Impact Factor ISSN: 1449-2288
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. Materials and Methods
3. Results and Discussion
4. Conclusion
Acknowledgements
References
Author biography

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2008; 4(2):63-70. doi:10.7150/ijbs.4.63 This issue Cite
Short Research Communication
Effect of UVA Fluence Rate on Indicators of Oxidative Stress in Human Dermal Fibroblasts
James D. Hoerter1,2, Christopher S. Ward1,3,*, Kyle D. Bale1,4,*, Admasu N. Gizachew1,*, Rachelle Graham1,5, Jaclyn Reynolds1, Melanie E. Ward1, Chesca Choi1, Jean-Leonard Kagabo1, Michael Sauer1, Tara Kuipers1, Timothy Hotchkiss1, Nate Banner1, Renee A. Chellson1, Theresa Ohaeri1, Langston Gant6, Leah Vanderhill6 ![]()
1. Ferris State University, Department of Biological Sciences, Big Rapids, MI 49307 USA
2. Dublin Institute of Technology, Radiation and Environmental Science Centre, Focas Institute, Dublin-8, Ireland (U.S. Fulbright Scholar, January-August, 2008)
3. Current address: Baylor College of Medicine, Department of Molecular and Human Genetics, Houston, TX 77030 USA
4. Current address: Cayman Chemical Company, Antibody Group, Ann Arbor, MI 48108 USA
5. Current address: National Cancer Institute, Center for Cancer Research, Pediatric Oncology Branch, Bethesda, MD 20892 USA
6. Mecosta-Osceola Intermediate School District, Math Science Technology Center, Big Rapids, MI 49307 USA
* Equal contribution
Received 2007-11-7; Accepted 2008-2-16; Published 2008-2-19
Abstract
During the course of a day human skin is exposed to solar UV radiation that fluctuates in fluence rate within the UVA (290-315 nm) and UVB (315-400 nm) spectrum. Variables affecting the fluence rate reaching skin cells include differences in UVA and UVB penetrating ability, presence or absence of sunscreens, atmospheric conditions, and season and geographical location where the exposure occurs. Our study determined the effect of UVA fluence rate in solar-simulated (SSR) and tanning-bed radiation (TBR) on four indicators of oxidative stress---protein oxidation, glutathione, heme oxygenase-1, and reactive oxygen species--in human dermal fibroblasts after receiving equivalent UVA and UVB doses. Our results show that the higher UVA fluence rate in TBR increases the level of all four indicators of oxidative stress. In sequential exposures when cells are exposed first to SSR, the lower UVA fluence rate in SSR induces a protective response that protects against oxidative stress following a second exposure to a higher UVA fluence rate. Our studies underscore the important role of UVA fluence rate in determining how human skin cells respond to a given dose of radiation containing both UVA and UVB radiation.
Keywords: UVA, UVB, tanning bed, solar simulated radiation, fluence rate, sunbed
1. Introduction
Within the last two decades the molecular and cellular response to UVB (290-315 nm) and UVA (315-400 nm) radiation in human cells both in vivo and in vitro have been studied extensively. Many studies used radiation sources with fluence rates and UVA/UVB ratios different from that of solar radiation. Furthermore, many of the experiments were designed under the assumption that the biological response to given dose of radiation depends only on the total cumulative dose and not on the fluence rate at which the dose is delivered [1,2]. Fluence rate refers to the radiant intensity or power (W) incident on a surface divided by the cross-sectional area of that surface (m2). However, recent studies suggest that the biological response to a given dose of UVA radiation can be influenced by the UVA fluence rate at which that dosage is delivered [3-4].
The purpose of our study was to determine the effect of UVA fluence rate on indicators of oxidative stress in human dermal fibroblasts (HDFs) when cells are exposed to solar-simulated (SSR) or tanning-bed radiation (TBR). Since UVA and UVB radiation each elicit a different time course of response and operate through different mechanisms (oxidative vs. direct DNA absorption), altering UVA fluence rate may either enhance or attenuate the biological response through synergistic interactions when cells are also receiving UVB radiation.
For our experiments we selected two radiation sources-- SSR and TBR--that human skin cells might be exposed to during a 24-hr period to determine if UVA fluence rate affects indicators of oxidative stress in HDFs. Four indicators of oxidative stress were used to measure changes in biological response in HDFs--protein oxidation (carbonyl groups), glutathione (GSH), heme oxygenase-1 (HO-1) and reactive oxygen species (ROS). Our hypothesis was that under a given UVA dose, radiations with lower fluence rates will have reduced protein oxidation/ROS levels and increased levels of protective agents (GSH, HO-1). Our rationale being that radiations with lower UVA fluence rates will have the effect of spreading the given dose of radiant energy over time, reducing ROS levels and biological damage due to a greater amount of time being available for induction of protective pathways for repair and defense. On the other hand, radiations with higher UVA fluence rates will concentrate the radiant energy over a shorter period of time, increasing oxidative stress and overwhelming defense mechanisms before protective mechanisms have time to take effect.
