Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2008; 4(2):87-95. doi:10.7150/ijbs.4.87 This issue Cite
Research Paper
Mutation at Tyrosine in AMLRY (GILRY Like) Motif of Yeast eRF1 on Nonsense Codons Suppression and Binding Affinity to eRF3
Akhmaloka 1, Prima Endang Susilowati1,2, Subandi 3, Fida Madayanti1 ![]()
1. Biochemistry Research Division, Faculty of Mathematics and Natural Sciences, Institut Teknologi Bandung, Jln Ganesha 10, Bandung, Indonesia.
2. Department of Chemistry, Faculty of Mathematics and Natural Sciences, Universty of Halouleo, Kendari, Indonesia.
3. Department of Chemistry, Faculty of Mathematics and Natural Sciences, State Universty of Malang, Malang, Indonesia.
Received 2008-2-17; Accepted 2008-4-20; Published 2008-4-21
Abstract
Termination translation in Saccharomyces cerevisiae is controlled by two interacting polypeptide chain release factors, eRF1 and eRF3. Two regions in human eRF1, position at 281-305 and position at 411-415, were proposed to be involved on the interaction to eRF3. In this study we have constructed and characterized yeast eRF1 mutant at position 410 (correspond to 415 human eRF1) from tyrosine to serine residue resulting eRF1(Y410S). The mutations did not affect the viability and temperature sensitivity of the cell. The stop codons suppression of the mutant was analyzed in vivo using PGK-stop codon-LACZ gene fusion and showed that the suppression of the mutant was significantly increased in all of codon terminations. The suppression on UAG codon was the highest increased among the stop codons by comparing the suppression of the wild type respectively. In vitro interaction between eRF1 (mutant and wild type) to eRF3 were carried out using eRF1-(His)6 and eRF1(Y410S)-(His)6 expressed in Escherichia coli and indigenous Saccharomyces cerevisiae eRF3. The results showed that the binding affinity of eRF1(Y410S) to eRF3 was decreased up to 20% of the wild type binding affinity. Computer modeling analysis using Swiss-Prot and Amber version 9.0 programs revealed that the overall structure of eRF1(Y410S) has no significant different with the wild type. However, substitution of tyrosine to serine triggered the structural change on the other motif of C-terminal domain of eRF1. The data suggested that increasing stop codon suppression and decreasing of the binding affinity of eRF1(Y410S) were probably due to the slight modification on the structure of the C-terminal domain.
Keywords: eRF1, eRF3, Saccharomyces cerevisiae, termination translation, nonsense codon suppression, binding affinity
INTRODUCTION
Protein biosynthesis is a complex phenomenon involving RNA-RNA, RNA-protein and protein-protein interactions. This process is carried out in three distinct steps: initiation, elongation and termination. While the first two steps have been extensively studied, our understanding of the termination process has lagged behind.
Protein biosynthesis terminates when one of the three stop codons enters the aminoacyl site (A-site) and signals polypeptide chain release from the peptidyl-tRNA located in the ribosomal P-site [1]. The process is facilitated by two general groups of accessory proteins: a class I release factor, codon-specific RFs (RF1 and RF2 in prokaryotes; eRF1 in eukaryotes), and a class II release factor, codon-non specific RFs (RF3 in prokaryotes and eRF3 in eukaryotes) that binds guanine nucleotides-binding proteins possessing GTPase activity [2, 3, 4]. Although the basic biological action of class-1 RFs is similar between prokaryotes and eukaryotes, they exhibit distinct structural and functional features. Yeast eukaryotic release factor 1 (eRF1) encoded by SUP45 gene, recognizes any of the three stop codons [5]. Eukaryotic release factor 3 (eRF3) in yeast encoded by SUP35 gene, stimulates the termination reaction in GTP dependent manner [6, 4]. Following stop codon recognition, eRF1 also induces polypeptide chain release by activating peptidyl transferase center of the ribosome.
Both eRF1 and eRF3 are essential for viability of yeast cells and deletion of the C-terminal part of each protein separately lead to lethality [1]. Crystal structure of human eRF1 has been determined and found that the protein is composed by three domains. The N-terminal domain (domain1) has been proposed to be responsible for stop codon recognition [7]. This proposal was supported by mutational approach [5] and crosslinking experiment [8]. The middle domain (domain 2) is responsible for peptidyl transferase hydrolytic activity and includes a GGQ motif that has been highly conserved through evolution [9]. Mutations in GGQ (Gly residues) are dominant-negative in vitro and lethal in vivo in S. cerevisiae cells [7]. The domain 3 corresponds to the C-terminal part of eRF1 that is necessary for the interaction with eRF3 although there are discrepancies in the precise location of the region of eRF1 that interacts with eRF3. Progressive deletion of the C-terminal of eRF1, 6-19 amino acids in S. cerevisiae [10] and 17 amino acids of Schizosaccharomyces pombe [2] resulted in a corresponding loss of eRF3 binding. In any case, the core eRF3-binding region identified for Homo sapiens eRF1 (by the yeast two-hybrid and deletion analysis), showed that two regions in each release factor are critical for mutual binding, position 281-305 and 411-415 (GILRY) of eRF1 and position 478-530 and 628-637 of eRF3 (11]. Although deletion of residues within domain 3 of eRF1 resulted in the loss of eRF3 interaction, however detail position and amino acid residues for the interaction are still unclear yet.
