Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2008; 4(2):103-110. doi:10.7150/ijbs.4.103 This issue Cite
Research Paper
Screening of Human Antibody Fab Fragment against HBsAg and the Construction of its dsFv Form
Leili Jia1,*, Jiyun Yu2,*, Hongbin Song1, Xuelin Liu1, Weina Ma1, Yuanyong Xu1, Chuanfu Zhang1, Shicun Dong1, Qiao Li2,3 ![]()
1. Center for Disease Control and Prevention, Academy of Military Medical Sciences, Beijing, China.
2. Institute of Basic Medical Sciences, Beijing, China
3. Department of Surgery, University of Michigan, Ann Arbor, Michigan, USA
* These two authors contributed equally to the work.
Received 2008-3-13; Accepted 2008-4-22; Published 2008-4-24
Abstract
The objective of this study was to pursue the techniques involving the screening of the human antibody Fab fragment against hepatitis B virus surface antigen (HBsAg) and the construction of its disulfide-stabilized Fv fragment (dsFv). The phage antibody Fab fragments against HBsAg were screened from the human combinatorial immunoglobulin library. Sequence analysis revealed that its heavy chain gene was complete, but the light chain gene was lost. To improve the affinity of the antibody by chain shuffling, a human antibody light chain gene repertoire was generated by reverse transcriptase-polymerase chain reaction (RT-PCR) from the human peripheral blood lymphocytes. A phage antibody sub-library was then constructed by inserting the light chain gene repertoire into the phagmid that contained the Fd gene. Five clones with appreciably higher absorbance than that of the original clones were obtained, which indicated that the affinity of the light chain-shuffled phage antibodies was improved. Then, the mutated genes of dsFv against HBsAg were constructed by using PCR-based point mutagenesis method. Purified VH and VL proteins were folded into a 25-kDa protein, designated as anti-HBsAg dsFv. ELISA and competition ELISA revealed that the dsFv maintained the specificity of the Fab by binding to HBsAg, even through with a lower binding activity. These results have facilitated the undertaking of further functional analyses of the constructed dsFv, and may therefore provide an improved technique for the production and application of dsFvs against HBsAg.
Keywords: HBsAg, Fab, dsFv, phage antibody, chain shuffling, point mutagenesis
Introduction
The field of recombinant antibodies is a fast developing area and has grabbed the attention of a large number of researchers [1]. In general, the goal is to produce human monoclonal antibodies for disease diagnosis and therapy, a goal that was often unattainable before. The technology of immunoglobulin combinatorial libraries for the generation of monoclonal antibodies has undergone considerable changes since it was first reported in 1989. As the phage display technology progresses, the phage antibody library has been widely used to isolate specific antibody fragments from large antibody gene pools. Compared with conventional hybridoma technologies, the human phage antibody technology has more advantages: 1. The process is efficient and a relatively large number of antibodies can be produced; 2. It has been suggested that it can mimic the key features of the humoral immune system in vitro by expressing the antibody fragment gene repertoires on the surfaces of the bacteriophage (phage display). As a result, human antibodies with high affinities can be produced without prior immunization or other conventional monoclonal antibody generation technology; 3. Human antibodies are useful in therapy in human. However, it is extremely difficult to make human monoclonal antibodies using conventional hybridoma technologies. The use of bacteriophage display libraries of Fab or scFv antibodies on their surfaces has been proven to be efficient for the isolation of a diverse set of human monoclonal antibodies from immune or nonimmune volunteers against a variety of infectious diseases [2],[3]. In comparison to a full-length antibody, Fab fragment can be easily expressed in bacterial expression systems [4]. Although native unstabilized Fv heterodimers have been made from antibodies [5], Fvs by themselves are generally unstable because the VH and VL domains of the heterodimer can rapidly dissociate [6]. This results in drastically reduced binding affinity. Another disadvantage of Fab fragments is the tendency of light chains to form homodimers, which are known as Bence Jones proteins [7]. Meanwhile, single-chain Fv fragments (scFvs) have a tendency to form aggregates and are relatively unstable over time [8]. Furthermore, some scFvs show reduced affinity of up to one order of magnitude as compared to the corresponding Fab fragments [9]. One approach to generate stable recombinant Fvs is to connect the VH and VL domains by an interdomain disulfide bond instead of a linker peptide. Disulfide-stabilized Fvs (dsFvs) have resolved most of the problems that are associated with scFvs. DsFvs are stable, often show full antigen binding activity, and sometimes even demonstrate better affinity than scFvs [10].
