 ;48:29-33
4. Kroncke KD, Kolb-Bachofen V, Berschick B. et al. Activated macrophages kill pancreatic syngeneic islet cells via arginine-dependent NO generation. Biochem Biophys Res Commun. 1991 ;175:752-8
5. Steiner L, Kroncke K, Fehsel K. et al. Endothelial cells as cytotoxic effector cells: cytokine-activated rat islet endothelial cells lyse syngeneic islet cells via NO. Diabetologia. 1997 ;40:150-5
6. Estevez AG, Jordan J. NO and superoxide, a deadly cocktail. Ann NY Acad Sci. 2002 ;962:207-11
7. Chung HT, Pae HO, Choi BM. et al. NO as a bioregulator of apoptosis. Biochem Biophys Res Commun. 2001 ;282:1075-9
8. Umansky V, Schirrmacher V. NO-induced apoptosis in tumor cells. Adv Cancer Res. 2001 ;82:107-31
9. Lau HK. Cytotoxicity of NO donors in smooth muscle cells is dependent on phenotype, and mainly due to apoptosis. Atherosclerosis. 2002 ;166:223-32
10. Barbu AR, Welsh N, Saldeen J. Cytokine-induced apoptosis and necrosis are preceded by disruption of the mitochondrial membrane potential in pancreatic RINm5F cells: prevention by Bcl-2. Mol Cell Endocrinol. 2002 ;190:75-82
11. Saldeen J, Tilmar L, Karlsson E. et al. Nicotinamide- and caspase-mediated inhibition of poly(ADP-ribose) polymerase are associated with p53-independent cell cycle (G2) arrest and apoptosis. Mol Cell Bioch. 2003 ;243:113-22
12. Cardozo AK, Ortis F, Storling J. et al. Cytokines downregulate the sarcoendoplasmic reticulum pump Ca2+ ATPase 2b and deplete endoplasmic reticulum Ca2+, leading to induction of endoplasmic reticulum stress in pancreatic β-cells. Diabetes. 2005 ;54:452-61
13. Larsen CM, Wadt KA, Juhl LF. et al. Interleukin-1beta-induced rat pancreatic islet NO synthesis requires both the p38 and extracellular signal-regulated kinase 1/2 mitogen-activated protein kinases. J Biol Chem. 1998 ;273:15294-300
14. Saldeen J, Lee JC, Welsh N. Role of p38 MAPK in cytokine-induced rat islet cell apoptosis. Biochem Pharmacol. 2001 ;61:561-9
15. Storling J, Binzer J, Andersson AK. et al. NO contributes to cytokine-induced apoptosis in pancreatic beta cells via potentiation of JNK activity and inhibition Akt. Diabetologia. 2005 ;48:2039-50
16. Ammendrup A, Maillard A, Nielsen K. et al. The c-Jun amino-terminal kinase pathway is preferentially activated by interleukin-1 and controls apoptosis in differentiating pancreatic β-cells. Diabetes. 2000 ;49:1468-76
17. Bonny C, Oberson A, Negri S. et al. Cell-permeable peptide inhibitors of JNK: novel blockers of β-cell death. Diabetes. 2001 ;50:77-82
18. Aikin R, Maysinger D, Rosenberg L. Cross-talk between PI3K/AKT and JNK mediates survival of isolated human islets. Endocrinology. 2004 ;145:4522-31
19. Eckhoff DE, Smyth CA, Eckstein C. et al. Suppression of the c-JNK N-terminal kinase pathway by 17-beta-estradiol can preserve human islet functional mass from proinflammatory cytokine-induced destruction. Surgery. 2003 ;134:169-79
20. Makeeva N, Myers JW, Welsh N. Role of MKK3 and p38 MAPK in cytokine-induced death of insulin producing cells. Biochem. J. 2006 ;393:129-39
21. Ge B, Gram H, Di Padova F. et al. MAPKK-independent activation of p38alpha mediated by TAB1-dependent autophosphorylation of p38alpha. Science. 2002 ;295:1291-4
22. Ge B, Xiong X, Jing Q. et al. TAB1beta (transforming growth factor-beta-activated protein kinase 1-binding protein 1 beta), a novel splicing variant of TAB1 that interacts with p38alpha but not TAK1. J Biol Chem. 2003 ;278:2286-93
