Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. Materials and Methods
3. Results
4. Discussion
Acknowledgements
References
Author biography

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2007; 3(6):393-401. doi:10.7150/ijbs.3.393 This issue Cite
Research Paper
Thermal injury-plus-sepsis contributes to a substantial deletion of intestinal mesenteric lymph node CD4+ T cell via apoptosis
Nadeem Fazal, Walid M Al-Ghoul ![]()
Department of Biological Sciences, Chicago State University, Chicago, IL, USA
Received 2007-8-2; Accepted 2007-8-30; Published 2007-9-12
Abstract
Thermal injury (TI) with septic complications continues to be a serious clinical problem. One of the main concerns in such patients is immunosuppression related to functional derangements in intestinal CD4+ T lymphocytes. Extensive previous studies in thermal injury/septic patients and animal models of thermal injury/sepsis have shown decreased responsiveness of intestinal CD4+ T cells to antigen/mitogen. This hyporesponsiveness could significantly contribute to increase injured host susceptibility to pathogens including those translocating from host's gut lumen. Our previous studies indicated that while thermal injury or sepsis alone lead to suppressed proliferation and IL-2 production of intestinal CD4+ T cells, this study showed a substantial deletion via apoptosis of the Mesenteric Lymph Nodes (MLN) CD4+ T cells. Hence, thermal injury-plus-sepsis contributes not only to suppressed CD4+ T proliferation/IL-2 production but also to a substantial modulation of CD4+ T cell survivability. These findings allow us to conclude that while thermal injury alone can produce attenuated cell mediated responses without an overt change in CD4+ T cell survival, thermal injury with septic complications causes CD4+ T cell death and an irreversible loss of cell-mediated responses. The latter happening could be responsible for high morbidity and mortality in the injured host afflicted with thermal injury plus a critical infection.
Keywords: Burn injury, infection, hoescht staining, immune suppression, adaptive immune response, activation-induced cell death
1. Introduction
Sepsis and the resultant multi-organ failure are the leading causes of death in intensive care units [1, 2]. Patients with sepsis have massive apoptosis in lymphoid organs such as thymus, spleen, lymph nodes, and gastrointestinal associated lymphoid tissue [3, 4, 5]. Apoptosis is notable in most subpopulations of lymphoid cells including B cells and CD4+ and CD8+ T cells [6-13]. The death of these cells may result in depletion of key immune effector cells and this effect may account for the immunosuppression that is a central pathogenic event. A number of laboratory and clinical studies have shown that extensive thermal injury induces a state of immune-insufficiency, and that the immune-refractoriness predisposes the injured host to critical morbidity and mortality [7, 9, 12-16]. To date, their involvement as critical regulatory molecules responsible for T cell suppression in TI and septic injuries continues to be a subject of extensive studies as evident from reports from several laboratories [17-20]. The principal outcome of such immune suppression is increased susceptibility of injured host to opportunistic pathogens causing high risk of death. Recent studies have shown that immune-insufficiency with TI is characterized by monocyte/macrophage hyperactivity and/or depressed adaptive cell-mediated immunity associated with T lymphocyte functional deficits [17-23].
In our previous studies, we assessed thermal injury (TI) induced T cell effector immune responses having the potential of adversely affecting T cell activation, IL-2 production and proliferation [17, 18, 22]. The extent of immune dysfunction is apparently dependent not only on TI severity but also on concomitant smoke inhalation or ethanol ingestion, as well as host's age, gender, and nutritional status [14, 16]. Despite rather extensive studies of T cell functional deficits in animal models of TI and in patients, the mechanisms of such deficits and a potential role of these deficits in the lethal outcome following TI particularly with the septic complications have remained unknown. Our present studies have assessed both the individual effects of TI and sepsis as well as an effect of superimposition of sepsis on TI, in rats, on apoptotic responses of MLN CD4+ T cells. While TI or sepsis individually produced low animal mortality, the combined injury resulted in exacerbation of both morbidity and mortality [17,19]. Thus, the focus in this study on the combined TI-plus-sepsis injury affords us an opportunity to assess potential non-lethal versus lethal implications of the T cell dysfunction.
We hypothesized while the T cell hyporesponsiveness (i.e., attenuated IL-2 production/ proliferation) with TI or sepsis injury alone (of a certain magnitude), accompanied by a low level of T cell apoptosis, could result in a reversible state of depressed immunity compatible with high animal survivability, a superimposition of the two injuries produces CD4+ T cell hypo-responsiveness accompanied by intensified T cell apoptosis contributing to a state of irreversible immune dysfunction, and to a pronounced animal morbidity and mortality. The immune system impaired by a primary sub-lethal injury makes the host more susceptible to a secondary infection/death. TI followed by CLP, a double injury, is useful in the study of sepsis, since it creates a prolonged period of infection as opposed to TI alone. This prolonged infection may offer a clearer understanding of the immune dysregulation mechanism as seen in clinical sepsis.
2. Materials and Methods
Animal model of thermal injury (TI)
Male Sprague Dawley rats (250-300gm) were housed and used in compliance with the regulation of the Animal Care Facility of Chicago State University, Chicago. Major thermal injury model has been previously described as a suitable model demonstrating the response associated with thermal injury [17-23]. Briefly, rats were anesthetized with sodium pentobarbital [40-50 mg/kg intraperitoneal (i.p)]; their dorsal body surface hair shaved off, and placed in an appropriately sized template device such that the shaved area of the skin on the animals' back was exposed. Adequacy of anesthesia was tested by the absence of withdrawal response to toe pinching. The template device was then lowered into a hot water bath (95-97oC) to immerse the exposed skin area in hot water for 10 seconds. With this technique, full thickness 3rd degree thermal injury comprising 30% of the total body surface were obtained. Sham rats were subjected to identical anesthesia and other treatments, except that they were immersed in 37oC water.