We also tested the hypothesis that when cells are given two sequential exposures, the first exposure with a lower fluence rate (SSR) will protect the cell from the oxidative damage of a subsequent exposure with a higher fluence rate (TBR). Our rationale being that the first irradiation with a lower fluence rate will permit more time for the induction of protective mechanisms permitting cells to withstand the oxidative stress of the second exposure.
2. Materials and Methods
Cell Culture
HDFs (GM00038) were obtained from the Coriell Institute for Medical Research (Coriell Cell Repository, Camden, NJ, USA). Cultures were incubated (5% CO2) at 35o C in T-75 tissue culture flasks in growth media consisting of Eagle's minimal essential medium (EM) supplemented with 10% fetal bovine serum and 2mM L-glutamine. EM was replaced every 72 hrs. Cells were passaged at 90% confluence by removing EM, rinsing cells with phosphate-buffered saline (PBS) and incubating with trypsin for 10 min at 35o C. Flasks were seeded at a density of 1 x 104 cells/cm2.
Irradiation and Survival
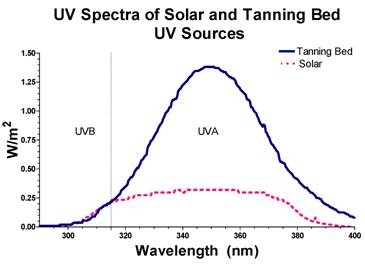
Prior to irradiation, cells were harvested, enumerated using a hemocytometer, diluted and suspended in PBS. The effect of leaving the cells in PBS during the irradiation time did not have a significant effect on survival. SSR was performed with a solar simulator (Solar Light, Glenside, PA , USA) at a fluence rate of 49 W/m2 for UVA and 1.7 W/m2 for UVB. UVA and UVB fluence rate and total dose was measured using a UV meter (PMA2100; Solar Light, Glenside, PA, USA) equipped with UVA (PMA2110) and UVB (PMA2106) probes. Fluence rate was measured through one plastic layer of a T-75 flask to determine the fluence rate received by cells on the bottom surface of the flask. TBR was performed with a commercial tanning bed (Wolff System Technology, Kennesaw, GA, USA) equipped with Dominion bulbs to deliver radiation to the bottom surface of the flask at a fluence rate of 135 W/m2 for UVA and 1.7 W/m2 for UVB. The top surface of the flask was covered with tin foil to prevent cells from receiving radiation from the top bank of tanning bulbs. During either SSR or TBR, HDFs received equivalent dosages of UVA (90 kJ/m2) and UVB (1.1 kJ/m2). For SSR, this was equivalent to about 31 min of exposure (10 min into the exposure, a glass plate was placed over the flask to attenuate the UVB for the remaining 21 min of exposure to SSR so that the cells would only receive 1.1 kJ/m2 of UVB). For TBR, this was equivalent to about 11 min of exposure. When cells were exposed to sequential cycles of TBR and SSR, each exposure contained equivalent dosages of UVA (90 kJ/m2) and UVB (1.1 kJ/m2). Sequential exposures were separated by a 24-hr period during which cells were returned to normal media. Temperature of the medium during irradiation never exceeded 37° C. The spectral distribution of irradiance in SSR and TBR is shown in Figure 1.
UVA and UVB mission spectra of the radiation emitted from the solar simulator (dotted line) and tanning-bed bulbs (dark black line) used in this study.
After each irradiation, PBS was replaced with EM. At 0, 14, 24 and 48 hrs post irradiation, percent cell survival was determined using the dye exclusion method (0.4% trypan blue). Assays for indicators of oxidative stress (protein oxidation, GSH and HO-1) were also conducted at 0, 14, 24 and 48 hrs post irradiation except when determining ROS levels in which case cells were analyzed immediately after irradiation. For sequential exposures, PBS was replaced with EM following the first irradiation. Cells were then incubated for 24 hrs and media replaced again with PBS before the second exposure. Sham-irradiated (control) cells were treated as described above but were not exposed to radiation. Cell survival and assays for indicators of oxidative stress were conducted at 0, 14, 24 and 48 hrs after the last exposure.