Preliminary study using computer modeling analysis on the structure of yeast eRF1, especially on the above two regions showed that tyrosine at position of 410 (Y410) and threonine at position of 295 (T295) of eRF1 exposed to the surface of molecules. Tyrosine at position of 410 is one of amino acid residues in AMLRY (GILRY like) motif of yeast eRF1 protein. Among of the amino acid residues on the AMLRY motif, tyrosine is one of best possible amino acid residues that contributing on its interaction since this residues containing hydroxyl group. In order to probe the role of tyrosine at AMLRY motif (Y410) of eRF1, here we reported the effect of mutation Y410 on the termination of protein biosynthesis. The nonsense codon suppressions have been used to assay the accuracy of termination process.
MATERIALS AND METHODS
Microbial strains, plasmids and growth conditions
S. cerevisiae strains used in this study are listed in Table 1. All strains were SUQ5 and [psi-]. Yeast cultures were growth in standard rich medium, YPD (1% (w/v) bacto peptone, 0.5% (w/v) yeast extract, and 2% (w/v) glucose); Y8 (1% (w/v) bacto peptone, 0.5% (w/v) yeast extract, and 8% (w/v) glucose) or minimal medium was used SM (0.67% (w/v) yeast nitrogen base, 2% (w/v) glucose, supplemented with appropriate amino acids) at 300C with gently shaking. For the plasmid shuffling, the strains were grown on medium YNB-FOA (0.67 % (w/v) yeast nitrogen base, 2% (w/v) glucose, 0.1% (w/v) 5-Fluorootic acid (5-FOA), 20 μg/mL uracil).
Yeast strains used in this study.
| Strains | Genotype |
|---|---|
| Yeast ΔLE2(803) | SUQ5, ade2-1,his3-11,15, ura3-1, leu2-1, can1-100, [psi-], SUP45::HIS3 |
| Yeast ΔLE2(SUP45) | SUQ5, ade2-1,his3-11,15, ura3-1, leu2-1, can1-100, [psi-], SUP45::HIS3, [LEU2-1-SUP45 wild-type] |
| Yeast ΔLE2(Y410S) | SUQ5, ade2-1,his3-11,15, ura3-1, leu2-1, can1-100, [psi-], SUP45::HIS3, [LEU2-1-sup45-Y410S] |
Plasmid pUKC1901, a shuttle vector between E. coli and S. cerevisiae cells, carrying sup45-Y410S gene was used for construction of other plasmids and ∆LE2-Y410S strain. The other plasmid, pUKC815, 817, 818, and 819 were used for nonsense codon readthrough assay on S. cerevisiae. All plasmids named by pUKC (Table 2) were gift from M.F. Tuite, University of Kent, United Kingdom.
Plasmid used in this study.
| Plasmid | Description |
|---|---|
| pUKC630 | E. coli expression vector carrying SUP45 gene with a hexa-histidine sequence under the control of T7 RNA polymerase promoter. |
| pEPES-Y410S | Similar plasmid with pUKC630 that SUP45 was replaced by sup45-Y410S |
| pUKC1901 | S. cerevisiae LEU2 based plasmid carrying the sup45-Y410S |
| pSPES-SUP45 | Similar plasmid with pUKC1901 that sup45-Y410S gene was replace by SUP45 |
| pUKC606 | Multicopy yeast vector carrying SUP35 gene with original promoter |
| pUKC815 | S. cerevisiae URA3 based plasmid carrying the PGK-LACZ gene fusion. |
| pUKC817 | S. cerevisiae URA3 based plasmid carrying the PGK-TAA- LACZ gene fusion. |
| pUKC818 | S. cerevisiae URA3 based plasmid carrying the PGK-TAG- LACZ gene fusion. |
| pUKC819 | S. cerevisiae URA3 based plasmid carrying the PGK-TGA- LACZ gene fusion. |
Plasmid Constructions
Two plasmids, namely pSPES-SUP45, and pEPES-Y410S were constructed in this study. To generate plasmids pSPES-SUP45, a 1400 bp BglII-HindIII fragments from plasmid pUKC630 containing the entire SUP45 gene was cloned into BglII and BamHI sites of plasmid pUKC1901, a shuttle vector of E. coli and S. cerevisiae. While plasmid pEPES-Y410S was constructed using 1400 bp of HindIII-BglII fragment from plasmid pUKC1901 ligated into HindIII and BglII restriction sites of plasmid pUKC630. All of the plasmids were used to transform E. coli DH5α for propagating the plasmid.
Plasmid Shuffling
The plasmid shuffling was carried out based on the standard method [12]. The haploid ∆LE2(803) (SUP45::HIS3, URA3, SUP45) was used in plasmid shuffling. The strain was transformed with (LEU2 sup45) plasmids (pSPES-SUP45 and pUKC1901). The transformants were selected on medium without uracil and leucine ( –Ura –Leu) and then checked by replicating onto 5-FOA medium to counter selects against URA3 plasmids. Growth was also assayed using serial dilution overnight culture with OD600 = 1. Serially (10-fold) diluted yeast cell cultures were spotted on plates containing 5-FOA to determine the ability of sup45 mutant alleles to support cell growth. The wild type of yeast SUP45 gene carried on URA3 plasmid eliminates since 5-FOA is toxic to cell expressing the URA3 gene. The resulting strains, ∆LE(SUP45) and ∆LE(Y410S), were used for further characterization.
