Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. Materials and Methods
3. Results and Discussion
4. Conclusion
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2007; 3(6):375-379. doi:10.7150/ijbs.3.375 This issue Cite
Short Research Communication
High level glucose increases mutagenesis in human lymphoblastoid cells
Ying Zhang 1, *, Junqing Zhou 1,*, Tieli Wang2, Lu Cai 3, 4 ![]()
1. Department of Environmental and Radiological Health Science, Colorado State University, Fort Collins, Colorado 80521, USA
2. Chemistry Department, College of Natural and Behavioral Sciences, California State University, Carson, CA 90747, USA
3. Departments of Medicine and Radiation Oncology, University of Louisville, Louisville, KY 40202, USA
4. Chinese-American Research Institute for Diabetic Complications, Wenzhou Medical College, Wenzhou 4325035, China
* Equal contribution.
Received 2007-8-6; Accepted 2007-8-31; Published 2007-9-4
Abstract
Epidemiological data have suggested an increased cancer rates in diabetic patients, for which the underlying mechanism is poorly understood. We studied whether high level of glucose (HG) treatment that mimic the hyperglycemic condition in diabetes mellitus is mutagenic. Mutagenesis studies were carried out at both hypoxanthine phosphoribosyltransferase (hprt) and thymidine kinase (tk) loci. Role of p53 in HG-induced mutagenesis was also investigated by using human lymphoblastoid cell lines derived from same donor but differs in p53 statuses; TK6 has wild-type p53, NH32 has null p53, and WTK1 has mutant p53 (ile237). In addition, we studied the influence of antioxidant treatment on HG-induced mutagenesis. Mutation fractions at both loci increased significantly in all three lines at 21 and 28 days after HG treatments. At tk locus, the increase of a class of mutants with normal growth rate is mainly responsible for the overall increased mutant fraction. Compared to TK6 cells, both NH32 and WTK1 cells showed an early onset of mutagenesis. Treatment of cells with antioxidant N-acetyl-L-cysteine partially reduced HG induced mutagenesis. This study is the first to indicate that HG is able to induce gene mutation which may be one of the important mechanisms of diabetes-associated carcinogenesis.
Keywords: Hyperglycemia, lymphoblastoid cell, gene mutations
1. Introduction
The prevalence of diabetes mellitus is increasing dramatically worldwide and various diabetic complications threat severely the life of diabetic patients. Epidemiological data have suggested an increased cancer rates in diabetic patients, including primary liver cancer [1, 2], pancreatic cancer [1, 3], colorectal [4, 5], endometrial [6, 7], breast [8, 9], and renal cancer [10]. However, the mechanism of diabetes-associated carcinogenesis is unclear.
Hyperglycemia in diabetic subjects is widely recognized as the major cause for diabetes complications, mainly via overgeneration of reactive oxygen species (ROS) [11]. Fact that diabetes or hyperglycemia causes DNA damage by oxidation to bases and the sugar-phosphates has been demonstrated recently [12, 13]. These DNA damages is likely responsible for the increased cancer in diabetes mellitus. Gene mutation is one of the important mechanisms of carcinogenesis [14]; however whether diabetes or hyperglycemia induces gene mutations has not been addressed yet. We hypothesize that hyperglycemia may induce gene mutation via ROS-induced DNA damage, and such gene mutations may be one of the important mechanisms of diabetes-associated carcinogenesis.
In this study, therefore, we have investigated the mutagenic effects in human lymphoblastoid cells treated with high level of glucose (HG) that mimic the hyperglycemia in diabetic subjects. We carried out mutagenesis studies at both X-linked hypoxanthine phosphoribosyltransferase (hprt) and autosomal thymidine kinase (tk) loci. Because p53 has been implicated in DNA damage response, mutagenesis and tumorigenesis [15], the status of p53 may also play a role in hyperglycemia mediated mutagenesis. We thus selected TK6, NH32, and WTK1 cells in this study since all these three lines were derived from same donor but differed in p53 status; TK6 has wild-type p53, NH32 has null p53, and WTK1 has mutant p53 (ile237) that is one of the most common mutations of p53 gene in human cancers and has been found to promote mutagenesis [16]. In addition, we studied the influence of antioxidant treatment on HG-induced mutagenesis.