Protein Oxidation
Proteins samples were treated according to the OxyBlot protocol (Chemicon, Temecula, CA, USA) to detect carbonyl groups. Briefly, 2μg of total extracted protein were incubated for 15 min at room temperature with 2,4-dinitrophenylhydrazine to form the carbonyl derivative, dinitrophenylhydrazone, before spot-blotting on a nitrocellulose membrane. Oxidized proteins were detected by anti-dinitrophenol antibodies. Blots were enhanced with Quentix signal enhancer (Pierce Biotechnology, Rockford, IL, USA) and blocked with 1% BSA. Secondary antibody and chemiluminescent detection was performed with WesternBreezeTM (Invitrogen, Carlsbad, CA, USA). Signal was detected on film, scanned and quantified by densitometry. Relative protein oxidation levels were normalized to the amount of protein spotted on the membrane and plotted as a fold increase over control levels.
Glutathione (GSH)
Protein samples were assayed for glutathione according to the method of Griffith [5]. Assays were performed in triplicate. Glutathione concentration was determined by extrapolating from the standard curve and normalizing to protein concentration. Glutathione concentration was plotted as a fold increase over control levels.
Heme oxygenase-1 (HO-1)
HO-1 was detected on spot blots using rabbit anti-HO-1 protocol (Abcam, Cambridge, UK). For the primary antibodies, 1/2000 rabbit anti-HO-1 and 1/5000 rabbit anti-β-actin were used for their respective blots. HO-1 positive control was also spotted. Spot blots were conducted in triplicate and run in parallel, one for HO-1 and the other for β-actin. Secondary antibody incubation, chemiluminescent signal detection, and quantification were performed as described above for protein oxidation. HO-1 expression was normalized to β-actin expression and plotted as a fold increase over control levels.
Fluorescent detection of mitochondrial superoxide and ROS
Immediately after irradiation, HDFs were treated with MitoSOX™ Red (Invitrogen, Carlsbad, CA, USA) and incubated in the dark at 370 C. After 10 min the cells were washed with PBS and viewed under a fluorescent microscope. MitoSOX™ Red reagent is readily oxidized by superoxide in the mitochondria and binds to nucleic acids, emitting a bright red fluorescence. Image-iTTM LIVE Green Reactive Oxygen Species kit (Invitrogen, Carlsbad, CA, USA) was used to detect ROS in live cells. In the presence of ROS, the reduced fluorescein compound is oxidized, emitting a bright green fluorescence wherever ROS is present in the cell. HDFs were viewed with an Olympus BH-2 epi-fluorescent microscope with fluorosceine and rhodamine filters. Fluorescent intensity of at least 100 individual cells, randomly selected in 10 different fields of view, was measured with a photodensitometer for each of four independent experiments to calculate average fluorescent intensity under each experimental condition. The slide that was treated to detect fluorescence was also stained to confirm the presence of cells.
3. Results and Discussion
As shown in Table 1, all fluence rates and dosages used in our experiments yielded nearly a 75% survival rate at all time points post irradiation with the exception of the TBR:SSR exposure sequence which was 68%. This suggests that under these dosages and radiation conditions, cells were capable of inducing oxidative defense pathways. No significant changes in survival rate were observed between SSR and TBR at 14, 24, or 48 hrs post irradiation suggesting that fluence rate at which a given dose of UVA/UVB radiation is received is not a variable significantly influencing cell survival with 48 hrs after receiving this dose of radiation. The data show a slight trend toward decreased survival in the TBR:SSR sequential exposure compared to SSR:TBR sequential exposure but was not found to be significant. However, much more detailed analysis over a more extended period of time is needed to determine if fluence rate significantly affects survival under different conditions (dosages and fractionated exposures) and for other indicators of radiation damage such as delayed apoptotic cell death and genomic instability.
Effect of fluence rate on percent (%) viability of HDFs at 0, 14, 24 and 48 hrs post irradiation following a single exposure (SSR or TBR) or following sequential exposures (SSR:TBR or TBR:SSR). Each exposure received equivalent dosages (90kJ/m2 UVA; 1.1 kJ/m2 UVB. Data represent mean ±S.E. of at least three independent experiments. No significant differences were detected either between single exposures (SSR and TBR) or between double exposures (SSR:TBR or TBR:SSR). The mock-treated controls were set at 100%.