Sepsis model of cecal ligation and puncture (CLP)
Sepsis was induced immediately following thermal injury i.e., under the same anesthesia. The CLP model was used to induce intra-abdominal peritonitis as previously described [17]. Earlier studies from our laboratory include positive blood cultures for polymicrobial organisms (aerobic and anaerobic bacteria) from CLP, but not sham-operated rats. An abdominal incision was performed and the cecum was identified, ligated, and punctured once with a 22-gauge needle. The abdomen was closed in two layers and 1 ml of 0.9% saline was subcutaneously administered. Sham-operated rats were treated identically except the cecum was not ligated or punctured.
Quantification of in situ apoptosis (TUNEL)
DeadEndTM colorimetric TUNEL system (Promega) was used to determine apoptosis in tissue sections of MLN and PP. This non-radioactive system is designed to provide simple, accurate and rapid detection of apoptotic cells in situ at the single cell level. This assay measures nuclear DNA fragmentation, an important biochemical indicator of apoptosis in many cell types.
T cell separation by magnetic microbead method (MACS)
MLN were aseptically isolated from rats and crushed under sterile stainless steel sieve. The crushed tissues were passed through cell strainer (35µm pore size) to remove clumps. The filtered cell suspension was spun at 300g for 10 min and pellet suspended in 1 ml RPMI-1640 media. Cell population counted with a trypan blue exclusion method to determine cell viability. Cells were adjusted to 107 cells in 80µl and incubated with 20µl of CD4+ T cell Magnetic Micro-beads (Miltenyi Inc.) at 4oC for 15 min. The cells were washed with elution buffer (Miltenyi, Inc.) by centrifugation at 300x g for 10 min. The micro-beads and cell mixture was passed through pre-wet columns. The columns were flushed with 0.5 ml of elution buffer 3 times. The cells passed through the columns (negative selection) were discarded. Columns were removed from magnetic field; the cells (positive selection) eluted by running 0.5 ml of elution buffer twice with gentle pressure from plunger. The eluted cells were washed with elution buffer by centrifugation at 300g for 10 min. Positive selection cells (106) were taken and incubated with FITC-labeled anti-CD4 antibody (Biosource Inc, CA) to determine positive yield by FACS analysis. This method repeatedly yielded 99% purity of CD4+ T cells.
Flow Cytometry (FACS)
Flow cytometry was used for the determination of T cell type, activation markers, and apoptosis by Annexin-V (Promega) labeling in the experiments. MLN CD4+ T cells were labeled with respective FITC/PE/APC rat-specific antibodies at 1-5 μg/ml per 106 cells. Isotype control antibodies and unstained cells were used as controls. Cells were fixed in 0.7 ml of 1% paraformaldehyde solution and acquired on a FACScan flow cytometer (Becton Dickinson). At least 10,000 events were acquired by using LYSYS II software (Becton Dickinson). Both forward-angle and right-angle light scatter signals were adjusted for optimum results for the detection of T cells. The forward-scatter threshold signal was adjusted to exclude debris and unlysed erythrocytes. All parameters were optimized on unstimulated purified T cells that have been loaded with respective antibody.
Activation-induced cell death (AICD)
To induce AICD, live CD4+ T cell were cultured in the presence of IL-2 (5 IU/ml) with or without plate-bound anti-CD3 (10 µg/ml). Following protocol was adapted; MLN CD4+ T cells were cultured with Con-A (48h), αCD3 (48h) and IL-2 (24h). At the end of assay, cells were harvested and stained with Hoechst 33342 to score for apoptotic nuclei, and for dye exclusion using trypan blue. Hoechst 33342 is a nucleic acid dye (DNA) that can pass through intact cell membranes, thereby allowing DNA content analyses of viable cells. This UV excitable dye Hoechst 33342 solution (500 μg/ml) was prepared by dissolving 3 mg of Hoechst 33342 in 6 ml HBSS (without phenol red) or PBS. CD4+ T cells (106) were resuspended in 1.8 ml 5% BCS (bovine calf serum)-HBSS (without phenol red). 200 μl of the 500 μg/ml Hoechst 33342 solutions were added to respective T cells post-stimulation and in coculture and incubated at 37oC for 2 hours. The cells were washed with 5% BCS-HBSS without phenol red (or PBS) and fluorescence microscopy was performed to assess apoptosis of CD4+ T cells.
Statistical analysis
Data are reported as the mean ± SEM. Data were analyzed using the statistical software program Prism (GraphPad Software). Data involving two groups only were analyzed by a Student's t test, whereas data involving more than two groups were analyzed using one-way ANOVA with Tukey's multiple comparison test. Significance was accepted at p < 0.05.