During the last two decades, liver transplantation for liver diseases related to hepatitis B virus (HBV) infection has been successful [11],[12]. Administration of high doses of HBIG and lamivudine for prophylaxis during liver transplantation has reduced the risk of the recurrence of HBV and therefore improved the survival of the patients undergoing transplants [13]. However, the cost of long-term prophylaxis with high doses of HBIG is extremely high, and lamivudine may lead to the selection of complex mutants [14]. The use of HBsAg is considered to be the necessary immunoprophylaxis in complex situations such as immunosuppressive therapy [15],[16].
In this study, a human immunoglobulin combinatorial library was generated by using a phage surface display expression system. Phage antibodies (Fab fragments) were screened against HBsAg. To improve the affinity of the antibody by chain shuffling, a human antibody light-chain gene repertoire was generated by reverse transcriptase-polymerase chain reaction (RT-PCR) from human peripheral blood lymphocytes. Then, a phage antibody sub-library was constructed by inserting the light-chain gene repertoire into the phagmid that contained the Fd gene. After high-affinity Fab fragment against HBsAg was produced, we constructed dsFvs against HBsAg by using the PCR-based point mutagenesis method. Fab against HBsAg and its dsFv form were expressed in E. coli. Such expressions exhibited strong bindings to HBsAg. These studies have laid the foundations for the production and application of Fab and dsFv against HBsAg.
Materials and Methods
Materials
A Fab phage display combinatorial library (1014 cfu/ml) was previously constructed in our laboratory. HBsAg was obtained from the Institute of Bioengineering, Academy of Military Medical Sciences (Beijing, China). E. coli XL1-Blue and helper phage VCSM13 (1012 cfu/ml) were purchased from Promega (Madison, WI, USA). The prokaryotic expression vector pET-20b (+) and competent E. coli BL21(DE3) were purchased from Novagen Inc. (Madison, WI, USA).
Panning of phage library
Three rounds of biopanning were done as previously described [17],[18]. After the last round of panning, the eluted phages were amplified by infection of E. coli XL1-Blue which were then cultured in a super-broth (SB) medium plate at 37°C overnight. Randomly selected clones were added in 2-ml SB medium and maintained at 37°C for 4 h. Fifty microliters of the helper phage VCS M13 (1012 cfu/ml) was then added separately. The cultures were incubated overnight at 37°C, followed by centrifugation at 10,800g, 4°C, for 10 min. Twenty five microliters of supernatant with an equal volume of PBS/T was transferred to the ELISA wells coated with the HBsAg antigen (10 μg/ml). The ELISA plate was then incubated at 37°C for 2 h and washed 3 times with PBS/T. After the addition of enzyme conjugated with the antibody (rabbit anti-VCSM13-alkaline phosphatase), the ELISA plate was incubated for another 1 h at 37°C and washed 3 more times with PBS/T. Fifty microliters of the substrate solution was then added to each well. The ELISA plate was maintained at room temperature and the OD value was read after 15 minutes. The absorbance was measured as the optical density at 490 nm [19].
For inhibition ELISA, positive colony phage-containing supernatant (25 μl) mixed with 25 μl of PBS/T containing HBsAg (10 μg/ml) was added to the ELISA plate wells coated with HBsAg. The control was the same phage-containing supernatant (25 μl) mixed with 25 μl of PBS/T without HBsAg. ELISA was performed as described above. The OD was read at 490 nm and its inhibition rate was calculated as follows: Inhibition rate (%) = 100 – (OD with HBsAg)/(OD without HBsAg).
Extraction and purification of plasmid cloned with phage antibody genes
XL1-Blue cells infected with positive phages were streaked onto a Luria-Bertarni (LB) plate and incubated overnight at 37°C. On the next day, single colony was selected to inoculate into 2 ml of the LB medium containing 50 mg/l of carbenicillin (Car) and 15 mg/l of tetracycline. The culture was stirred overnight at 37°C. The cells were then used for the extraction and purification of plasmid by Miniprep Kit (50) of QIAGEN (Hamburg, Germany).
By means of agarose gel electrophoresis, we determined whether the plasmid contained heavy-chain or light-chain genes of the antibody of interest after digestion with Spe I/Xho I and Sac I/Xba I respectively.