| Radiation Source | 0 hrs % ±S.E. | 14 hrs % ±S.E. | 24hrs % ±S.E. | 48hr % ±S.E |
|---|---|---|---|---|
| Control | 100 | 100 | 100 | 100 |
| SSR | 93 ± 1.6 | 92 ± 5.6 | 89 ± 3.2 | 86 ± 7.1 |
| TBR | 89 ± 5.1 | 91 ± 4.5 | 86 ± 6.2 | 79 ± 4.3 |
| SSR:TBR | 88 ± 7.1 | 83 ± 8.3 | 84 ± 6.0 | 79 ± 5.3 |
| TBR:SSR | 78 ± 3.3 | 79 ± 5.4 | 74 ± 2.6 | 68 ± 4.2 |
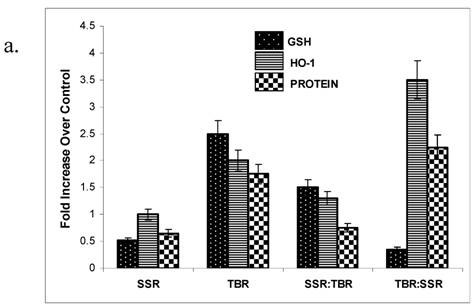
Physiological doses of UVA radiation administered at an irradiance of 300 W/m2 have previously been shown to induce oxidative damage to proteins in human skin fibroblasts [6-7]. Carbonylated proteins are formed early and tend to be more stable than other indicators of oxidative damage [8] and are known to be an effective indicator of environmental oxidative damage to human skin [9]. We selected protein oxidation as an indicator of UV-induced oxidative damage because of its potential impact on a multitude of biological pathways of defense and repair [10]. We found a significantly higher amount of protein oxidation in HDFs after TBR over that observed in cells exposed to SSR (Figure 2a,b).
a. GSH, HO-1 and protein oxidation levels in HDFs following a single exposure (SSR or TBR) or following sequential exposures (SSR:TBR or TBR:SSR). Activity levels were measured 14 hrs post irradiation and plotted as a fold increase over control levels (set at 1.0). Data represents the mean of at least three independent experiments. In each experiment, assays were performed in triplicate. Error bars represent SEM. All indicators were significantly different (p<0.05) between the single exposures (SSR vs. TBR) and between the two double exposures (SSR:TBR vs. TBR:SSR) using Student's t-test. 2b. Representative spot blots of protein oxidation in HDFs in control at 14 hrs, 24 hrs and 48 hrs after receiving single exposures of either SSR or TBR. 2c. Representative spot blots of protein oxidation in HDFs in control and at 14 hrs, 24 hrs and 48 hrs after receiving double exposures (SSR:TBR or TBR:SSR). 2d. Representative spot blots of HO-1 in HDFs in control and at 14 hrs, 24 hrs and 48 hrs after receiving single exposures (SSR or TBR). 2e. Representative spot blots of HO-activity in HDFs in control and at 14 hrs, 24 hrs and 48 hrs after receiving double exposures (SSR:TBR or TBR:SSR).
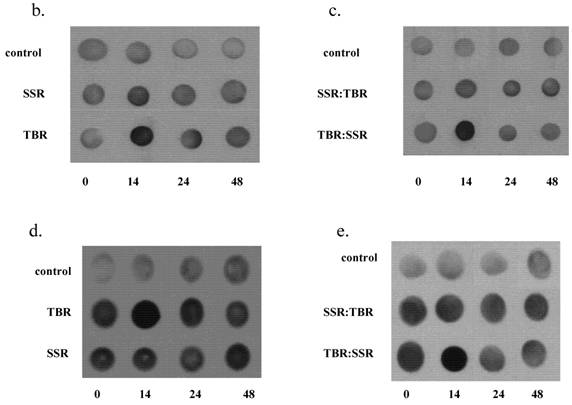
One explanation for our data showing a reduction in the amount of protein oxidation in cells exposed to SSR as well as in cells exposed to the sequential exposure (SSR:TBR) is that protein repair pathways are induced by the lower UVA fluence rate and protect cells from a second exposure. This increases the repair capability of the cells and increases their protection against higher UVA fluence rates (TBR). In other words, a prior dose of UVA radiation delivered at low fluence rate (SSR) protects cells from protein oxidation following a second exposure to UVA radiation delivered at a higher fluence rate (TBR).
No significant differences were found in protein oxidation between cells exposed to TBR and SSR at 24 hrs, suggesting that protein oxidation repair pathways are upregulated sometime between 14 and 24 hrs leading to the removal of oxidized proteins. Our data correlate with a study showing that repetitive SSR (0.5 MED for 10 consecutive days) of human epidermis and single UVA exposures (10 kJ/m2) of human keratinocytes upregulate the expression of epidermal methionine-S-sulfoxide (MSRA), the only enzyme thus far identified in human skin that is capable of repairing oxidative protein damage and preventing the appearance of protein carbonyls [11-12].