β-galactosidase assays
Centromeric plasmids pUKC815, 817, 818 and 819 [13] were transformed into suppressor strains (∆LE(SUP45), and ∆LE(Y410S)). The transformants were grown in plasmid-selective medium and 3 X 106 cells were inoculated into 5 mL YPD respectively. Triplicate samples from cultures grown to OD600 ~ 1.0 were treated essentially as described by Coligan et al. [14], with the following modifications. The cells were suspended in 500 µL Z buffer, 10 µL 0.1% SDS and 20 µL chloroform. The samples were then vortexed for 15 s and equilibrated for 15 min in a 30oC water bath. After adding 100 µL 4 mg/mL O-nitrophenyl- β-D-galactosidase (ONPG), samples were vortexed for 5 s and the reaction were carried out for 30 min at 30oC water bath before stopping the reaction by adding 500 µL 1M sodium carbonate. After centrifugation at 5000 g, the supernatants were measured at both 420 nm and 550 nm wavelengths and the Miller unit of each samples were calculated as (OD420) – (OD550 x 1.75). The readthrough for each of the three stop codons was calculated as the percentage of LacZ expression relative to the construct lacking a stop codon (pUKC815) in the same medium.
Expression of yeast eRF1-(His)6 and eRF1(Y410S)-(His)6 in E. coli
The recombinant plasmid, pUKC630 and pEPES-Y410S containing His-tag gene at the upstream region of SUP45 or sup45-Y410S coding sequences were introduced into E. coli BL2(DE3) as host strain for eRF1 expression system. For expression of the gene, the transformants were grown aerobically at 370C in LB containing ampicillin until the cells density reached 2x108 cell/mL or OD600 at around 0.6. The cultures were induced by addition of 0.1 mM IPTG and incubated at 250C for 4 h with aeration. The cultures were then centrifuged at 5000 g and the pellets were re-suspended in solubilization buffer (50mM Tris-HCl pH 7.4; 200 mM NaCl). Lysis cells were performed by sonication. The samples were centrifuged at 10000 g and analyzed by sodium dodecyl sulfate polyacrylamid gel electrophoresis (SDS-PAGE). The SDS-PAGE was performed with a 12.5% (w/v) acrylamide gel, and the proteins were stained with commassie blue G-250 [15].
Preparation of yeast post-mitochondrial supernatants (PMS)
Yeast cultures were grown to a cell density of 3.5x 107 cells/ml. Harvested cells were washed with lysis buffer (25 mM Tris-HCl, pH 7.2, 5 mM MgCl2, 5 mM 2-mercaptoethanol and 25 mM KCl) containing 1 mM phenylmethylsulphonyl fluoride (PMSF), 1 mM benzamidine, 10 µM leupeptin and 10 µM pepstatin A to limit proteolytic degradation. Following washing in lysis buffer, harvested cells were resuspended in a minimum volume of the same buffer. Cells were lysed by vortexing with glass beads and cell debris removed by centrifugation at 13 000 rpm for 15 min in a benchtop microcentrifuge, producing a postmitochondrial supernatant (PMS).
Immobilization of eRF1-(His)6
The following operations were performed at room temperature unless otherwise stated and columns were allowed to flow by gravity. A 500 µL column of Ni-NTA resin (Qiagen) was equilibrated with 30 mM bis-Tris-HCl, pH 6.8, 0.8 M KCl, 5 mM 2-mercaptoethanol, 30 mM imidazole (buffer A). E. coli crude extract containing eRF1-(His)6 and eRF1(Y410S)-(His)6 were passed three times through the resins separately, which were then washed with 10 column vol. buffer A followed by 10 vol. buffer containing 30 mM bis-Tris-HCl, pH 6.2, 1 M KCI, 5 mM 2-mercaptoethanol, 20% glycerol, 30 mM imidazole. This procedure resulted in partial purification and immobilization of eRF1-(His)6 and eRF1(Y410S)-(His)6 on the Ni-NTA resins. In order to assess the binding of eRF3 to the immobilized eRF1-(His)6 and eRF1(Y410S)-(His)6, the resins were equilibrated with 10 column vol. lysis buffer containing 25 mM KCI. A PMS prepared from cells overexpressing eRF3 (transformed with plasmid pUKC606) was then incubated with a suspension of the resin in 25 mM KCI lysis buffer for 2 h on a shaking platform at 4°C. After this time the resin was centrifuged briefly, the supernatant removed and retained and the resins returned to columns for washing with 10 column vol. 25 mM KCI lysis buffer containing 0.15% w/v Tween-20 detergent.
Computer modelling
The initial three dimensional structure of eRF1 was constructed by homology protein structure modeling from Swiss-Prot using predict protein program [16]. The comparative modeling program used three steps: alignment of amino acid sequence of yeast eRF1 with the template of human eRF1 [7], PDB number 1DT9.pdb., structure prediction based on primary sequences homology, and validation of the structure using WHAT_IF program in Swiss-Prot.