Heavy-chain gene sequencing
After digestion with Spe I/Xho I, the plasmid pBluescript KS was purified and recovered as described previously [20]. The heavy chain was then ligated into the plasmid pBluescript KS with T4 DNA ligase. XL1-Blue cells transformed with the ligation product were streaked on the LB plate containing X-gal and IPTG, and were incubated overnight at 37°C. The white colony identified to be positive by PCR was inoculated into the LB medium and then incubated overnight at 37°C. To obtain the heavy-chain gene sequences, primers from the 5' vector sequence (5'-CTCGAGCAGTCTGGGGCTG-3') and the CH1 constant region of the antibody (5'-CTCGGTGTTGCTGGGCTT-3') were used. The heavy-chain gene was sequenced by dideoxy-mediated chain-termination method with the 373A automatic DNA sequence analysis machine (Applied Biosystems, Weiterstadt, Germany).
Construction and biopanning of phage antibody sub-library
A volunteer was immunized with the hepatitis B vaccine and 1 ml of blood was obtained. The white blood cells were pelleted, and the lymphocytes were separated using Histopaque gradients (Sigma, St. Louis, M S, USA). The mRNA was extracted from the lymphocytes with the mRNA extraction kit (Invitrogen, Carlsbad, CA, USA). The first-strand cDNA was then synthesized with a primer of oligo (dT)15 using a reverse transcriptase first-strand cDNA synthesis kit (GIBCO-BRL, Rockville, MD, USA). All the light-chain genes (κ and λ) were amplified from the cDNA product by PCR using the panel of 5' forward primers and 3' reverse primers described previously [21]. The variable regions of the κ-light chain (Vκ) were amplified using VK1 and VK2, and the variable regions of the λ-light chain (Vλ) were amplified using VL1 and VL2. The constant regions of the κ-light chain (Cκ) were amplified using VK2 and CLK, and the constant regions of the λ-light chain (Cλ) were amplified using VL2 and CLL. The conditions for PCR were as follows: 35 cycles at 90°C for 40 s, 52°C for 40 s, and 72°C for 60 s, followed by a final extension for 10 min at 72°C.
The recombination of heavy and light chains was performed in a manner similar to that used to construct the library. The phagemid pComb3 that contains the Fd gene was obtained as mentioned above. The shuffling of the immunoglobulin chains was performed as described previously [22].
Expression and renaturation of Fab fragment against HBsAg
Based on the gene sequences of Fab as constructed above, the PCR primers were designed by using the computer software Primer Premier 5.0, and the relative restriction endonuclease sites were introduced into the primers for the construction of prokaryotic nonfusion expression vectors of pETFd and pETL. All of the PCR primers were synthesized by Shengong Inc. (Shanghai, China) with the following sequences: Fd back: 5'-gcggaattcatatggttaaactgctggaacagtctggggctga-3'; Fd forward: 5'-cggaattcattaagaggtcttgtcacaggactttggttcaactttc-3'; L back: 5'-gcggaattcatatggaactggttatgactcagtctccagacacc-3'; and L forward: 5'-cggaattcattaacattcacctctgttgaagctcttc-3'. The underlined sequences represent the restriction sites for Nde I and EcoR I. The 5' primer was used to provide the cloning site Nde I, and the 3' primer was used to provide the cloning site EcoR I. PCR was performed as described above, and its product was cloned into pGEM-T (Promega, Madison, WI, USA). The PCR-amplified Fab genes were cleaved with Nhe I and EcoR I and the purified fragments were then ligated at a ratio of 8:1 to cleaved and purified pET-20b(+). The ligated phasmids pETFd and pETL were transformed into E. coli BL21(DE3). Single colonies were cultured in 2 ml of LB medium with 50 μg/ml of Car and stirred overnight at 37°C. For Fab expression, 50 ml of LB with 50 μg/ml of Car was inoculated with 500 μl of the overnight culture of the recombinant clone and grown at 37°C till the OD600 value was approximately 0.6. By then, the expression of Fab was induced by 0.4 mmol/l of IPTG. After 3 h of growth at 37°C, the cells were harvested by centrifugation at 6,000 rpm for 10 min. Ten (10) μl of supernatant was mixed with 10 μl of 2 × SDS-sample buffer (5% SDS, 10% glycerol, 0.002% bromophenol blue, 0.0062 M Tris-HCl, pH 7.0), supplemented with 2% mercaptoethanol. The extracted Fab fragment protein was renaturated as described [23].
Construction of mutational dsFvs against HBsAg
The selection of the location of VH and VL (H44/L100) has been described [24]. Primers VH5, VHM, VH3, VL5 and VL3 used for all the PCR analyses are described previously [20].