Glutathione (GSH) is a major endogenous antioxidant that protects cells from toxic free radicals. GSH has a high redox potential, making it a potent antioxidant. Our results showing at 14 hrs a significantly higher GSH level (2.5 fold over control) in HDFs treated with TBR over that observed after SSR (Fig 2a), suggest that UVA radiation with a higher fluence rate invokes a stronger antioxidant response, leading to higher levels of intracellular glutathione. The significantly higher levels of ROS and mitochondrial superoxide that we observed in HDFs after TBR (Figure 3) over that observed following SSR, suggest that ROS may play an important role in triggering the increase in GSH in order to quench the reactive intermediates that may otherwise lead to spontaneous mutations [13]. One possible hypothesis to explain why there is less GSH in the TBR:SSR exposure sequence is that when TBR is administered first, there is a longer period of time for the oxidation of GSH before the levels are experimentally determined. Another possible explanation is that when TBR is the first exposure there is more time for GSH to leak outside the cells due to membrane damage [14]. No significant differences in GSH were detected between the different exposure regimes after 14 hrs post irradiation, suggesting that a delayed induction of GSH does not occur after either SSR or TBR within a 24-hr period.
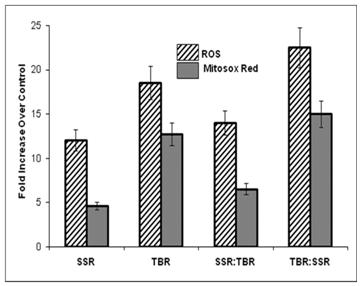
Fluorescent intensity expressed as a fold increase over control levels in HDFs treated to detect ROS and mitochondrial superoxide immediately following single (SSR or TBR) or sequential exposures (SSR:TBR or TBR:SSR). Fluorescent intensity of at least 100 individual cells, randomly selected in 10 different fields of view, was measured with a photodensitometer for each of four independent experiments to calculate average intensity for single and double exposure sequences. All exposures showed significantly higher fluorescent intensity over that observed in controls (p<0.05). Both ROS and mitochondrial superoxide were significantly different (p<0.05) between the two single exposures (SSR vs. TBR) and between the two double exposures (SSR:TBR vs. TBR:SSR) using Student's t-test.
HO-1 is known to be expressed in fibroblasts and melanocytes as a result of oxidative stress via UVA [15-17]. Our data show that at 14 hrs post irradiation, cells exposed to higher fluence rates (TBR) have significantly higher HO-1 activity (2.1 fold over controls) over that observed in cells exposed to SSR (Fig 2a,d,e). One possible explanation for our results is that higher UVA fluence rates trigger signaling pathways or transcription factors such as Nrf-2 which are known to induce many antioxidant and detoxification genes including HO-1 [18], but low-fluence UVA has the opposite effect. When cells receive their first UVA exposure at a lower fluence rate, it attenuates or suppresses oxidative signals that normally induce HO-1 within 14 hrs Recently, it has been shown that increased UVA/UVB ratios attenuate apoptotic effects of UVB-induced apoptosis via HO-1 [19]. Our data suggest that UVA with a higher fluence rate may also protect against apoptosis via increased HO-1. Our data have important implications for people who expose their skin to tanning-bed radiation either before or after recreating in the Sun. One possible consequence of recreating in the Sun within 14 hrs after exposing skin to TBR is that apoptosis will be inhibited via increased HO-1, allowing the accumulation of UVB-induced mutations and increasing the risk of skin cancer [20]. No significant differences in HO-1 were detected between the different exposure regimes after 14 hrs post irradiation, suggesting that a delayed induction of HO-1 does not occur either after SSR or TBR within a 24-hr period.
It is generally accepted that the damaging effects of UVA are mediated by the activities of ROS. Following UVA irradiation, elevated levels of the superoxide anion (O2.- ) have been previously detected by chemiluminescence in the skin of live mice following UVA irradiation with the initial burst of chemiluminescence signal dependent on the UVA fluence rate [21-22]. As shown in Figure 3, our data provide additional evidence that fluence rate at which a given dose of UVA irradiation is administered to live HDFs is an important variable influencing the levels of ROS generated. Specifically, increasing the fluence rate of a given dose of UVA radiation leads to significantly higher levels of intracellular ROS over that observed with low-fluence UVA.