Amber version 9.0 program package [17] was used for the molecular simulation. The structure was subjected to energy minimization calculation by steepest descent method with 500 iterations followed by the conjugate gradient method with 4500 iteration to be used as starting lowest energy structure. Simulation image of the protein was generated using Visual Molecular Dynamic (VMD) software [18]. The Root Mean Square Deviation (RSMD) was calculated for the backbone atoms with reference to starting structure of time 0.
RESULTS
Construction characterization of yeast strains, ∆LE2(SUP45) and ∆LE2(Y410S)
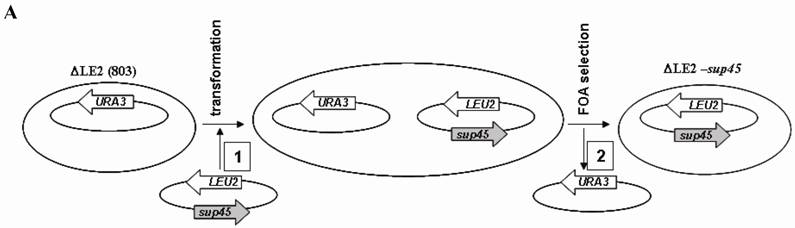
∆LE2(SUP45) and ∆LE2(Y410S) are haploid yeast strains that carrying chromosomal SUP45 disrupted by HIS3 gene (SUP45::HIS3), SUP45 and sup45 gene in plasmid pSEPES-SUP45 and pUKC1901 respectively. The strains were constructed from ∆LE2(803), haploid strain which carrying SUP45::HIS3 and SUP45 gene in CEN URA3 based plasmid. ∆LE2(803) strain was transformed with LEU2 based plasmid carrying SUP45 or sup45-Y410S mutant. The transformants were then subjected to plasmid shuffle analysis to verify weather strains containing sup45-Y410S could lose the viability (Fig 1A). Transformants carrying sup45-Y410S allele and SUP45 (as control) were able to grow in the presence of 5-FOA, indicating that all tested mutation can replace the wild type of SUP45 for the viability (data not show). However, plasmid shuffle was less efficient with sup45 mutants than with the wild type SUP45.
Shuffling strategy and the phenotype of the transformants. A. Strategy used for plasmid shuffling. (1), LEU2 based plasmid was introduced to ∆LE2(803). (2), double transformant was iduced on media containing 5-FOA. B. Phenotypes of transformants. ∆LE2(SUP45) and ∆LE2(Y410S) grown on rich medium (Y8), SM medium without leucine (-Leu), SM medium without uracil (-Ura).
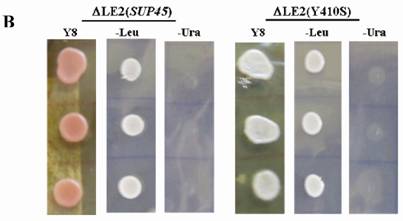
The viable transformants on media containing 5-FOA were characterized, including genotypes, temperature sensitivity and the allosuppressor phenotypes to UAA codon in ade2-1 gene. All of viable cells carrying SUP45 and sup45-Y410S could grow on rich medium (Y8) and synthetic medium without leucine (-Leu), however unable to grow on synthetic medium without uracil (-Ura) (Fig 1B). The viability of the transformants on media –Leu and unviable on media without uracil showed that URA3 based plasmid had been replaced by LEU2 based plasmid. The data confirmed that the plasmid shuffling had been successfully carried out. On rich medium (Y8), the color of transformants carrying wild type SUP45 gene appeared pink color, while transformants carrying mutation on sup45-Y410S showed white color (Fig 1B). This indicating, that sup45-Y410S exhibited allosuppressor phenotype. Furthermore, none of the transformants showed temperature sensitive mutants (data not shown).
Efficiency of ∆LE2(Y410S) on stop codon suppression
Stop codon readthrough on ∆LE2(Y410S) and ∆LE2(SUP45) were measured based on the ability of the cells to terminate the translational process on fusion gene between PGK-termination codon-LACZ carried by centromeric (single copy), URA3 based plasmid [26]. The strains were transformed by each of serial plasmids (pUKC815, 817, 818, and 819). Plasmid pUKC817, 818, and 819 carry UAA, UAG, and UGA termination codons respectively, while plasmid pUKC815 was used as control plasmid without codon termination.
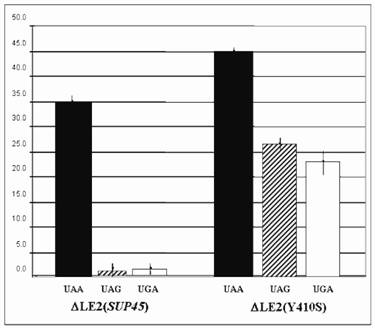
The suppression of ∆LE2(Y410S) on all stop codons were significantly increased compared to that the wild type (∆LE2(SUP45)) (Fig 2). The suppression of ∆LE2(Y410S) on UAG codon was the highest than that other codons, over 17 times fold than the ∆LE2(SUP45). While, the suppression for UGA codon was over 12 times higher than the wild type. Total suppression on UAA codon revealed the highest compared to that the other codons in both mutant and the wild type, this was not surprising since the strains used in this study carrying SUQ5, which is a weak tRNA suppressor for UAA codon, genetic background.