2. Materials and Methods
Cell culture
Human lymphoblast cell lines TK6, NH32, and WTK1 are derived from Epstein-Barr virus transformed WI-L2 line. TK6 contains a wild-type p53 gene, NH32 contains null p53 mutation, and WTK1 harbors hemizygous or homozygous p53 mutation (p53ile237) and contains no wild-type p53, thus, mutation assay data obtained from these cell lines can provide useful information about the role of p53 in mutagenesis [17]. All lines were derived from a male and there is only one copy of X-linked hprt gene, which makes both lines applicable to carry out mutagenesis study at hprt locus. In addition, all lines are heterozygous for the autosomal tk gene on chromosome 17q21-23, which makes them widely used in mutagenesis studies at autosomal locus [18]. All cell lines were maintained as exponentially growing cultures in RPMI 1640 medium supplemented with 10% heat inactivated horse serum, 100u/ml penicillin, and 100μg/ml streptomycin. Cultures were incubated at 37oC in 5% CO2 and 100% humidity and maintained at densities of 1-10 × 105 cells/ml. Cultures were treated with either 22 mmol/l D-glucose (Sigma) to mimic hyperglycemia condition in patients, or 5.5 mmol/l glucose as control. To exclude a hyperosmolar effect, we added extra 16.5 mmol/l mannitol (Sigma) in control cultures. N-acetyl-L-cysteine (L-NAC) was purchased from Sigma and added to culture at final concentration of 10mM/l. NAC treatments were carried out daily for 2h. After culturing with glucose for varying time periods, the cultures were washed with normal medium and the cells were subjected to mutation assay.
Mutation assay
We use both hprt and tk as marker genes to study mutagenesis. Both genes code for non-essential salvage pathway enzymes involved in nucleotide metabolism and are similar in that any mechanism which eliminates gene function will produce a mutant, thus, the mutants will be selected easily by colony forming assay when cells are cultured in selective agents. We used our standard protocol to carry out mutagenesis study [18]. Briefly, prior to be used in mutation assay, cells were grown in CHAT medium (complete RPMI 1640 medium with 10-5 M deoxycytidine, 2×10-4M hypoxanthine, 2×10-7 M aminopterin, and 1.75×10-5 M thymidine) for 2 days to reduce the background hprt- and tk-/- mutants. The cells were then allowed to recover for 1 day in THC (10-5 M deoxycytidine, 2×10-4 M hypoxanthine, and 1.75×10-5 M thymidine). At various days after glucose treatment, cultures were grown in normal medium for either 3 or 6 days to allow the expression of mutant phenotype; 3 days for tk-/- and 6 days for hprt- mutants. After expression, cells were seeded into 96-well plates at various densities in the presence of selective agents; 0.5μg/ml 6-thioguinine to select hprt- mutants or 2μg/ml trifluorothymidine (TFT) to select tk-/- mutants. For hprt- mutant selection, cells were seeded at a density of 20000 cells/well in 0.2ml medium/well. For tk-/- mutant selection, TK6 and NH32 cells were seeded at a density of 20000 cells/well in 0.2ml medium. WTK1 cells were seeded at a density of 20000 cells/well in 0.2ml medium. Cells from each sample were also seeded at 1 cell/well in the absence of selective agents to determine plating efficiency (PE). The colonies cultured with 6-thioguinine were determined to be hprt mutants and scored at 11 days after seeding. The colonies cultured with TFT scored at 11 days are classified to be tk normal growth mutants (tk-NG). For plates for selecting tk-/- mutants, 0.02ml of 20μg/ml TFT was refed to restore the concentration of TFT since TFT is unstable in medium and extended incubation causes TFT to degrade, resulting in an outgrowth of wild type colonies which escape initial selection. After refeeding, the plates were incubated for another 10 days, and the newly formed colonies were classified to be tk slow growth (tk-SG) mutants [19]. The sum of tk-NG and tk-SG were determined to be overall mutants.