We found this same direct correlation (higher ROS with increased fluence rate) using two different methods (MitoSOX™ Red and Image-iTTM LIVE Green ROS) for fluorescent detection of ROS. This correlation is further supported by another study showing free radical levels in skin (measured using the ascorbate radical) are constant over time when irradiated with SSR (18 W/m2), but when irradiated at a higher fluence rate (39 W/m2), radical production exceeds its recycling to the antioxidant ascorbate, suggesting that higher fluence rates render skin unprotected against further oxidative stress [23]. Shown in Figure 4 are representative pictures of HDFs viewed under a fluorescence microscope immediately after exposure to single or sequential exposures of SSR and TBR and treated with highly selective probes for ROS.
Representative photomicrographs of HDFs viewed under a fluorescent microscope for ROS (green) and mitochondrial superoxide (red) immediately after a single exposure (SSR or TBR) or after a sequential exposure (SSR:TBR or TBR:SSR).
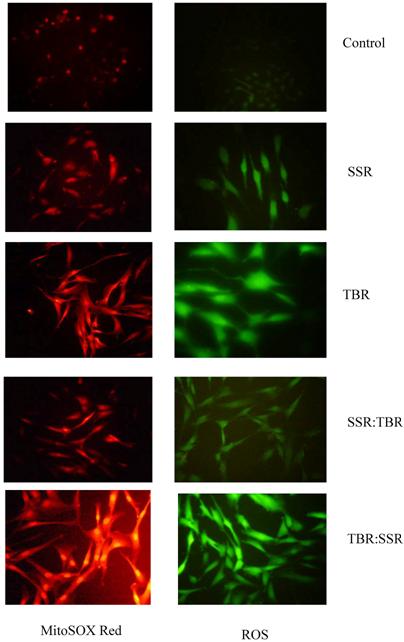
One possible hypothesis to explain the increased levels of ROS following TBR is that UVA with a higher fluence rate is more effective in photodegradation of ferritin. This would increase the availability of intracellular iron for redox cycling in the generation of ROS and inhibit the cell's ability to sequester and store excess iron. The surge in HO-1 activity that we observed in cells after TBR will further increase intracellular iron via its release from endogenous heme sources [24]. This also can explain why we observed an increase in protein oxidation after cells receive UVA radiation with a higher fluence rate.
However, our results are in contradiction to a recent study that does not show an increase, but a decrease in ROS in keratinocytes after receiving radiation with a higher UVA fluence rate [3]. Possible reasons for this discrepancy may due to the following: (1) inherent differences between fibroblasts and keratinocytes in their protective responses and/or basal levels of intracellular antioxidants, (2) differences in UVA dosage (200 kJ/m2 vs. 90kJ/m2 in our study), (3) differences in fluence rates (our study compared 49 W/m2 and 139W/m2; their study compared 80 W/m2 and 224 W/m2), (4) different techniques for detecting ROS, or more importantly to (5) differences in radiation wavelengths (both UVA and UVB in our study vs. UVA only in their study). Since our irradiation conditions simulate possible conditions when humans recreate outside and/or use tanning beds, the results of our study may be more environmentally relevant for predicting how changes in the UVA fluence rate affect pathways related to oxidation damage and defense. We included UVB in all our experiments because we wanted to mimic the radiation spectrum of the Sun and a representative tanning bulb. Cells have evolved mechanisms to response to both UVA and UVB radiation simultaneously, and when UVA and UVB responses are measured separately, this may prevent the occurrence of important synergistic interactions in the oxidative damage and defense pathways.
The significantly higher ROS levels in TBR:SSR exposure sequence over that observed in the opposite exposure sequence (SSR:TBR) is consistent with the reduced levels of GSH and with the higher protein oxidation levels also observed in the TBR: SSR exposure sequence. These cells will have reduced antioxidant capability, contributing to increased levels of ROS levels. Our data suggest that the ROS levels are determined by the first exposure. In other words, when HDFs received TBR for the first exposure, the second exposure of SSR will not significantly increase ROS levels over that found after a single exposure to TBR, and when HDFs received SSR for the first exposure, the second exposure of TBR will not significantly increase ROS levels over that found after a single exposure to SSR. Our data support the hypothesis that radiation with lower UVA fluence rates (SSR) induces an immediate antioxidant response that protects cells against subsequent exposure to higher fluences of UVA radiation. The antioxidant response will quench the increased levels of ROS. Since only modest increases in GSH (0.5 fold) and HO-1 (1.0 fold) over basal levels were found after SSR, this suggests that GSH and HO-1 are not playing a major role in this immediate protective response.
The implication of our ROS findings is that under equal doses, UVA radiation from tanning beds yields greater ROS-mediated damage in HDFs than that after solar radiation, and that a prior exposure of SSR may reduce the harmful effects of a subsequent exposure to TBR. This suggests one possible strategy for reducing the potential for ROS-mediated damage in skin cells following TBR--use sequential exposures of TBR containing low- and high-fluence UVA irradiation.