The construction of the dsFv VH mutant gene by using the megaprimer PCR method has been described [25]. Briefly, VH5 and VHM were used to amplify the primer VH5-M and then VH5-M and VH3 were used to amplify the dsFv VH mutant gene. The dsFv VL fragment was amplified by using conventional PCR using the primers VL5 and VL3. The conditions for both PCRs were 30 cycles at 94°C for 1 min, 55°C for 1 min, 72°C for 1 min, followed by a final extension for 10 min at 72°C. The PCR products of the mutated VH and VL genes were cloned into plasmid pGEM-T and sequenced using the dideoxynucleotide method.
Expression and renaturation of dsFvs against HBsAg
The plasmids pGEM-VH and pGEM-VL constructed as described above were digested by Nhe I and EcoR I, and the fragments were inserted into pET-20b(+). Then, the positive clones were transformed into E. coli BL21(DE3) and the expressions of the VH and VL genes were analyzed by the SDS-PAGE analysis as described above. The dsFv-VH and dsFv-VL proteins, which were extracted from the inclusion body, were renaturated and identified as described above.
Comparison of binding activities between Fab fragments and dsFvs
The primers VH1a, PVH3, Vκ1a and PVL3 used for the insertion of dsFv genes into pComb3 were described previously [20]. VH was amplified using VH1a and PVH3. VL was amplified using Vκ1a and PVL3 and the VL genes were ligated into the phagemid pComb3. Then, the recombinant plasmids pComb3, which include the heavy and light chains of Fab and dsFv, were transformed into XL1-Blue. The preparation of the phage antibodies of Fab and dsFv and the ELISA and inhibition ELISA were then performed as described above.
Results
Isolation of Fab fragment with binding activity toward HBsAg
After three rounds of panning, 30 colonies were randomly selected for the collection of Fab. The supernatant that contained the Fab was directly subjected to the binding activity analysis by means of ELISA. The positive ODs (2.1 times or higher than that of the negative control) was 69%. Among the 30 clones analyzed, 8 clones with OD value 10 times higher than that of the negative control demonstrated specific binding activities toward HBsAg. The colony Fab-1 with the highest OD (OD490 = 0.72 ± 0.02) was confirmed by inhibition ELISA. The inhibition rate was 86% (OD490 = 0.10 ± 0.01).
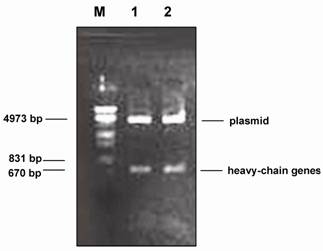
A fragment of approximately 670 bp was obtained when the plasmid DNA containing the gene of Fab-1 was digested with the restriction enzyme Spe I and Xho I (Fig.1). This result suggested that the plasmid DNA contained a heavy-chain gene of the human phage antibody. This is based on the fact that the construction of the human phage antibody libraries is on the pComb3 vector in which the heavy-chain gene of the human antibody is cloned between the restriction enzyme sites of Spe I and Xho I. However, the light-chain gene fragment was not detected when the positive plasmid DNA was digested with the restriction enzymes Sac I and Xba I (data not shown). The heavy-chain genes were then sequenced and the results of the DNA sequences revealed that the heavy chain contained the entire variable (VHI) and constant (CHI) regions. When compared with the sequences of Genbank [26], the variable heavy-chain (VH) gene of Fab-1 was more homologous with that of the VHI subgroup (>75%) than with that of the other group (<60%). We therefore conclude that the heavy-chain gene of Fab-1 belongs to the subfamily of the VHI gene.
Restriction enzyme (Spe I and Xho I) digestion of plasmid DNA containing the gene of the phage antibody against HBsAg. M: DNA molecular weight marker (λ DNA/EcoR I + Hind III). 1 and 2: plasmid DNA containing Fd gene (Fab-1).
Construction and panning of the light-chain library
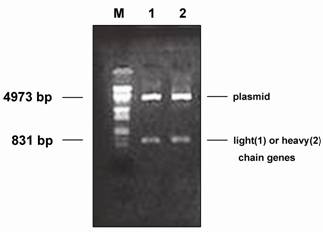
By means of chain shuffling, a light-chain sub-library was constructed. Briefly, the PCR-amplified κ- and λ-genes were cleaved with Sac I and Xba I and the purified fragments were ligated to cleaved and purified pComb3. The ligated material was then transformed into electrocompetent E. coli XL1-Blue. Bacteriophage stock solutions were prepared following super infection with VCSM13 helper phage by culturing overnight at 30°C. Clones producing HBsAg-specific Fab were selected and identified, and sequence analysis of the Fab segments was performed as described in the Materials and Methods. After 3 rounds of selection, 5 clones with absorbance higher than that of phage Fab-1 at 490 nm in ELISA were obtained. All the clones contained heavy and light chains after enzyme digestion (Fig.2). DNA sequencing showed that 3 out of the 5 VL genes were of the κ-type, and the remaining 2 were of the λ-type.