Statistical Analysis
Statistical analyses were done on the means of the data obtained from three independent experiments. All results were presented as means ± SD. Significance was assessed by using student's t test at p < 0.05.
3. Results and Discussion
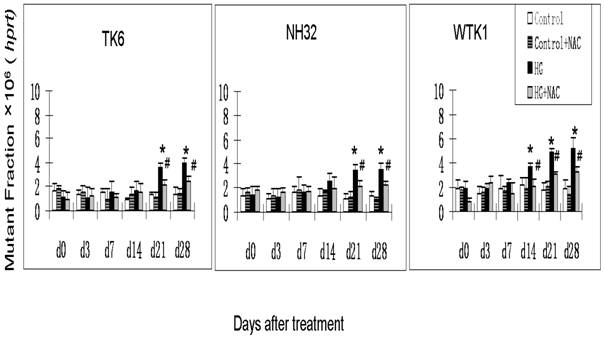
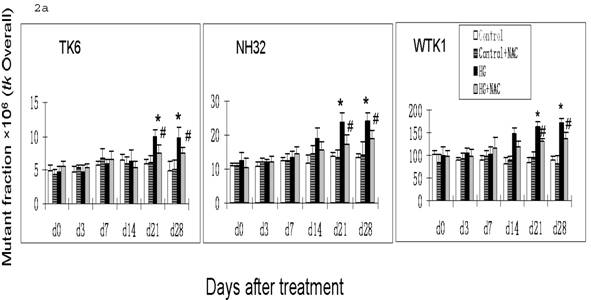
Previous studies have demonstrated that DNA damage-caused mutations at autosomal tk locus include tk-NG mutants that arise 11 days after plating with similar growth rate to their parental cells, and tk-SG mutants that arise 21 days after plating with slower growth rate than their parental cells [19]. At hprt locus, all mutants arise at 11 days after plating with similar growth rate to their parental cells. It is well-known that the different molecular natures of DNA damage led to the formation of various mutants; large genetic alterations that affect cell-growth-related genes around tk locus are responsible for the slow growth phenotype in tk-SG mutants, whereas small genetic alterations that does not affect cell-growth-related genes around tk locus and hprt locus are responsible for the phenotype of the normal growth in hprt and tk-NG mutants [19]. We, therefore, measured the MFs for both hprt and tk loci at different days after HG-treatment in three cell lines. As seen in Fig. 1 and Fig. 2a, all cell lines showed an increase in the MFs at hprt locus and at tk locus at 21 and 28 days after HG treatment, compared with control (p<0.05). A trend of increase without statistical significance in NH32 cells (p>0.05) and significant increase in WTK cells (p<0.05) were also found at 14 days after HG-treatment. Induction of the two locus mutations (autosomal tk and X-linked hprt) in the three cell lines exposed to HG for 21 days strongly indicates the mutagenic effect of HG.
HG-induced mutagenesis at hprt locus. At indicated days after HG treatment, TK6, NH32, and WTK1 cells were collected for mutagenesis study. * p<0.05 compared to control, # p<0.05 compared to HG-treatment.
HG-induced mutagenesis at tk locus. At indicated days after HG treatment, TK6, NH32, and WTK1 cells were collected for mutagenesis study. Panel a is for overall MF, panel b is for tk-NG MF, and panel c is for tk-SG MF. * p<0.05 compared to control, # p<0.05 compared to HG-treatment.