3. Results
Sepsis causes increased in situ apoptosis of immune cells of mesenteric lymph nodes
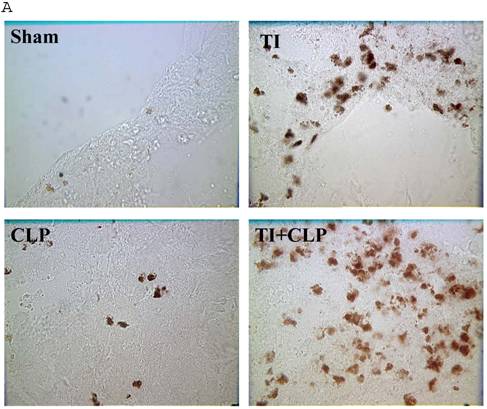
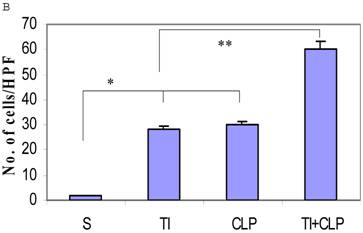
The rationale for in situ apoptosis was to determine apoptosis that occurred in vivo in a lymphatic organ that hosts unactivated naïve CD4+ T cells. Cryosections of MLN were prepared from sham, TI, CLP and TI + CLP rats. DEAD-cell kit, Promega (TUNEL Assay) was used for in situ analysis of apoptotic cells in MLN sections. The results as shown in Figures 1A & B showed a significant increase in percentage of apoptotic cells in TI which further increased after superimposition of CLP injury. There was a similar increase albeit of lesser intensity in apoptosis of CLP and TI groups separately, whereas no apoptosis was seen in cryosections obtained from MLN of sham animals. These results provided us a lead that cellular apoptosis was a dominant feature occurring in intestinal lymphoid cellular tissues such as MLN following TI plus sepsis injury. Further studies were then designed to investigate the specific immune cells undergoing cell-death in these MLN of injured animals.
Representative cryosections of in situ apoptosis (TUNEL) of mesenteric lymph nodes (MLN) obtained from sham, TI, CLP and TI+CLP animals (Fig. 1A). At least 3 slides with 3 sections per animal for each injury group of animals were examined and data analyzed. Fig. 1B represents a histogram of collective data obtained from 3 experiments representing mean with SE (n=6). Numbers of apoptotic cells were counted per high field of microscope. * p <0.05 between sham vs. TI, ** p<0.05 TI vs. TI+CLP.
Activated CD4+ T cells are the major cells undergoing apoptosis
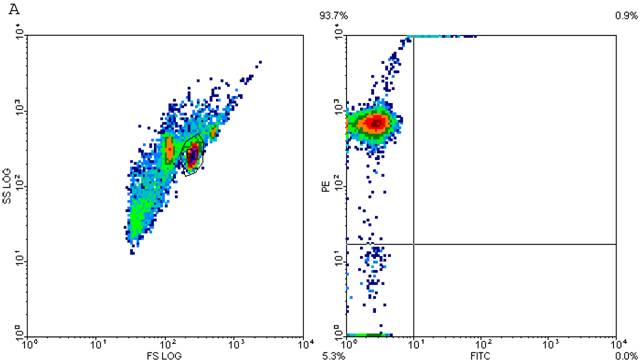
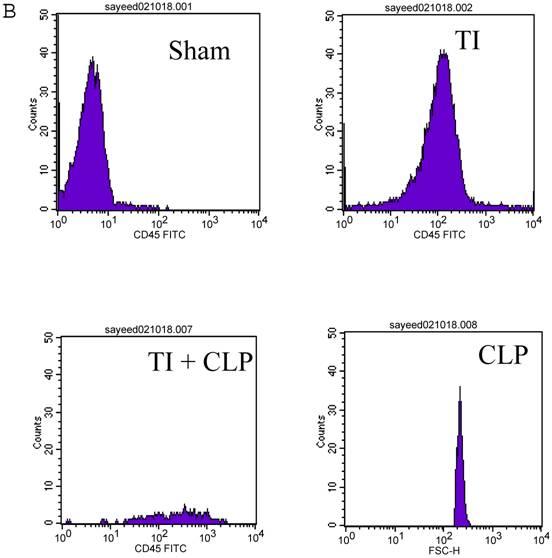
CD4+ T cells are known to maximally populate MLN, as these are the cells mainly responsible for the immune response that is prevalent at these sites in TI and sepsis. We studied the CD4+ T cells of MLN undergoing cell-death following TI and sepsis injury and/or both. Our studies confirmed that MLN of injured animals are loaded with CD4+ T cells and 94% purity gated population was achieved by MACS as shown in Fig. 2A. We assume that 5% contaminated cells may belong to other immune cells i.e., macrophages, dendritic cells, NK cells, B cells etc. It would have been important to eliminate them for culture studies but since we were studying morphological features and specific cell surface markers we did not attempt to remove these cells. After concluding that MLN of injured animals were loaded with CD4+ T cells, we determined if MLN CD4+ T cells were of activated or un-activated (naïve) type by using a surface marker i.e., CD45RC a high molecular-weight isoform of CD45 (Leukocyte Common Antigen). CD45RC-positive was taken as naïve or unactivated cell and CD45RC-negative as activated or memory T cell in rat specie. The results in Figure 2B indicated a shift of all MLN CD4+ T cells of TI towards activated or memory type as indicated by uptake of CD45 marker whereas in TI plus sepsis there was a significant loss of CD45 uptake showing that CD4+ T cells following TI injury have become activated or memory subtype.