Restriction enzyme digestion of pComb3 containing light- and heavy-chain genes. M: DNA molecular weight marker (λ DNA/EcoR I + Hind III). 1: light-chain gene digested from pComb3 with Sac I and Xba I. 2: heavy-chain gene digested from pComb3 with Spe I and Xho I.
Expression and renaturation of Fab fragment protein
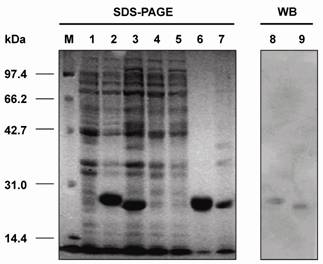
Protein separation was run under reduced conditions on 12.5% polyacrylamide gels with a 4% stacking gel. The cell pellet was re-suspended in 5 ml of 50 mM Tris-HCl (pH 8.0)/2 mM EDTA and maintained at 30°C for 15 min after addition of 100 μg/ml antalzyme/0.5 ml Triton X-100. Clarified periplasmic extract was obtained by centrifugation of the re-suspended product at 12,000 rpm for 10 min. Then, the periplasmic extract was mixed with 50 μl of 2 × SDS-sample buffer and the pellet was re-suspended in 100 μl of 1 × SDS-sample buffer for SDS-PAGE analysis. SDS-PAGE analysis revealed that the Fd fragment and L chain were expressed in the inclusion body form with molecular weights of 27 kDa and 25 kDa, respectively (Fig.3). Further more, Western blot revealed that both proteins could react with the anti-human Fab antibody (Fig.3). This indicates that the Fd fragment and L chain belonged to human antibodies.
Detection of the Fd fragment and L chain of the antibody against HBsAg expressed in E. coli BL21(DE3) by SDS-PAGE and Western blot analyses. M: protein molecular weight marker; 1: BL21(DE3) cell lysate; 2: cell lysate of pETFd -transformed BL21(DE3); 3: cell lysate of pETL -transformed BL21(DE3); 4: supernatant from cell lysate of pETFd-transformed BL21(DE3); 5: supernatant from cell lysate of pETL-transformed BL21(DE3); 6: inclusion body extracted from pETFd -transformed BL21(DE3); 7: inclusion body extracted from pETL-transformed BL21(DE3); 8 and 9: Fd and L chain inclusion body detected by anti-human Fab.
A total of 50-ml culture of E. coli containing pETFd or pETL was induced for the protein expression under the same condition as mentioned above. The inclusion body was isolated and solubilized by 8 mol/l urea. Fd and L dissolved in urea were mixed in an equal molar concentration. The renaturation buffer (0.1 mol/l Tris-HCl, 0.5 mol/l L-argine, 8 mmol/l GSSG, and 2 mmol/l EDTA) was then added and incubated at 10°C for 24–36 h. After that, Fd and L were refolded to yield Fab. Then, 25 μl of the folding solution was mixed with 25 μl of 2 × SDS-sample buffer and the mixture was detected by the SDS-PAGE analysis. In the renaturation buffer, a new protein Fab was formed at approximately 52-kDa after the Fd fragment and L chain were refolded. This result was revealed by the SDS-PAGE analysis as shown in Fig.4.
Renaturation of Fab fragment proteins. 1: inclusion body extracted from cells transformed with pETFd; 2: inclusion body extracted from cells transformed with pETL; 3: protein molecular weight markers; 4: proteins of L chain and Fd fragment before renaturation; 5: renatured products of L chain and Fd fragment proteins; 6: Fab incubated with β-mercaptoethanol.
Construction of dsFv
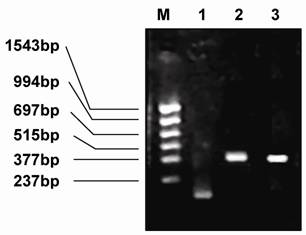
The megaprimer fragment (161 bp), which was located at the mutant point of position 44 of VH, was amplified using the primers VH5 and VHM. The VL mutant gene (386 bp), which was found at position 100 of VL, was amplified using the primers VH5 and VHM (Fig.5). The two sites—VH44 (mutation from Gly to Cys) and VL100 (mutation from Gln to Cys)—were identified by sequencing, and both were successfully mutated.