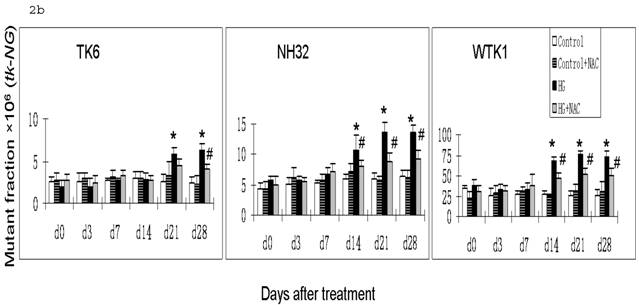
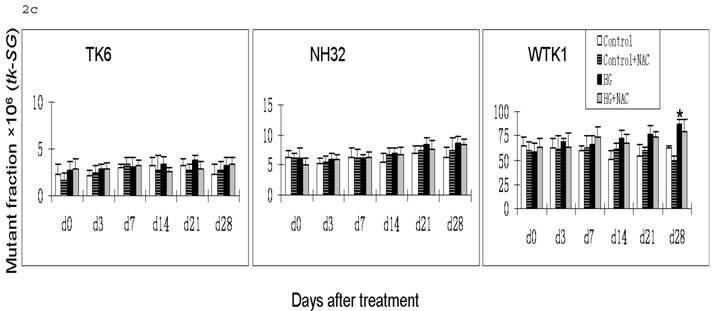
Gene mutations can be formed at different scales, either small deletions of the entire genes of hprt or tk or larger deletions that span one or more genes. The latter will not be detectable because the affected cells would not survive. Therefore, large genetic alterations along X-linked hprt locus are lethal and escape the mutagenic assay. However, because of the presence of the homologous chromosome and genes, large multilocus deletions of autochromosome genes such as tk locus remain detectable in mutagenic assay [19]. Therefore we further examined the effects of HG on mutations of tk-NG and tk-SG separately (Fig. 2b,c). It is shown that there was no increased tk-SG in TK6 and NH32 cells even at 28 days after HG treatment, while MFs of tk-SG was slightly increased without statistical significance at day 14 and 21 (p>0.05) and significantly increased at day 28 (p<0.05) after HG treatment (Fig. 2c), suggesting that increased tk-NG is mainly responsible for the overall increased MF at tk locus in TK6 and NH32 cells, while both tk-NG and tk-SG mutations occur in WTK1 cells in response to HG.
Apparently, the difference for the onset time in hprt and tk-NG mutation, and mutation types among three cells lines is related with p53. In contrast to p53 wild type TK6 cells, both NH32 with a null p53 and WTK1 cells with a mutant p53 showed an early onset of HG-induced mutagenesis. In addition, WTK1 cells showed an increased MF for tk-SG at 28 days after HG treatment, which is different from TK6 and NH32 cells. p53 is a transcription factor that serves as a potent tumor suppressor. Inactivating mutations in the p53 protein are found in over 50% of cancers, suggesting loss of p53 function provides a selective advantage to tumor cells that may contain gene mutation [20]. Mutation of p53 sensitizes cells to endogenous and environmental mutagens. For example, compared to TK6, WTK1 cells are approximately 10 folds more, and NH32 cells are approximately 1.5 folds high sensitivity to ionizing radiation-induced mutagenesis [17, 18]. Upon DNA damage, cells utilize multiple DNA repair pathways to mend damaged DNA including nucleotide excision repair, mismatch repair, base excision repair, homologous recombination, and non-homologous end joining pathways. p53 involves in all these DNA damage repair pathways by either direct association with or transcriptional modulation of the key players in the above repair pathways [20]. Impairment of the above pathways results in unfaithful repair of DNA damage that may lead to increased mutagenesis. These may explain why NH32 and WTK1 cells display an early onset and high incidence of gene mutations (Fig. 1, Fig. 2b) since the lack or mutant of p53 function may impair DNA repair mechanisms. However, it should be mentioned that the DNA repair by homologous recombinational activity was found remarkably increased in WTK1 cells due to the mutant p53 function, i.e.: about 7-folds higher than that in TK6 cells. The increased homologous recombinational DNA repair activity would improve the large scale DNA damage via loss of heterozygosity and be thus related with slow growth phenotype [16]. This suggests that in WTK1 cells, the mutant p53 significantly increased abnormal homologous recombinational repair in response to HG, leading to an increased large-scale DNA damage, such as tk-SG.
It has been noted that ROS-induced DNA damage is mainly small scale, such as base pair substitution, small scale insertion and deletions [21]. We have shown that cells produce ROS in response to HG treatment [22, 23]. In consistent with these studies, we demonstrated in the present study that there is predominant increase in tk-NG (Fig. 2b) that most likely contributes to the increase in tk-overall gene mutations (Fig. 2a), suggesting the possible role of HG-induced ROS in the induction of gene mutations. To further define the direct link of HG-induced ROS to gene mutations, we demonstrated that the addition of ROS scavenger NAC in the cultured cells exposed to HG partially, but significantly, prevented HG-induced hprt and tk-NG MFs (Fig. 1 and Fig. 2).