4. Conclusion
Our studies underscore the complexity of those factors contributing to the signaling pathways regulating the biological response to a given dose of UVA/UVB radiation, and further support the important role that UVA fluence rate plays in determining that response. Cells residing in different layers of the skin are continuously subjected to fluctuating UVA fluence rates due to differences in the penetrating ability of UVA. Other variables altering UVA fluence rate include sunscreens that differ in their ability to block the UVA wavelengths, and seasons of the year when exposure occurs [25]. Tanning-bed bulbs are another source of radiation containing different UVA fluence rates. UVA fluence rate needs to be considered in addition to overall dosage when designing radiation protocols so that an accurate biological response pattern may be obtained [26]. Because our data suggest that fluence rate impacts how a cell will respond to a given dose of radiation, we encourage all investigators who are utilizing cell culture, in vitro skin constructs, or live animals for investigating the biological response to a given dose of UVA radiation to design radiation protocols that will accurately reflect the fluence rate that cells receive in vivo under a given set of environmental conditions. This will enable investigators to compare experimental results and obtain data that will more accurately reflect the biological responses in vivo, providing a standard to evaluate potential therapeutic strategies for the prevention of UVA/UVB radiation damage. Since UVA has been implicated in the etiology of malignant melanoma [27] our data suggest that sunscreens and tanning beds that change the UVA fluence rate may affect oxidative pathways known to be involved in photocarcinogenesis.
Acknowledgements
This publication was made possible by grant number R15ES013232 from the National Institute of Environmental Health Sciences (NIEHS) and by an Exceptional Merit Grant from Ferris State University Foundation to James D. Hoerter. Special appreciation is extended to Ann Hoerter for her assistance in preparing the figures and to Sandra Balkema for her editorial suggestions.
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
1. Bunsen RW, Roscoe HE. Photochemische untersuchungen. Ann Phys. 1859;108(2):193
2. Diffey BL. What is light? Photodermatol Photoimmunol Photomed. 2002;18(2):68-74
3. Shorrocks J, Paul ND, McMillan TJ. The Dose Rate of UVA Treatment Influences the Cellular Response of HaCaT Keratinocytes. J Invest Dermatol. 2007 [Epub ahead of print]
4. Merwald H, Klosner G, Kokesch C, Der-Petrossian M, Honigsmann H, Trautinger F. UVA-induced oxidative damage and cytotoxicity depend on the mode of exposure. J Photochem Photobiol B. 2005;79(3):197-207
5. Griffith OW. Determination of glutathione and glutathione disulfide using glutathione reductase and 2-vinylpyridine. Anal Biochem. 1980;106(1):207-12
6. Vile GF, Basu-Modak S, Waltner C, Tyrrell RM. Heme oxygenase 1 mediates an adaptive response to oxidative stress in human skin fibroblasts. Proc Natl Acad Sci U S A. 1994;91(7):2607-10
7. Vile GF, Tyrrell RM. UVA radiation-induced oxidative damage to lipids and proteins in vitro and in human skin fibroblasts is dependent on iron and singlet oxygen. Free Radic Biol Med. 1995;18(4):721-30
8. le-Donne I, Rossi R, Giustarini D, Milzani A, Colombo R. Protein carbonyl groups as biomarkers of oxidative stress. Clin Chim Acta. 2003;329(1-2):23-38
9. Thiele JJ, Traber MG, Re R, Espuno N, Yan LJ, Cross CE. et al. Macromolecular carbonyls in human stratum corneum: a biomarker for environmental oxidant exposure? FEBS Lett. 1998;422(3):403-6
10. Davies MJ. Singlet oxygen-mediated damage to proteins and its consequences. Biochem Biophys Res Commun. 2003;305(3):761-70
11. Picot CR, Petropoulos I, Perichon M, Moreau M, Nizard C, Friguet B. Overexpression of MsrA protects WI-38 SV40 human fibroblasts against H2O2-mediated oxidative stress. Free Radic Biol Med. 2005;39(10):1332-41
12. Ogawa F, Sander CS, Hansel A, Oehrl W, Kasperczyk H, Elsner P. et al. The repair enzyme peptide methionine-S-sulfoxide reductase is expressed in human epidermis and upregulated by UVA radiation. J Invest Dermatol. 2006;126(5):1128-34
13. Applegate LA, Lautier D, Frenk E, Tyrrell RM. Endogenous glutathione levels modulate the frequency of both spontaneous and long wavelength ultraviolet induced mutations in human cells. Carcinogenesis. 1992;13(9):1557-60