Electrophoresis of the PCR products of the megaprimer, VH, and VL. M: PCR markers, 1: megaprimer, 2: PCR product of VH, 3: PCR product of VL.
Expression and renaturation of dsFv
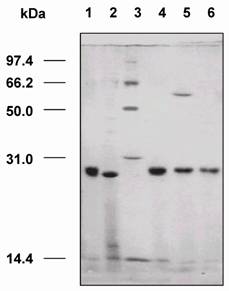
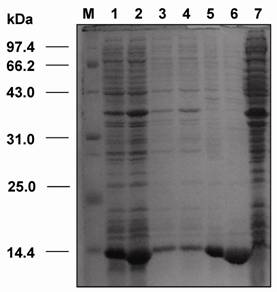
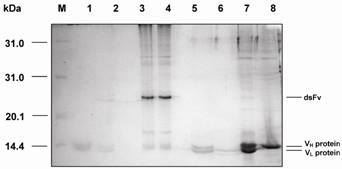
SDS-PAGE analysis revealed that the dsFv was expressed as an inclusion body protein (25 kDa) (Fig.6). The protein could be reduced to VH and VL after treatment with β-mercaptoethanol or boiling for 5 min (Fig.7).
SDS-PAGE analysis of the expressed VH and VL genes of dsFv against HBsAg in E. coli. M: protein molecular weight markers; 1: total fractions of cells expressing VH gene; 2: total fractions of cells expressing VL gene; 3: soluble fractions of cells expressing VH gene; 4: soluble fractions of cells expressing VL gene; 5: insoluble fractions of cells expressing VH gene; 6: insoluble fractions of cells expressing VL gene; 7: total cell proteins.
SDS-PAGE analysis of VH and VL proteins folding into dsFv. M: protein molecular weight markers; 1: dsFv boiled for 5 min; 2: dsFv boiled for 10 min; 3: dsFv treated at 37°C for 15 min; 4: VH and VL proteins folding into dsFv; 5: VH and VL proteins diluted into refolding buffer; 6: VL protein; 7: dsFv incubated with β-mercaptoethanol; 8: VH protein.
Comparison of antigen binding activities between Fab fragment and dsFv by ELISA and inhibition ELISA
ELISA and competition/inhibition ELISA were performed as described in Materials and Methods. Results revealed that while dsFv showed lower binding activity to HBsAg (0.56±0.08) than that of Fab (1.22±0.13), it maintained the specificity of the Fab to bind to HBsAg (Table 1).
ELISA and inhibition ELISA
| samples | ELISA(X±SD) | inhibition ELISA(X±SD) | inhibition(%) |
|---|---|---|---|
| Fab | 1.22±0.13 | 0.42±0.14 | 65.6 |
| dsFv | 0.56±0.08 | 0.16±0.11 | 71.4 |
| positive control | 1.65±0.09 | - | - |
| negative control | 0.08±0.16 | - | - |
Discussion
Human-engineered antibodies are the third generation of antibodies which have potential therapeutic applications in human diseases. Some of the engineered antibodies have been approved by FDA in the USA for clinical uses. The phage antibody library technology makes possible not only the production of large numbers of human antibodies that are difficult to obtain using the hybridoma method but also the screening of many specific antibodies against various antigens. In this regard, both pIII and pVIII phage display formats have been used for the construction of various libraries. There was also report showing that pVII and pIX could be used to individually display fusion proteins, notably the VH and VL of the antibody Fv fragment [27]. Large and nonimmune or ‘‘naïve’’ antibody-phage libraries displayed on pIII have proven to be a method to readily isolate high-affinity and specific human antibodies against a variety of target antigens. Although Human antibody Fab fragments that bind to HBsAg were generated by using a recombinant phage surface-display expression system [28], a significant disadvantage of Fab fragments is the formation of light chain homodimers [7]. In this study, we successfully constructed pIII phage antibody libraries using semi-nested PCR, remarkably increased the size of the libraries, and screened the phage antibody Fab fragments that can bind HBsAg specifically from the antibody libraries. Importantly, the more stable dsFv fragment against HBsAg was constructed from its Fab form by “chain shuffling” technique. This work thus represents significant improvements over the techniques described previously by the other laboratories [29]. The results of restriction enzyme digestion revealed that this specific antibody contained a heavy chain gene. Sequence analysis showed that the heavy chain comprised the entire variable (VH1) and constant (CH1) regions. Although the restriction enzyme digestion demonstrated that its light chain gene was lost, the phage antibody with the entire heavy chain gene exhibited definite binding activity to HBsAg. Generally, it is considered that the variable heavy chain region of the antibody plays a leading role, while the variable light chain region plays a subordinate role in the binding process of antibody to antigen. It is possible that the antibody, which undergoes significant conformational changes, ameliorates its binding to the antigen (“induced fit” antibody). Therefore, we rescreened the light chain gene while the heavy chain gene was fixed to imitate the antibody maturing procedure in vitro in order to increase the antigen binding affinity by the “chain shuffling” method. Fortunately, we screened the light chain gene by using this method and therefore obtained the genes of the entire phage antibody (Fab fragment) against HBsAg. This antibody significantly increased the binding activity to HBsAg. The VL and VH proteins were expressed and refolded into active molecules in vitro. Furthermore, we successfully applied this technology to the expression of dsFvs against HBsAg.