Flow cytometric analysis of CD45RC marker surface expression on MLN CD4+ T cells. Representative data from four experimental groups i.e. sham, TI, CLP and TI+CLP are shown in the figure (Fig. 2A). CD45RC-positive cells (naïve) and CD45RC-negative (memory/activated) are given in (Fig. 2A). Fig. 2B is a histogram of collective data obtained from 3 experiments showing mean with SE (n=6). * p <0.05 between sham vs. TI, ** p<0.05 TI vs. TI+CLP.
IL-2 independent Fas-mediated apoptosis of freshly isolated CD4+ T cells
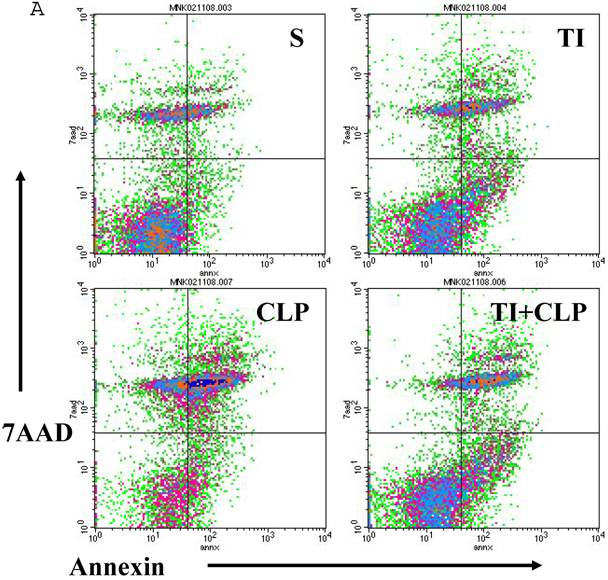
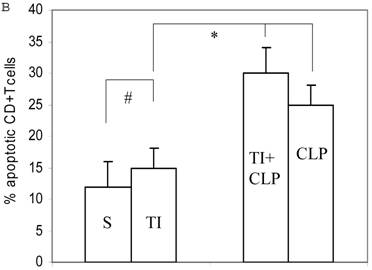
Apoptosis of mature T cells occurs in at least two major forms: antigen-driven and lymphokine withdrawal. These forms of death are controlled in response to local levels of IL-2 and antigen in a feedback mechanism. Antigen-induced (Active) apoptosis is due to Fas ligand and TNF. Active antigen-induced T cell death constrains the immune response under conditions of high IL-2 and antigen, whereas passive lymphokine withdrawal T cell death removes excessive T cells and occurs under conditions of low IL-2 and antigen stimulation. Therefore, we determined the effect of IL-2 on this population of CD4+ MLN T cells. In this series of experiments, we studied Fas-induced apoptosis of TI and/or sepsis CD4+ T cells. MLN CD4+ T cell were prepared from sham, TI, CLP and TI + CLP rats. The highest percentage of annexin V-positive apoptotic cells was observed at 3h after Fas cross-linking. The results as shown in Figures 3A & B showed a 10-12% percentage CD4+ T cells undergoing apoptosis in both sham and TI cells, but there is no statistically difference (p>0.05) in these two groups. CLP alone caused 25% of CD4+ T cells and TI plus sepsis a 30% increase in apoptosis which is statistically significant (p<0.05) than sham or TI group. Earlier studies have proposed a possibility that if Fas-cross linking be increased to 20h, the percentage of apoptotic cells may fall gradually; possibly owing to gradual deletion of Fas-sensitive cells however we did not investigate later time points in the present study.
Apoptosis of MLN CD4+ T cells as determined by flow cytometric analysis of Annexin -positive cells. In this series of experiments apoptosis of CD4+ T cells was induced by incubation with anti-Fas Antibody (0.5 μg/ml) for 3h. Four experimental groups i.e. sham, TI, CLP and TI+CLP are shown in the figure 3A. Histogram data represents mean with SE (n=6) from 3 different experiments (Fig. 3B), # p >0.05 between sham vs. TI, ** p<0.05 TI vs. TI+CLP.
IL-dependent activation-induced cell death (AICD) of MLN CD4+ T cells in culture
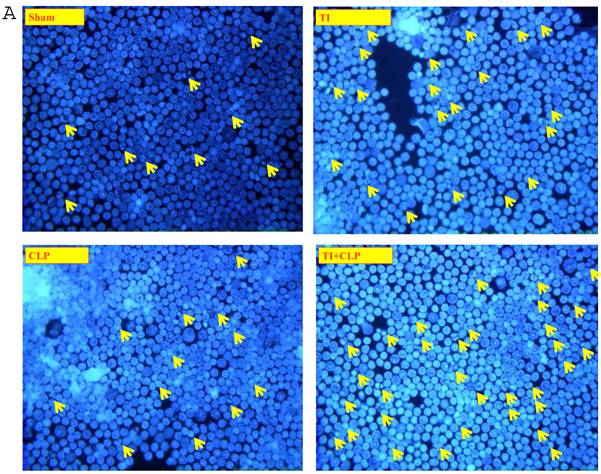
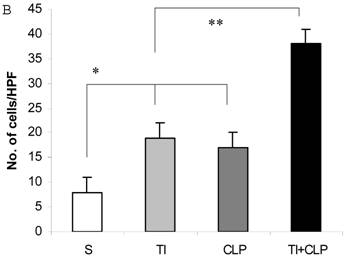
It has been suggested that cells that have entered the cell cycle are more vulnerable to apoptosis from external stimuli. Because cells were not proliferating following sepsis, it was possible that T cells become activated during sepsis, enter the cell cycle, but die before they can complete a round of proliferation. In order to closely mimic the complex milieu of adaptive immune response occurring at intestinal MLN where continuous antigenic stimulus of naïve CD4+ T cell is occurring, a three-step procedure was adapted [8] in the following experiments. Excessive antigenic stimulation was achieved by polyclonally activating T cells for 48h with Con-A (Step-1), followed by αCD3-induced activation (Step-2), and supplementation by growth factor i.e., IL-2 (Step-3). CD4+ T cells were obtained by MACS microbead method from sham, TI, CLP and TI+CLP rats and cultured with Con-A (48h), αCD3 (48h) and IL-2 (24h). CD4+ T cells were then stained with Hoescht stain and morphological features of apoptosis were examined under microscope, i.e., nuclear condensation, plasma membrane shrinkage, membrane blebbing, and absence of lysis of cell body (Fig. 4A). The results in Figures 4 (A&B) showed that thermal injury induced significant increase in apoptosis of CD4+ T cells, which further increased when sepsis injury was superimposed on TI. The accumulated data of number of CD4+ T cells going in apoptosis per high power field (HPF) in figure 2B represented mean with SE (n=6) obtained from 3 different experiments. This study showed that CD4+ T cells are very susceptible to apoptosis once TI injury sets in and this process is aggravated following dual insult i.e. TI plus sepsis.