β- galactosidase assay for ∆LE2(SUP45) and ∆LE2(Y410S). Vertical axis for % of suppression compared to β- galactosidase activity transformant carrying plasmid pUKC815.
Binding Affinity of eRF1(Y410S) to eRF3
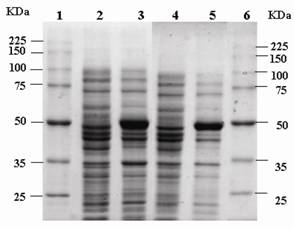
eRF1 has been demonstrated to interacts with eRF3 in vitro [19]. In order to investigate the influence of mutation at Y410S on the interaction to eRF3, the ability of eRF1(Y410S) to precipitate eRF3 was tested and compared to that the ability of eRF1 wild type. Over expression of eRF1-(His)6 and eRF1(Y410S)-(His)6 were carried out in E. coli BL2(DE3) using plasmid pUKC630 and pEPES-Y410S (Fig 3). Four E. coli crude extracts, two containing eRF1-(His)6 and other containing eRF1(Y410S)-(His)6, in high salt concentration (0.8 M KCl) lysis buffer, were repeatedly passed through Ni-NTA-agarose resins separately, ensuring preferential binding of the His-tag proteins to the resins and limiting non-specific binding of other proteins. The majority of non-specifically bound proteins were removed by washing the resins under stringent condition of low pH and high salt and glycerol. This resulted in preparation of partially purified eRF1-(His)6 and eRF1(Y410S)-(His)6 bound to nickel resins.
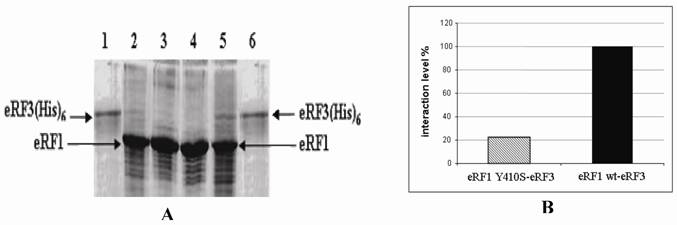
SDS-PAGE electrophoregram of crude extract of E. coli overexpressed eRF1-(His)6 and eRF1(Y410S)-(His)6.; It is in first without IPTG induction and then with IPTG induction for both couple of lanes 2-3 and 4-5; lanes 1 and 6 are protein molecular markers.
In order to probe the eRF1-eRF3 interaction, the eRF1-(His)6 and eRF1(Y410S)-(His)6 resins were tested for the ability to precipitate eRF3 from solution. eRF3 was overexpressed in yeast strain ∆LE2(803) transformed with pUKC606, from which a PMS was prepared. The eRF3-rich PMS was then incubated with either the eRF1-(His)6 resin or eRF1(Y410S)-(His)6 resin for 2 h in a low salt buffer to promote protein-protein interactions. Both eRF1-(His)6 and eRF1(Y410S)-(His)6 resins were then returned to columns to facilitate washing. The control resins were not mixed with eRF3-rich PMS.
Samples of eRF3-rich PMS were analyzed by SDS-PAGE. The results showed that there was no eRF3 detected on the control resins. While for the sample resins, there were some amounts of eRF3 protein. The eRF3 bound to eRF1(Y410S)-(His)6 was less quantity than that on eRF1-(His)6 (Fig 4A). Quantitative analysis of eRF3 bound to the resins were carried out by using densitometric analysis to measure the ratio of eRF3 : eRF1-(His)6 and eRF3 : eRF1(Y410S)-(His)6. The result showed that the binding affinity of eRF3 to eRF1(Y410S)-(His)6 was 20% of the eRF3 to the wild type (Fig 4B).
SDS-PAGE electrophoregram of eRF3 bound to eRF1(His)6 resin and densitometric quantitation. A. Electrophoregram of eRF3 bound to eRF1(His)6 resin. Lane 1 and 6, purified eRF3 carrying His tag overexpressed in yeast. Lane 2 and 3, eRF1-Y410S(His)6 resins with (sample) and without (control) eRF3 bound. Lane 4 and 5, eRF1(His)6 resins without (control) and with (sample) eRF3 bound. B. Comparison between the ratio of eRF3 : eRF1(His)6 and eRF3 : eRF1-Y410S(His)6. The ratio between eRF3 : eRF1(His)6 used as control (100%).