DsFvs (disulfide-stabilized Fvs) are significantly more stable in human serum and under various conditions. Therefore, we constructed the mutated genes of dsFvs against HBsAg by using the PCR-based point mutagenesis method. The design of the disulfide connection was made by employing molecular modeling techniques. Previous studies [30] have identified two positions located in the interface between VH and VL: the best locations for disulfide stabilization are position 44 in framework region 2 (FR2) of VH and position 100 of FR4 of VL or position 105 of FR4 in VH and position 43 of PR2 in VL. The mutated genes of VH and VL were cloned into plasmid pET-20b and sequenced with the dideoxynucleotide method. The results showed that the cysteines were successfully introduced at position 44aa of VH and position 100aa of VL. The recombinant proteins of VH and VL were separately expressed in E. coli BL21(DE3). The VH and VL inclusion bodies were solubilized and combined in equal molar ratios in the refolding solution. Then, VH and VL were folded into a 25-kDa protein, designated as anti-HBsAg dsFv, which exhibited strong binding toward HBsAg.
ELISA and competition/inhibition ELISA in this study indicated that the constructed dsFv maintained the specificity of Fab, but it exhibited lower binding activity toward HBsAg than Fab. Reiter et al [31] reported that each Fv is influenced differently with regard to its binding affinity to antigens after disulfide stabilization. While the antigen specificity may not be altered, disulfide stabilization can increase or decrease the binding affinity of individual Fvs. However, the loss of binding affinity can be compensated by the significantly improved stability of dsFvs [31].
Acknowledgements
This work was supported by grants from the Chinese Ministry of Science and Technology 863 project (No.2007AA02Z144) and the Beijing Natural Science Foundation (No.7002033).
Conflict of Interests
The authors have declared that no conflict of interests exists.
References
1. Sidhu SS, Koide S. Phage display for engineering and analyzing protein interaction interfaces. Curr Opin Struct Biol. 2007;17:481-7
2. Brekke O.H, Loset G.A. New technologies in therapeutic antibody development. Curr Opin Pharmacol. 2003;3:544-50
3. Thullier P, Lafaye P, Megret F. et al. A recombinant Fab neutralizes dengue virus in vitro. J Biotechnol. 1999;69:183-90
4. Skerra A. Bacterial expression of immunoglobulin fragments. Curr Opin Immunol. 1993;5:256-62
5. Skerra A, Pluckthun A. Assembly of a functional immunoglobulin Fv fragment in Escherichia coli. Science. 1988;240:1038-41
6. Glockshuber R, Malia M, Pfitzinger I. et al. A comparison of strategies to stabilize immunoglobulin Fv-fragments. Biochemistry. 1990;29:1362-7
7. Kirsch M, Zaman M, Meier D. et al. Parameters affecting the display of antibodies on phage. J Immunol Meth. 2005;301:173-85
8. Marks JD, Hoogenboom HR, Griffiths AD. et al. Molecular evolution of proteins on filamentous phage. J Biol Chem. 1992;267:16007-10
9. Bird RE, Walker BW. Single chain variable regions. Trends Biotech. 1991;9:132-7
10. Webber K. O, Reiter Y, Brinkmann U, et al. Preparation and characterization of a disulfide-stabilized Fv fragment of the anti-Tac antibody: comparison with its single-chain analog. Mol Immunol. 1995;32:249-58
11. Eisenbach C, Sauer P, Mehrabi A. et al. Prevention of hepatitis B virus recurrence after liver transplantation. Clin Transplant. 2006;20:111-6
12. Lavanchy D. Hepatitis B virus epidemiology, disease burden, treatment, and current and emerging prevention and control measures. J Viral Hepat. 2004;11:97-107