Representative figures showing apoptosis (Hoescht staining) of MLN CD4+ T cells obtained by Microbead separation (MACS) from four experimental groups i.e. sham, TI, CLP and TI+CLP (Figure 2A). The yellow colored arrows indicate apoptotic CD4+ T cells with characteristic features of apoptosis (condensation of nuclei and membrane blebbing etc.). Fig. 2B is a histogram of collective data from 3 experiments showing mean with SE (n=6). Number of apoptotic cells as determined by Hoescht stain was counted per high power field (HPF) of microscope. * p <0.05 between sham vs. TI, ** p<0.05 TI vs. TI+CLP.
4. Discussion
Present studies confirmed that thermal injury-plus-sepsis contribute to a substantial deletion of MLN CD4+T cell via apoptosis leading to irreversible immune dysfunction. The differences in the T cell deficits after the individual TI or sepsis injuries and the combined injury condition seemed well correlated with differences in mortality in these injured animal groups as determined in our previous studies [17, 18]. From these observations, it seemed reasonable to conclude that intestinal CD4+ T cell incompetence with thermal injury and/or sepsis alone was mostly reversible which allowed for an eventual restoration of T cell homeostasis and competence compatible with high animal survival. On the other hand, the exacerbation of animal mortality with thermal injury-plus-sepsis seemed related to a sustained loss of immune-competence and a critical deletion of antigen triggered T cells.
The results showed that injury like TI or sepsis triggers an apoptotic program that may be responsible for altered immune response observed at these sites. Since T cell-mediated adaptive immune response requires several days to become effective and has been considered to be slowly activated host defense mechanism a day 3 post-injury had been found to be optimum time point in earlier as well as subsequent studies. In this study, sepsis was induced through CLP model where it has been known that bacteria isolated from the blood cultures of septic rats were predominantly extracellular rather than intracellular pathogens, hence, a CD4+ T cell rather than CD8+ T cell response would be required to control infection. Therefore, our study focused on CD4+ T cells. There is much evidence that CD4+ T cells could only be detected and initially activated in the lymph nodes, spleen and mucosal lymphoid tissues. Moreover, determination of apoptosis of blood CD4+ T cells may not represent the true scenario of what is actually happening in the tissues or body after injury. It has also been suggested that apoptotic cells may have already been eliminated from respective lymphatic sites, i.e., phagocytosis by macrophages, etc., which are used to obtain purified T cells therefore we designed this study to use a lymphatic tissue such as MLN which is right next to portal of entry of intestinal bacteria and chose a time point i.e., 3 days post-injury to represent a window for studying a typical immune response and/or any dysfunction that is predominant at this immune sites. In the present study, the migration of naïve and effector T cells was also investigated separately in MLN of sham and TI rats. This supported earlier data showing that lymphocyte accumulation in MLN was part of an active rather than passive process. Intact CD4+ T cells are retained in MLN; where they encounter antigens from gut. In addition to focusing on the reversibility of thermal injury/sepsis induced T cell deficits, the experiments in this series determined what potentially are the key factors contributing to derangements in T cell apoptosis, and then evaluate if those factors play a role in the reversibility of the derangements. The question we asked was if T cell apoptosis and cell responsivity alterations are reversible in the thermal injury and sepsis groups and not in the TI+sepsis group of animals. Our previous studies [17, 18] determined IL-2 production and proliferation responses in MLN CD4+ T cells cocultured with same MLN's APCs harvested from sham, thermal injury, sepsis, and TI+sepsis animals sacrificed on days 3 post-injury. As has been implicated by several recent studies [11-22], the suppression/hypo-responsiveness of T cells following septic injury conditions can also be understandably related to a disturbance in the programming of their cell death. Although apoptosis of T cells occurs, during repeated and persistent activation, along with the IL-2 dependent proliferation of the cells, proliferation presumably exceeds apoptosis. On the other hand, apoptosis of T cells likely begins to exceed proliferation as cell activation begins to wane. The latter dominance of apoptosis over proliferation is apparently responsible for the homeostatic control of the activated T cells via the process of cell deletion. Inappropriate augmentation of T cells' apoptosis during/following their activation after TI/sepsis could contribute to loss of the potentially responsive T cells.