14. Moysan A, Marquis I, Gaboriau F, Santus R, Dubertret L, Morliere P. Ultraviolet A-induced lipid peroxidation and antioxidant defense systems in cultured human skin fibroblasts. J Invest Dermatol. 1993;100(5):692-8
15. Applegate LA, Noel A, Vile G, Frenk E, Tyrrell RM. Two genes contribute to different extents to the heme oxygenase enzyme activity measured in cultured human skin fibroblasts and keratinocytes: implications for protection against oxidant stress. Photochem Photobiol. 1995;61(3):285-91
16. Keyse SM, Tyrrell RM. Heme oxygenase is the major 32-kDa stress protein induced in human skin fibroblasts by UVA radiation, hydrogen peroxide, and sodium arsenite. Proc Natl Acad Sci U S A. 1989;86(1):99-103
17. Tyrrell RM. Solar ultraviolet A radiation: an oxidizing skin carcinogen that activates heme oxygenase-1. Antioxid Redox Signal. 2004;6(5):835-40
18. Hirota A, Kawachi Y, Itoh K, Nakamura Y, Xu X, Banno T. et al. Ultraviolet A irradiation induces NF-E2-related factor 2 activation in dermal fibroblasts: protective role in UVA-induced apoptosis. J Invest Dermatol. 2005;124(4):825-32
19. Ibuki Y, Allanson M, Dixon KM, Reeve VE. Radiation sources providing increased UVA/UVB ratios attenuate the apoptotic effects of the UVB waveband UVA-dose-dependently in hairless mouse skin. J Invest Dermatol. 2007;127(9):2236-44
20. Runger TM. How different wavelengths of the ultraviolet spectrum contribute to skin carcinogenesis: the role of cellular damage responses. J Invest Dermatol. 2007;127(9):2103-5
21. Evelson P, Ordonez CP, Llesuy S, Boveris A. Oxidative stress and in vivo chemiluminescence in mouse skin exposed to UVA radiation. J Photochem Photobiol B. 1997;38(2-3):215-9
22. Yasui H, Sakurai H. Chemiluminescent detection and imaging of reactive oxygen species in live mouse skin exposed to UVA. Biochem Biophys Res Commun. 2000;269(1):131-6
23. Haywood R. Relevance of sunscreen application method, visible light and sunlight intensity to free-radical protection: A study of ex vivo human skin. Photochem Photobiol. 2006;82(4):1123-31
24. Vile GF, Tyrrell RM. Oxidative stress resulting from ultraviolet A irradiation of human skin fibroblasts leads to a heme oxygenase-dependent increase in ferritin. J Biol Chem. 1993;268(20):14678-81
25. Reeve VE, Domanski D, Slater M. Radiation sources providing increased UVA/UVB ratios induce photoprotection dependent on the UVA dose in hairless mice. Photochem Photobiol. 2006;82(2):406-11
26. Tyrrell RM. Ultraviolet radiation and free radical damage to skin. Biochem Soc Symp. 1995;61:47-53
27. Wang SQ, Setlow R, Berwick M, Polsky D, Marghoob AA, Kopf AW. et al. Ultraviolet A and melanoma: a review. J Am Acad Dermatol. 2001;44(5):837-46
Author biography
James Hoerter is professor of biological sciences at Ferris State University. He earned his Ph.D. in genetics from Pennsylvania State University. In addition to his teaching responsibilities, he directs a research program on the molecular effects of UVA on human skin cells as it relates to photocarcinogenesis. He has received several grants from the National Science Foundation (Research Opportunity Awards) and from the National Institutes of Health (Academic Research Enhancement Awards) to support and promote research activities at the undergraduate level. He is a strong advocate for using research as a teaching method to help undergraduate students learn the scientific method. Under his supervision, he encourages undergraduate students to discover the excitement of research by having them actively participate in research by presenting seminars, designing and conducting experiments, and collecting and analyzing data. Under an NSF-funded curriculum project, Research Link 2000, he helped many colleges and universities integrate undergraduate research into the curriculum. In 2008 he received a U.S. Fulbright Research Fellowship at the Dublin Institute of Technology, Research and Environmental Science Centre, Focas Institute, Dublin, Ireland.
![]() Correspondence to: Dr. James D. Hoerter, Department of Biological Sciences, Ferris State University, Big Rapids, MI 49307, USA, Phone: 231-591-2563, Fax: 231-591-2540, Email: hoerterjedu.
Correspondence to: Dr. James D. Hoerter, Department of Biological Sciences, Ferris State University, Big Rapids, MI 49307, USA, Phone: 231-591-2563, Fax: 231-591-2540, Email: hoerterjedu.