13. Lu SC, Yan LN, Li B. et al. Lamivudine prophylaxis of liver allograft HBV reinfection in HBV related cirrhotic patients after liver transplantation. Hepatobiliary Pancreat Dis Int. 2004;3:26-32
14. Schiff E, Lai CL, Hadziyannis S. et al. Adefovir dipivoxil for wait-listed and post-liver transplantation patients with lamivudineresistant hepatitis B: final long-term results. Liver Transpl. 2007;13:349-60
15. Ning D, Junjian X, Qing Z. et al. Production of recombinant humanized anti- HBsAg Fab fragment from Pichia pastoris by frmentation. J Biochem Mol Biol. 2005;38:294-9
16. Chisari FV, Ferrari C. Hepatitis B virus immunopathology. Springer Semin Immunopathol. 1995;17:261-81
17. Rosenblum JS, Barbas CF. Synthetic antibodies in antibody engineering, 2nd ed. New York: Oxford University Press. 1995:89-116
18. Barbas CF, Lerner RA. Combinatorial immunoglobin libraries on the surface of phage (phAb): rapid selection of antigen-specific Fabs. Comp Methods Enzymol. 1991;2:119
19. Begum N, Horiuchi S, Tanaka Y. et al. New approach for generation of neutralizing antibody against human T-cell leukaemia virus type-I (HTLV-I) using phage clones. Vaccine. 2002;20:1281-9
20. Song HB, Mao CS, Chen W. et al. Construction and DNA sequencing of dsFv mutated gene of human antibody to HBsAg. J Cell Mol Immunol. 1998;14:241-3
21. Bachlder R, Bilancieri J, Lin WY. et al. A human recombinant Fab identifies a human immunodeficiency virus type 1-induced conformational change in cell surface-expressed CD4. J Virol. 1995;69:5734-42
22. Ohlin M, Owman H, Mach M. et al. Light chain shuffling of a high affinity antibody results in a drift in epitope recognition. Mol Immunol. 1996;33:47-56
23. Buchner J, Rudolph R. Renaturation, purification and characterization of recombinant Fab-fragment produced in Escherichia coli. Biotechnology. 1991;9:157-62
24. Kabat EA, Wu TT, Perry HM. et al. Sequences of proteins of immunological interest, 5th ed. US: NIH publication. 1991:91
25. Barik S, Galinski M. "Mega-primer" method of PCR: Increased template concentration improves yield. BioTechniques. 1991;10:489-95
26. Bio-Rad Laboratories. Methods in electroporation: Bacterial-transformation and pulse controller instrument manual. Herculs, CA: Bio-Rad Laboratories. 1989
27. Changshou Gao, Shenlan Mao, Gunnar Kaufmann. et al. A method for the generation of combinatorial antibody libraries using pIX phage display. PNAS. 2002;99:12612-6
28. Zebedee SL. et al. Human combinatorial antibody libraries to hepatitis B surface antigen. Proc Nati Acad Sci USA. 1992;89:3175-9
29. Kang AS, Burten DR, Lerner RA. Combinatorial immunoglobulin libraries in phage λ. Methods Enzymol. 1991;2:111-8
30. Reiter Y, Briter Y, Brinkmann U, Lee B, Pastan I. Engineering antibody Fv fragments for cancer detection and therapy: disulfide-stabilized Fv fragments. Nature Biotechnol. 1996;14:1239-45
31. Reiter Y, Pastan I. Antibody engineering of recombinant Fv immunotoxins for improved targeting of cancer: disulfide-stabilized Fv immunotoxins. Clin Cancer Res. 1996;2:245-52
Author contact
![]() Correspondence to: Hongbin Song or Qiao Li, Center for Disease Control and Prevention, Academy of Military Medical Sciences, Beijing 100071, China. Tel: 86-10-66933358; Fax: 86-10-66933358; Email: hongbinsongnet; or Department of Surgery, University of Michigan, Ann Arbor; MI 48109-0666, USA. Tel: 734-615-1977, Fax: 734-763-4135; Email: qiaoliedu
Correspondence to: Hongbin Song or Qiao Li, Center for Disease Control and Prevention, Academy of Military Medical Sciences, Beijing 100071, China. Tel: 86-10-66933358; Fax: 86-10-66933358; Email: hongbinsongnet; or Department of Surgery, University of Michigan, Ann Arbor; MI 48109-0666, USA. Tel: 734-615-1977, Fax: 734-763-4135; Email: qiaoliedu