One mechanism for T cell death is activation-induced cell death (AICD) in which activation through the TCR with Ag or polyclonal activators (e.g., superantigens) results in apoptosis [11]. AICD can occur through a cell autonomous manner that is determined by a number of factors such as the nature and intensity of TCR signaling [11, 15, 16] and the availability of cytokine growth factors. It has been suggested that a common feature linking many of the causes of AICD is an aberration in the cell cycle program [8]. Although lymphocyte apoptosis is a common feature in sepsis [8], the precise death-inducing stimuli and other critical elements remain unknown. During sepsis the immune system is exposed to bacterial products as well as Ags from damaged tissues. Thus, one possible mechanism for the induction of T cell apoptosis and the loss of immune function in sepsis is AICD in which engagement of the TCR by Ags or polyclonal activators leads to cellular proliferation and death [8-11]. The purpose of this study was to determine whether activation and proliferation were responsible for sepsis-induced apoptosis and immune suppression. Our results show that although sepsis induces early T cell activation it does not induce proliferation in these cells. Collectively, these results suggest that the mechanism of killing of lymphocytes in sepsis does not require cognate Ag receptor interaction and is probably distinct from classical AICD. Thus the loss of immune functions is not directly related to the death of Ag-specific T cells but may be the result of a more generalized, nonspecific loss of immune function.
It is also to be noted that these experiments were performed on freshly isolated MLN CD4+ T cells. CD4+ T cells were separated by magnetic microbead cell sorting (MACS) from injured and control groups of rats and analyzed for apoptosis of CD4+ T cells. The Fas antigen (Apo-1, CD95) is a member of a family of cell surface receptors that includes tumor necrosis factor receptor (TNF-R), nerve growth factor receptor (NGF-R), CD40, CD27, CD30, and 4-1BB. Fas, is a cell surface apoptosis-signaling molecule that is widely expressed. In this series of experiments apoptosis of CD4+ T cells was induced by incubation with anti-Fas Antibody (0.5 μg/ml) for 3h. We found that overall apoptosis of MLN CD4+ T cell, in situ, was higher in thermally injured rats. Furthermore, there was an increase in Fas-mediated, IL-2 independent, apoptosis (AICD) in the T cells from the thermally injured rats. Although its mechanism of action is not known, IL-2 is both a promoter of AICD and a cell survival factor. Thus, an interpretation of our data could be that apoptosis in thermal injury rat CD4+ T cell was due to augmentation of AICD (triggered by Fas ligand) despite a deficiency in IL-2 production by thermal injury rat T cells possibly because of an increased AICD sensitivity to IL-2. Further, there may be an increase in mitochondrial Ca2+ pathway of passive apoptosis in the face of IL-2 deficiency (a phenomenon comparable to IL-2 withdrawal). AICD was assessed both by mediating it with anti-Fas, and by using the 3-step IL-2 dependent AICD protocol. Since increased AICD with thermal injury/sepsis could also be related to increased expressions of receptors Fas and TNFR, their surface expressions was assessed using anti-Fas and anti-TNFR antibodies and FACS analyses.
Our studies and other have also reported functional abnormalities relating to macrophages, dendritic cells, and T lymphocytes [17, 25, 31]. Recently, some studies have also elaborated the role of other immune cells, such as gamma-delta T cells, regulatory T cell subsets, and CD1d-restricted NKT cells [19, 21, 26]. An effective immune response is orchestrated by a mixture of immune cells with diverse functions likewise, a dysfunctional immune response following TI and/or sepsis is also known to cause alteration in a group of such immune cells including antigen presenting cells like dendritic cells, NK. We limited our studies to CD4+ T cells as they represented major effector cells of adaptive immune response. Previous studies have documented a profound loss of function of different phenotypes of DC, macrophages, neutrophils, B cells and co-stimulatory molecules in sepsis model both in humans and animals [25-32]. Based upon our data we speculate that the loss of CD4+ T cell wing of adaptive immune response is vital to all other effector immune responses and their contribution to an effective immune response is vital. One potential limitation of the present study is our inability to expand our experiments due to paucity of rat activation markers of T cells available in the market. However, in future we propose experiments on intestinal CD4+ T cells on days 4, 5, 6 and 7 post-injury, for AICD determinations, Fas and TNFR expressions, and measurements of mitochondrial δψ, Cytochrome C, mitochondrial membrane morphological changes, caspase 9, and expressions of Bad, Bid, Bcl-xL. All of these measurements will be carried out in freshly isolated cells. Dissipation in mitochondrial membrane potential with thermal injury/sepsis along with morphological change in the membrane and increased loss of cytochrome would support the concept of amplification of the mitochondrial loop. The investigation of role of mitochondria in CD4+ T cell apoptosis is planned to identify a key mechanism of apoptosis and a determinant of reversibility or irreversibility of apoptotic changes with thermal injury/sepsis injury.
Acknowledgements
This work was supported by NIH MBRS SCORE Grant No. 3 S06 GM008043-34S1 project # 6, and NSF MRI DBI-0520869.
Conflict of interest
The authors declare that they do not have any conflict of interest in this study.