Structural modeling of eRF1(Y410S)
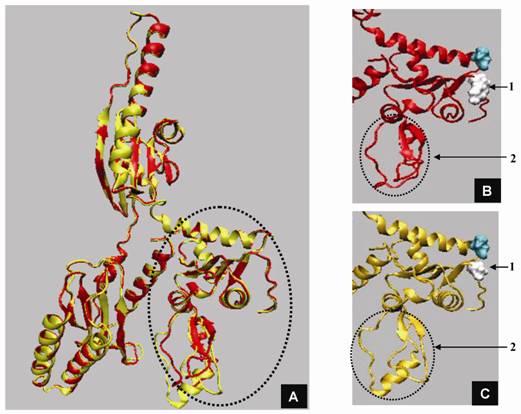
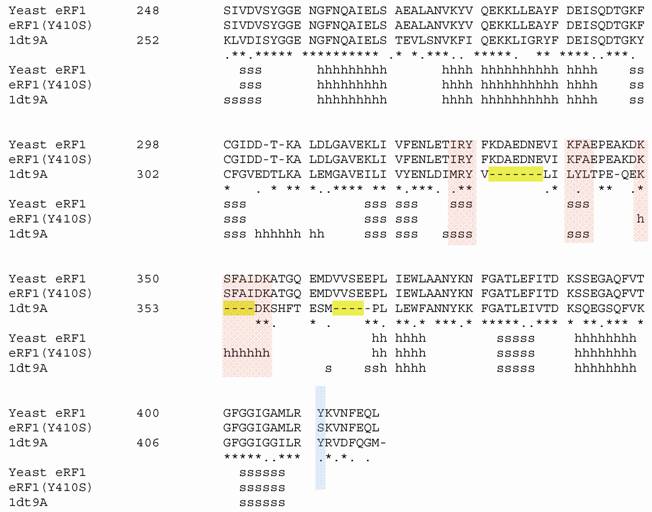
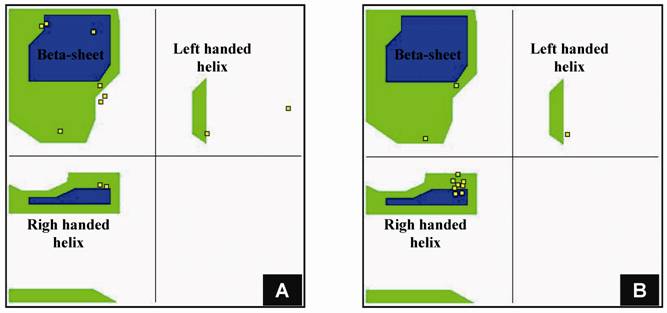
Structural modeling of eRF1 mutant and the wild type were performed using Amber 9 program followed by visualization using View Molecular Dynamic (VMD). Based on the Root Mean Square Deviation (RMSD) value, the overall structure of eRF1(Y410S) showed no significant different compared to the wild type (Fig 5A). However detail analysis on the structure of eRF1 third domain showed that eRF1(Y410S) contained secondary structure change (Fig 5 B, C). The structure of amino acid residues change on eRF1(Y410S) was not in the same motif with the position of mutation (Fig 5C, 1), but in the other motif (Fig 5C, 2). Comparison among the C-terminal domain of human eRF1 (1DT9), yeast eRF1 and eRF1(Y410S) showed that there was 7 amino acid residues from position at 343 - 349 (yeast position) changed from non-helix to helix forms (Fig 6). Ramanchandran Plot between yeast eRF1 and eRF1(Y410S) at the above position (343 - 349) revealed that the residues in eRF1(Y410S) tend to be more stable in helical form (Fig 7).
Three dimensional structure of eRF1 and eRF1-Y410S modelling. A. Superposition of eRF1 (red) and eRF1-Y410S (yellow) structures; domain 3 of eRF1 (circle). B and C. Close up structure of eRF1 and eRF1-Y410 domain 3; position of mutation (1), position of structural change before (B) and after (C) mutation (2, and 3).
Homological of C-terminal among human, yeast and Y410S of eRF1. 1dt9A, human eRF1; s for beta sheet; h for helix; red bar showed secondary structure change; blue bar showed position of mutation.
Ramachandran plot for 11 amino acid residues including 7 amino acid residues changed from non-helix to helix form. A for yeast eRF1; B for eRF1(Y410S).
DISCUSSION
Domain3 of eRF1 was proposed to be responsible for the interaction with eRF3 protein respectively [2, 20, 21]. Serial deletion on this region have been reported to have defect on its interaction [10], however there was no information concerning the effect of amino acid substitution in the region. We have constructed and characterized sup45 mutant which has mutation in codon no 410 for tyrosine to serine. Tyrosine at position 415 in human eRF1 is one of the amino acids that are critical for mutual binding lied on GILRY motif of human eRF1 [11].
SUP45 is essential gene in haploid S. cerevisiae cell [1]. Disruption of the gene causes lethality in haploid yeast cell. In order to assay mutation in the gene in vivo, the mutant gene was introduced to ∆LE2(803) which carrying disruption of chromosomal SUP45::HIS3 and SUP45 gene in single copy URA3 based plasmid. The transformants containing double plasmid (URA3 and LEU2 based plasmids) were subjected for plasmid shuffle [22] by growing the cell on medium containing 5-FOA. The wild type yeast SUP45 gene carried on the URA3 plasmid eliminated because 5-FOA is toxic to cell expressing the URA3 gene [23].