The exact explanation of why NAC partially prevents HG-induced gene mutations is unclear based on the present study, however, it may be related to the fact that gene mutation is related both DNA damage and repair process, i.e.: either increase in DNA damage or decrease in DNA repair ability. Diabetic hyperglycemia or HG has been extensively documented to inactivate protein action such as antioxidants and enzymes by glycation or nitration [24, 25]. NAC may be unable to prevent protein glycation, and thus unable to prevent the contribution of glycation-inactivated DNA repair enzymes to HG-induced DNA damage and consequently gene mutations. In addition, glyoxal as an intermediate of HG metabolism was found to induce DNA base damage via ROS-independent pathways [13]. Therefore, the present study shows only partial prevention of NAC against HG-induced gene mutations (Fig. 1 and Fig. 2).
4. Conclusion
In summary, we have shown that HG treatment leads to increased mutagenesis in human lymphoblastoid cells. ROS may play partial role in HG-induced mutagenesis, and null or mutant p53 function makes cells more susceptible to HG-induced mutagenic effect. Thus, the present study provides the first and direct evidence for HG-induced gene mutations, which provides novel insight into understanding the mechanism of cancer risk in diabetes mellitus.
Acknowledgements
This study is supported in part by research grants from American Diabetes Association (02-07-JF-02; 05-07-CD-02), Philip Morris USA, Inc. (020187), and a start-up fund for the Chinese-American Research Institute for Diabetic Complications from Wenzhou Medical College (to L.C.).
Conflict of interest
The authors have declared that no conflict of interest exists.
References
1. Wideroff L, Gridley G, Mellemkjaer L, Chow W.H, Linet M, Keehn S. et al. Cancer incidence in a population-based cohort of patients hospitalized with diabetes mellitus in Denmark. J Natl Cancer Inst. 1997;89(18):1360-5
2. La Vecchia C, Negri E, Decarli A, Franceschi S. Diabetes mellitus and the risk of primary liver cancer. Int J Cancer. 1997;73(2):204-7
3. Chow W.H, Gridley G, Nyren O, Linet M.S, Ekbom A, Fraumeni J.F. et al. Risk of pancreatic cancer following diabetes mellitus: a nationwide cohort study in Sweden. J Natl Cancer Inst. 1995;87(12):930-1
4. La Vecchia C, Negri E, Decarli A, Franceschi S. Diabetes mellitus and colorectal cancer risk. Cancer Epidemiol Biomarkers Prev. 1997;6(12):1007-10
5. Le Marchand L, Wilkens L.R, Kolonel L.N, Hankin J.H, Lyu L.C. Associations of sedentary lifestyle, obesity, smoking, alcohol use, and diabetes with the risk of colorectal cancer. Cancer Res. 1997;57(21):4787-94
6. Brinton L.A, Berman M.L, Mortel R, Twiggs L.B, Barrett R.J, Wilbanks G.D. et al. Reproductive, menstrual, and medical risk factors for endometrial cancer: results from a case-control study. Am J Obstet Gynecol. 1992;167(5):1317-25
7. La Vecchia C, Negri E, Franceschi S, D'Avanzo B, Boyle P. A case-control study of diabetes mellitus and cancer risk. Br J Cancer. 1994;70(5):950-3
8. Sellers T.A, Sprafka J.M, Gapstur S.M, Rich S.S, Potter J.D, Ross J.A. et al. Does body fat distribution promote familial aggregation of adult onset diabetes mellitus and postmenopausal breast cancer? Epidemiology. 1994;5(1):102-8
9. Weiderpass E, Gridley G, Persson I, Nyren O, Ekbom A, Adami H.O. Risk of endometrial and breast cancer in patients with diabetes mellitus. Int J Cancer. 1997;71(3):360-3