References
1. Vandijck DM, Brusselaers N, Blot SI. Septicemia as a cause of death in burns. Burns. 2007;33(4):538-9
2. Purdue GF. American Burn Association Presidential Address 2006 on Nutrition: yesterday, today, and tomorrow. J Burn Care Res. 2007;28(1):1-5
3. Hotchkiss RS, Swanson PE, Freeman BD, Tinsley KW, Cobb JP, Matuschak GM, Buchman TG, Karl IE. Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunctions. Crit. Care Med. 1999;27:1230-1251
4. Lavrentieva A, Kontakiotis T, Lazaridis L, Tsotsolis N, Koumis J, Kyriazis G, Bitzani M. Inflammatory markers in patients with severe burn injury. What is the best indicator of sepsis? Burns. 2007;33(2):189-94
5. Bandyopadhyay G, De A, Laudanski K, Li F, Lentz C, Bankey P, Miller-Graziano C. Negative signaling contributes to T-cell anergy in trauma patients. Crit Care Med. 2007;35(3):794-801
6. Mateo V, Menager M, de Saint-Basile G, Stolzenberg MC, Roquelaure B, Andre N, Florkin B, le Deist F, Picard C, Fischer A, Rieux-Laucat F. Perforin-dependent apoptosis functionally compensates Fas-deficiency in Activation-induced cell-death of human T-lymphocytes. Blood. 2007 [Epub ahead of print]
7. Scumpia PO, Delano MJ, Kelly-Scumpia KM, Weinstein JS, Wynn JL, Winfield RD, Xia C, Chung CS, Ayala A, Atkinson MA, Reeves WH, Clare-Salzler MJ, Moldawer LL. Treatment with GITR agonistic antibody corrects adaptive immune dysfunction in sepsis. Blood. 2007 [Epub ahead of print]
8. Green DR, Droin N, Pinkoski M. Activation-induced cell death in T cells. Immunol. Rev. 2003;193:70-81
9. Hotchkiss R.S, Swanson PE, Knudson CM, Chang KC, Cobb JP, Osborne DF, Zollner KM, Buchman TG, Korsmeyer SJ, Karl IE. Overexpression of Bcl-2 in transgenic mice decreases apoptosis and improves survival in sepsis. J. Immunol. 1999;162:4148-4156
10. Hotchkiss R.S, Tinsley KW, Swanson PE, Chang KC, Cobb JP, Buchman TG, Korsmeyer SJ, Karl IE. Prevention of lymphocyte cell death in sepsis improves survival in mice. Proc. Natl. Acad. Sci. USA. 1999;96:14541-14546
11. Hotchkiss R.S, Chang KC, Grayson MH, Tinsley KW, Dunne BS, Davis CG, Osborne DF, Karl IE. Adoptive transfer of apoptotic splenocytes worsens survival, whereas adoptive transfer of necrotic splenocytes improves survival in sepsis. Proc. Natl. Acad. Sci. USA. 2003;100:6724-6729
12. Hotchkiss R.S, Tinsley KW, Swanson PE, Schmieg RE, Hui JJ, Chang KC, Osborne DF, Freeman BD, Cobb JP, Buchman TG, Karl IE. Sepsis-induced apoptosis causes progressive profound depletion of B and CD4+ T lymphocytes in humans. J. Immunol. 2001;166:6952-6963
13. Purcell E.M, Dolan SM, Kriynovich S, Mannick JA, Lederer JA. Burn injury induces an early activation response by lymph node CD4+ T cells. Shock. 2006;25:135-140
14. Enoh VT, Lin SH, Lin CY, Toliver-Kinsky T, Murphey ED, Varma TK, Sherwood ER. Mice depleted of alphabeta but not gammadelta T cells are resistant to mortality caused by cecal ligation and puncture. Shock. 2007;27(5):507-19
15. Tinsley K.W, Grayson MH, Swanson PE, Drewry AM, Chang KC, Karl IE, Hotchkiss RS. Sepsis induces apoptosis and profound depletion of splenic interdigitating and follicular dendritic cells. J. Immunol. 2003;171:909-914
16. Wisnoski N, Chung CS, Chen Y, Huang X, Ayala A. The contribution of CD4+ CD25+ T-regulatory-cells to immune suppression in sepsis. Shock. 2007;27(3):251-7
17. Fazal N, Raziuddin S, Khan M, Al-Ghoul WA. Antigen presenting cells (APCs) from thermally injured and/or septic rats modulate CD4+ T cell responses of naïve rat. Biochim Biophys Acta. 30. 2006;1762(1):46-53
18. Fazal N, Choudhry M.A, Sayeed M.M. Inhibition of T cell MAPKs (Erk 1/2, p38) with thermal injury is related to downregulation of Ca2+ signaling. Biochim Biophys Acta. 2005;1741(1-2):113-9
19. Samonte V.A, Goto M, Ravindranath T.M, Fazal N, Holloway V.M, Goyal A, Gamelli R.L, Sayeed M.M. Exacerbation of intestinal permeability in rats after a two-hit injury: burn and Enterococcus faecalis infection. Crit Care Med. 2004;32(11):2267-73
20. Al-Ghoul WM, Khan M, Fazal N, Sayeed MM. Mechanisms of Postburn Intestinal Barrier Dysfunction in the Rat: Roles of Epithelial Cell Renewal, E-Cadherin and Neutrophil Extravasation. Crit Care Med. 2004;32(8):1730-1739
21. Choudhry MA, Haque F, Khan M, Fazal N, Al-Ghoul WM, Ravindranath T, Gamelli RL, Sayeed MM. Enteral nutritional supplementation prevents mesenteric lymph node T cell suppression in burn injury. Crit Care Med. 2003;31(6):1764-70
22. Fazal N, Al-Ghoul WM, Schmidt MJ, Choudhry MA, Sayeed MM. Lyn- and ERK-mediated vs. Ca2+ -mediated neutrophil O2- responses with thermal injury. Am J Physiol Cell Physiol. 2002;283(5):C1469-79