Mutation of sup45-Y410S showed an allosuppressor phenotype (Fig 1B). ∆LE2(803) carries ade2 and SUQ5 in addition of SUP45::HIS3 and SUP45 CEN URA3 based plasmid. SUQ5 is a weak tRNA suppressor [24]. This tRNA suppressor does not suppress nonsense codon on ade2 mutant on [psi-] genetic background [25]. However, mutation on sup45 gene could enhance the activity of SUQ5 [26] to suppress nonsense mutation on ade2 gene. Mutation on sup45-Y410S enhanced all of stop codon suppressions (Fig 2). However the quantitative values were variation depend on the type of stop codons. UAA codon was the most readthrough in all strains examined including the wild type. This was not surprising since all strains carrying SUQ5 genetic background. SUQ5 is codon specific tRNA suppressor for UAA codon [13]. While on UAG and UGA codons, the suppression of the wild type strain was very low, almost no suppression (Fig 2). However for the mutant strains, the suppression of UAG and UGA were significantly increased. The UAG codon was the mostly leaky codon in the mutant. This was unexpected since eRF1 reported to recognize all of stop codons in the same manner [27, 28, 6].
Termination translation in eukaryotic system that involved interaction between eRF1 and eRF3 is a complex phenomenon. Detail mechanism of the process is still unclear yet. eRF1 was reported to be phosphorylated by CK2 protein but the product did not significantly affect directly to translation termination [29]. Urakov et al. [30] demonstrated that IttIp could modulate the efficiency of termination translation in yeast. Meanwhile, recent report showed that eRF1 protein participates not only on termination translation but also in mRNA degradation and translation initiation via interaction with other proteins [22].
In order to characterize the causes of an elevation of suppression in sup45-Y410S, in vitro interaction of eRF1(Y410S) to eRF3 was examined. The result showed that there was significant reduction on the binding affinity of eRF1(Y410S) to eRF3 (Fig 4). As consequences the suppressor was preferable to recognize the nonsense codons and thus increase the read-through. Further analysis on three dimensional structures of eRF1(Y410S) showed there was alteration on secondary structure of some amino acids residues from non-helix to helix form (Fig 6). The structural modification in eRF1(Y410S) did not directly affect on the structure of AMLRY motif but triggered structural modification of neighboring motifs (Fig 5C). This modification might affect the interaction of eRF1(Y410S) to eRF3 and thus increasing the suppression of codon terminations.
The AMLRY motif lies at position 406- 410 residues of yeast eRF1 which has 437 amino acid residues. Amino acid substitution in the end of C- or N-terminal of protein usually does not affect the overall structure of the protein since these residues will be stabilized by water solvent. Substitution of tyrosine at position 410 to serine did not significantly affect the overall structure of eRF1 but triggered alteration on the secondary structure of amino acid residues lied at position 343-349. This was quite surprising since the distance of the residues is a far way. Tyrosine and serine both contained hydroxyl group, however, the proton on serine could not ionized while tyrosine does, in addition tyrosine contains bulky functional group and electron dense. These differences in electrostatic and electronic features might affect the probability of secondary structure of surrounding amino acid residues, however simulation result showed that substitution of tyrosine to phenylalanine did not affect the secondary structure of amino acid residues at the above position (unpublished data) so it is unlikely that the bulky functional group and the electron dense caused the structural change. The most possible explanation is that the differences on the properties of hydroxyl group was the key factor for the structural change on the region and thus substitution of tyrosine to serine reduced the energetic state and stabilized secondary structure to form helix (Fig 7).
All the data obtained suggests that tyrosine at AMLRY motif is important amino acid residue for the interaction to eRF3, however the hydroxyl group of tyrosine seems does not directly involve on the binding to eRF3. The intact of three dimensional structure of domain 3 of eRF1 is suggested as a key factor for the interaction to eRF3.
Acknowledgements
We would like to thank Prof M. F. Tuite, University of Kent, for plasmid and yeast cell gifts. This research was supported by Fundamental Research Grant from Directorate for Higher Education, Department of National Education, Indonesia to AKH, and BPPS scholarship to PES.
CONFLICT OF INTEREST
The authors have declared that no conflict of interest exists.