10. Lindblad P, Chow W.H, Chan J, Bergstrom A, Wolk A, Gridley G. et al. The role of diabetes mellitus in the etiology of renal cell cancer. Diabetologia. 1999;42(1):107-12
11. Cai L, Kang Y.J. Oxidative stress and diabetic cardiomyopathy: a brief review. Cardiovasc Toxicol. 2001;1(3):181-93
12. Blasiak J, Arabski M, Krupa R, Wozniak K, Zadrozny M, Kasznicki J. et al. DNA damage and repair in type 2 diabetes mellitus. Mutat Res. 2004;554(1-2):297-304
13. Shimoi K, Okitsu A, Green M.H, Lowe J.E, Ohta T, Kaji K. et al. Oxidative DNA damage induced by high glucose and its suppression in human umbilical vein endothelial cells. Mutat Res. 2001;480:371-8
14. Toyokuni S. Novel aspects of oxidative stress-associated carcinogenesis. Antioxid Redox Signal. 2006;8(7-8):1373-7
15. Gatz S.A, Wiesmuller L. p53 in recombination and repair. Cell Death Differ. 2006;13(6):1003-16
16. Xia F, Amundson S.A, Nickoloff J.A, Liber H.L. Different capacities for recombination in closely related human lymphoblastoid cell lines with different mutational responses to X-irradiation. Mol Cell Biol. 1994;14(9):5850-7
17. Peng Y, Zhang Q, Nagasawa H, Okayasu R, Liber H.L, Bedford J.S. Silencing expression of the catalytic subunit of DNA-dependent protein kinase by small interfering RNA sensitizes human cells for radiation-induced chromosome damage, cell killing, and mutation. Cancer Res. 2002;62(22):6400-4
18. Zhang Y, Lim C.U. K, Williams E.S, Zhou J, Zhang Q, Fox M.H, et al. NBS1 Knockdown by Small Interfering RNA Increases Ionizing Radiation Mutagenesis and Telomere Association in Human Cells. Cancer Res. 2005;65(13):5544-53
19. Liber H.L, Yandell D.W, Little J.B. A comparison of mutation induction at the tk and hprt loci in human lymphoblastoid cells; quantitative differences are due to an additional class of mutations at the autosomal tk locus. Mutat Res. 1989;216(1):9-17
20. Helton E.S, Chen X. p53 modulation of the DNA damage response. J Cell Biochem. 2007;100(4):883-96
21. Diaz-Llera S, Podlutsky A, Osterholm A.M, Hou S.M, Lambert B. Hydrogen peroxide induced mutations at the HPRT locus in primary human T-lymphocytes. Mutat Res. 2000;469(1):51-61
22. Cai L, Li W, Wang G, Guo L, Jiang Y, Kang Y.J. Hyperglycemia-induced apoptosis in mouse myocardium: mitochondrial cytochrome C-mediated caspase-3 activation pathway. Diabetes. 2002;51(6):1938-48
23. Cai L, Wang J, Li Y, Sun X, Wang L, Zhou Z. et al. Inhibition of superoxide generation and associated nitrosative damage is involved in metallothionein prevention of diabetic cardiomyopathy. Diabetes. 2005;54(6):1829-37
24. Zeng J, Dunlop R.A, Rodgers K.J, Davies M.J. Evidence for inactivation of cysteine proteases by reactive carbonyls via glycation of active site thiols. Biochem J. 2006;398(2):197-206
25. Turko I.V, Marcondes S, Murad F. Diabetes-associated nitration of tyrosine and inactivation of succinyl-CoA:3-oxoacid CoA-transferase. Am J Physiol Heart Circ Physiol. 2001;281(6):H2289-94
Author contact
![]() Correspondence to: Dr. Ying Zhang, Department of Environmental and Radiological Health Science, Colorado State University, Fort Collins, CO 80521, USA, Phone: 970-491-0574, Fax: 970-491-0623, e-mail: ying.zhangedu or Dr. Lu Cai, Email: l0cai001edu
Correspondence to: Dr. Ying Zhang, Department of Environmental and Radiological Health Science, Colorado State University, Fort Collins, CO 80521, USA, Phone: 970-491-0574, Fax: 970-491-0623, e-mail: ying.zhangedu or Dr. Lu Cai, Email: l0cai001edu