23. Choudhry MA, Mao H, Haque F, Khan M, Fazal N, Sayeed MM. Role of NFAT and AP-1 in PGE2-mediated T cell suppression in burn injury. Shock. 2002;18(3):212-6
24. Choudhry MA, Fazal N, Goto M, Gamelli RL, Sayeed MM. Gut-associated lymphoid T cell suppression enhances bacterial translocation in alcohol and burn injury. Am J Physiol Gastrointest Liver Physiol. 2002;282(6):G937-47
25. Ravindranath T, Al-Ghoul W, Namak S, Fazal N, Durazo-Arvizu R, Choudhry M, Sayeed MM. Effects of burn with and without Escherichia coli infection in rats on intestinal vs. splenic T-cell responses. Crit Care Med. 2001;29(12):2245-50
26. Fazal N, Al-Ghoul WM, Choudhry MA, Sayeed MM. PAF receptor antagonist modulates neutrophil responses with thermal injury, in vivo. Am J Physiol. 2001;281:C1310-7
27. Fukuzuka Edwards CK3rd, Clare-Salzler M et al. Glucocorticoid-induced, caspase-dependent organ apoptosis early after burn injury. Am J Physiol. 2000;278:R1005-R1018
28. Hotchkiss RS, Swanson PE, Freeman BD. et al. Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunctions. Crit Care Med. 1999;27:1230-51
29. Ayala A, Chung CS, Xu YX. et al. Increased inducible apoptosis in CD4+ T lymphocytes during polymicrobial sepsis is mediated by Fas ligand and not endotoxin. Immunol. 1999;97:45-55
30. Ju ST, Panka DJ, Cui H. et al. Fas(CD95)/FasL interactions requires for programmed cell death after T-cell activation. Nature. 1995;373:444-448
31. Ramirez F, Mason D. Recirculatory and sessile CD4+ T lymphocytes differ on CD45RC expression. J Immunol. 2000;165:1816-1823
32. Zhang J. et al. Fas-mediated apoptosis and activation-induced T-cell proliferation are defective in mice lacking FADD/Mort1. Nature. 1998;392(6673):296-300
Author biography
Nadeem Fazal, MD, PhD is a Research Professor at Chicago State University. He completed his medical degree from King Edward Medical University, Lahore and started his career as Microbiologist at Department of Pathology, Rawalpindi Medical College, The University of Punjab. He obtained PhD (Mechanisms of human anti-mycobacterial immunity) from The Medical School, University of Birmingham, England. He did postdoctoral training and fellowship from Stritch School of Medicine, Loyola University Chicago, USA. During his tenure at Loyola University Chicago as Assistant Professor he studied pathogenesis of sepsis and intestinal bacterial translocation following acute injury such as shock/burn/trauma. He published his work of host cellular immune responses to infection including dendritic cell, T cell, macrophages and neutrophils and their intracellular signaling pathways in peer reviewed journals. Dr. Fazal has recently accepted position as Professor of Microbiology at King Faisal University. New contact at King Faisal University is: Professor Nadeem Fazal, Department of Microbiology, College of Medicine, King Faisal University, P.O. Box 2114, Dammam 31451, Saudi Arabia. Tel Office: 8577000, Fax: 00966-3-8575329.
Walid Al-Ghoul, PhD, is an associate professor at Chicago State University. He earned his joint PhD degree from Rutgers, the State University of New Jersey and University of Medicine and Dentistry of New Jersey (1991). He has extensive research training at the postdoctoral and research assistant professor levels in the areas of pathophysiology of spinal cord injury and epilepsy (University of North Carolina, 1990-97), molecular pharmacology of circadian function (Northwestern University, 1997-00), and the pathophysiology of burn injury (Loyola University-Chicago, 2000-03). He currently holds an associate professor position at Chicago State University with a joint appointment at Rush University Medical Center's Division of Digestive Disease of the Department of internal medicine. In addition to his teaching responsibilities at Chicago State University, Dr Al-Ghoul has an active research program in the investigation of inflammatory drug targets in intestinal sepsis and stress-mediated gut mucosa pathophysiology. In addition to MBRS grant support, Dr Al-Ghoul's research has also been supported by the National Science Foundation Major Research Instrumentation (NIH-MRI) for acquiring a state-of-the-art Fluorescent Activated Cell Sorter (FACS) that is one of a few such powerful cell sorting instruments in the Midwest.
![]() Correspondence to: Nadeem Fazal, MD, PhD, Department of Biological Sciences, Chicago State University, 9501 South King Drive, Chicago, IL 60628. Email: nfazaledu. Tel: +1 773-821-4989; Fax: +1 773-995-3759
Correspondence to: Nadeem Fazal, MD, PhD, Department of Biological Sciences, Chicago State University, 9501 South King Drive, Chicago, IL 60628. Email: nfazaledu. Tel: +1 773-821-4989; Fax: +1 773-995-3759