References
1. Inge-Vechtomov SG, Zhouravleva G, Philippe M. Eukaryotic release factor (eRFs) history (review). Biology of the Cell. 2003;95:195-209
2. Ito K, Uno M, Nakamura Y. Single amino acid substitution in prokaryotic polypeptide release factor 2 permits it to terminate translation at all three termination codons. Proc Natl Acad Sci USA. 1998;95:8165-8169
3. Kisselev L, Ehrebreg M, Frolova L. Termination of translation: interplay of mRNA, rRNAs and release factor? Embo J. 2003;22:175-182
4. Salas-Marco J, Bedwell DM. GTP hydrolysis by eRF3 facilitates stop codon decoding during eukaryotic translation termination. MCB. 2004;24:7769-7778
5. Bertram G, Bell HA, Ritchie D. et al. Termination eukaryote translation: domain 1 of release factor eRF1 function in stop codon recognition. RNA. 2000;6:1236-1247
6. Frolova LY, Le GX, Zhouravleva G. et al. Eukaryotic polypeptide chain release factor eRF3 is an eRF1 and ribosom-dependent guanosine triphosphatase. RNA. 1996;2:234-341
7. Song H, Mugnier P, Webb HM. et al. The crystal structure of human eukaryotic release factor eRF1-mechanism of stop codon recognition and peptidyl-tRNA hydrolysis. Cell. 2000;100:311-321
8. Chavatte L, Frolova LY, Kisselev L. et al. The polypeptide chain release factor eRF1 specifically contacts the UGA stop codon located in the A site of eukaryotic ribosomes. Eur. J. Biochem. 2001;268:2896-2904
9. Frolova LY, Tsivkovskii RY, Sivolobova GF. et al. Mutations in the highly conserved GGQ motif of class 1 polypeptide release factor abolish ability of human eRF1 to trigger peptidyl-tRNA hydrolysis. RNA. 1999;5:1014-1020
10. Eurwilaichitr L, Graves FM, Stansfield I. et al. The C terminus of eRF1 defines a functionally important domain for translation termination in Saccharomyces cerevisiae. Mol. Microbial. 1999;32:485-496
11. Merkulova TI, Frolova LY, Lazar M. et al. C-terminal domains of human translation termination factors eRF1 and eRF3 mediated their in vivo interaction. FEBS Letter. 1999;443:41-47
12. Kaiser C, Michaelis S, Mitchell A. Methods in yeast genetics; Cold Spring Harbor Laboratory Course Manual. New York, USA: Cold Spring Harbor laboratory Press. 1994
13. Stansfield I, Akhmaloka, Tuite M.F. A mutant allele of the sup45 (sal4) gene of Saccharomyces cerevisiae shows temperature-dependent allosuppressor and omnipotent suppressor phenotypes. Curr Gennet. 1995;27:417-426
14. Coligan J, Dunn BM, Ploegh HL. et al. Current Protocols in Protein Science. New York, USA: Wiley. 1995
15. Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual. Cold Spring Harbor, USA: Cold Spring Harbor Press. 1989
16. Rost B, Yachdav G, Liu J. The Predict Protein server. Nucleic Acids Research. 2004;32:W321-W326
17. Case DA, Darden TA, Cheatham TE. et al. AMBER 9. San Fancisco, USA: University of California. 2006
18. Humphrey W, Dalke A, Schulten K. VMD-visual molecular dynamic. J. Molec. Graphics. 1996;14:33-38
19. Stansfield I, Jones K, Kusnirov VV. et al. The product of the SUP45 (eRF1) and SUP35 genes interact to mediate translation termination in Saccharomyces cerevisiae. EMBO J. 1995;14:4365-4373
20. Ebihara K, Nakamura Y. C-terminal interaction of translational release factor eRF1 and eRF3 of fission yeast: G-domain uncoupled binding and the role of conserved amino acid. RNA. 1999;5:739-750
21. Frolova LY, Merkulova TI, Kisselev LL. Translation termination in eukaryotes: polypeptide release factor eRF1 is composed of functionally and structurally distinct domains. RNA. 2000;6:381-390
22. Chabelskaya S, Gryzina V, Moskalenko S. et al. Inactivation of NMD increases viability of sup45 nonsense mutants in Saccharomyces cerevisiae. BMC Molecular Biology. 2007;8:71
23. Boeke JD, LaCroute F, Fink GR. A positive selection for mutants lacking orotidine-5'-phospate decarboxylase activity in yeast: 5-flouro-orotic acid resistence. Mol Genet. 1984;197:345-346
24. Cox B.S. Allosuppressors in yeast. Ganet Res. 1977;30:187-205
25. Inge-Vechtomov SG, Tikhodeev ON, Karpova TS. Selective system for obtaining recessive ribosomal suppressors in yeast Saccharomyces cerevisiae. Genetika. 1988;24:1159-1165
26. Stansfield I, Eurwilaichitr L, Akhmaloka. et al. Depletion in the levels of the release factor eRF1 cause a reduction in the efficiency of translation termination in yeast. Mol. Microbiol. 1996;20:1135-1143
27. Frolova LY, Le GX, Rasmussen HH, Cheperegin S. et al. A highly conserved eukaryotic protein family possessing properties of polypeptide chain release factor. Nature. 1994;372:701-703
28. Zhouravleva G, Frolova LY, Le GX. et al. Termination of translation in eukaryotes in governed by two interacting polypeptide chain release factor, eRF1 and eRF3. EMBO J. 1995;14:4065-4373
29. Kallmeyer AK, Keeling KM, Bedwell DM. Eukayotic release Factor 1 phosphorylation by CK2 protein kinase is dynamic but has little effect on the efficiency of translation termination in Saccharomyces cerevisiae. Eukaryotic Cell. 2006;5:1378-1387
30. Urakov VN, Valouev IA, Lewitin EI. et al. IttIp, a novel protein inhibiting translation termination in Saccharomyces cerevisiae. BMC Molecular Biology. 2001;2:9
Author contact
![]() Correspondence to: Akhmaloka PhD, Biochemistry Research Division, Faculty of Mathematics and Natural Sciences, Institut Teknologi Bandung, Telp 62-22-2515032, fax 62-22-2502360, E-mail: lokaitb.ac.id
Correspondence to: Akhmaloka PhD, Biochemistry Research Division, Faculty of Mathematics and Natural Sciences, Institut Teknologi Bandung, Telp 62-22-2515032, fax 62-22-2502360, E-mail: lokaitb.ac.id