23. Shibuya H, Yamaguchi K, Shirakabe K. et al. TAB1: an activator of the TAK1 MAPKKK in TGF-beta signal transduction. Science. 1996 ;272:1179-82
24. Ono K, Ohtomo T, Sato S. et al. An evolutionarily conserved motif in the TAB1 C-terminal region is necessary for interaction with and activation of TAK1 MAPKKK. J Biol Chem. 2001 ;276:24396-400
25. Welsh N. Assessment of apoptosis and necrosis in isolated islets of Langerhans: methodological considerations. Curr Top Biochem Res. 2000 ;2:189-200
26. Flodstrom M, Tyrberg B, Eizirik DL. et al. Reduced sensitivity of inducible NO synthase-deficient mice to multiple low-dose streptozotocin-induced diabetes. Diabetes. 1999 ;48:706-13
27. Mandrup-Poulsen T. β-cell apoptosis: stimuli and signaling. Diabetes. 2001 ;50:S58-S63
28. Kay TW, Thomas HE, Harrison LC. et al. The β-cells in autoimmune diabetes: many mechanisms and pathways of loss. Trends Endocrinol Metab. 2000 ;11:11-5
29. Thomas HE, Kay TW. How β cells die in type 1 diabetes. Curr Direct Autoimmun. 2001 ;4:144-70
30. Takamura T, Kato I, Kimura N. et al. Transgenic mice overexpressing type 2 nitric-oxide synthase in pancreatic beta cells develop insulin-dependent diabetes without insulitis. J Biol Chem. 1998 ;273:2493-6
31. Rabinovitch A, Suarez-Pinzon WL, Strynadka K. et al. Human pancreatic islet β-cell destruction by cytokines is independent of NO production. J Clin Endocrinol Metab. 1994 ;79:1058-62
32. Eizirik DL, Sandler S, Welsh N. et al. Cytokines suppress human islet function irrespective of their effects on NO generation. J Clin Invest. 1994 ;93:1968-74
33. Scarim AL, Nishimoto SY, Weber SM. et al. Role for c-Jun N-terminal kinase in β-cell recovery from NO-mediated damage. Endocrinology. 2003 ;144:3415-22
34. Bernabe JC, Tejedo JR, Rincon P. et al. Sodium nitroprusside-induced mitochondrial apoptotic events in insulin-secreting RIN5mF cells are associated with MAP kinases activation. Exp Cell Res. 2001 ;269:222-9
35. Cheung P, Campbell D, Nebreda A. et al. Feedback control of the protein kinase TAK1 by SAPK2α/p38α. EMBO J. 2003 ;21:793-805
36. Cuenda A, Rouse J, Doza YN. et al. SB 203580 is a specific inhibitor of a MAP kinase homologue which is stimulated by cellular stresses and interleukin-1. FEBS Lett. 1995 ;364:229-33
37. Jeon HK, Choi S, Jung NP. Association of the ERK1/2 and p38 kinase pathways with NO-induced apoptosis and cell cycle arrest in colon cancer cells. Cell Biol Tox. 2005 ;21:115-25
38. Kang YI, Chae SW. JNK/SAPK is required in NO induced apoptosis in osteoblasts. Arch Pharm Res. 2003 ;26:937-42
39. Ajizian SJ, English BK, Meals EA. Specific inhibitors of p38 and extracellular signal-regulated kinase mitogen-activated protein kinase pathways block inducible NO synthase and tumor-necrosis factor accumulation in murine macrophages stimulated with lipopolysaccharide and interferon-γ. J Infec Dis. 1999 ;179:939-44
Author contact
![]() Correspondence to: Nils Welsh, Department of Medical Cell Biology, Husargatan 3, P.O. Box 571, S-75123 Uppsala, Sweden. Telephone: +46-184714212. E-mail: Nils.Welshuu.se
Correspondence to: Nils Welsh, Department of Medical Cell Biology, Husargatan 3, P.O. Box 571, S-75123 Uppsala, Sweden. Telephone: +46-184714212. E-mail: Nils.Welshuu.se