Impact Factor ISSN: 1449-2288
- Issue 6; 2026
- Issue 5; 2026
- Issue 4; 2026
- Issue 3; 2026
- Issue 2; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
RESULTS
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2008; 4(4):223-235. doi:10.7150/ijbs.4.223 This issue Cite
Research Paper
Dysregulated Mitochondrial Genes and Networks with Drug Targets in Postmortem Brain of Patients with Posttraumatic Stress Disorder (PTSD) Revealed by Human Mitochondria-Focused cDNA Microarrays
Yan A. Su1, Jun Wu1, Lei Zhang2, Qiuyang Zhang1, David M. Su1, Ping He3, Bi-Dar Wang1, He Li2, Maree J. Webster4, Traumatic Stress Brain Study Group, Owen M. Rennert5, Robert J. Ursano2 ![]()
1. Department of Biochemistry and Molecular Biology and the Catherine Birch McCormick Genomics Center, The George Washington University School of Medicine and Health Sciences, Washington, DC, 20037
2. Department of Psychiatry and Center for the Study of Traumatic Stress, Uniformed Services University of the Health Sciences, Bethesda, MD 20814
3. Division of Hematology, Center for Biological Evaluation and Research, Food and Drug Administration, Bethesda, MD 20892
4. Stanley Medical Research Institute, Chevy Chase, MD 20894
5. Laboratory of Clinical Genomics, National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD 20892
Received 2008-6-24; Accepted 2008-8-2; Published 2008-8-5
Abstract
Posttraumatic stress disorder (PTSD) is associated with decreased activity in the dorsolateral prefrontal cortex (DLPFC), the brain region that regulates working memory and preparation and selection of fear responses. We investigated gene expression profiles in DLPFC Brodmann area (BA) 46 of postmortem patients with (n=6) and without PTSD (n=6) using human mitochondria-focused cDNA microarrays. Our study revealed PTSD-specific expression fingerprints of 800 informative mitochondria-focused genes across all of these 12 BA46 samples, and 119 (±>1.25, p<0.05) and 42 (±>1.60, p<0.05) dysregulated genes between the PTSD and control samples. Quantitative RT-PCR validated the microarray results. These fingerprints can essentially distinguish the PTSD DLPFC BA46 brains from controls. Of the 119 dysregulated genes (±≥125%, p<0.05), the highest percentages were associated with mitochondrial dysfunction (4.8%, p=6.61x10-6), oxidative phosphorylation (3.8%, p=9.04x10-4), cell survival-apoptosis (25.2%, p<0.05) and neurological diseases (23.5%, p<0.05). Fifty (50) dysregulated genes were present in the molecular networks that are known to be involved in neuronal function-survival and contain 7 targets for neuropsychiatric drugs. Thirty (30) of the dysregulated genes are associated with a number of neuropsychiatric disorders. Our results indicate mitochondrial dysfunction in the PTSD DLPFC BA46 and provide the expression fingerprints that may ultimately serve as biomarkers for PTSD diagnosis and the drugs and molecular targets that may prove useful for development of remedies for prevention and treatment of PTSD.
Keywords: PTSD, Brain dorsolateral prefrontal cortex BA46, Gene expression pattern, Biomarker, Mitochondria, Canonical pathways, Molecular networks, Neuropsychiatric drug targets
Introduction
Posttraumatic Stress Disorder (PTSD) is a complex mental disorder that can develop in response to a traumatic event such as a motor vehicle accident, rape, combat exposure or natural disaster. The National Vietnam Veterans Readjustment Study estimated that 53.4% of US male veterans of the Vietnam war developed full or partial PTSD and 15.2% still suffered from the disorder in 1990 [1]. The application of conservative procedures for analysis led Dohrenwend et al to document that 18.7% of male veterans developed war-related PTSD during their life time and 9.1% were still suffering from PTSD 11 to 12 years after the war [2]. In the general population, the estimated lifetime prevalence of PTSD is 7.8% in adult Americans, with women having a 2.3-fold higher prevalence than men [3]. PTSD symptoms include intrusive thoughts, hyperarousal, nightmares, flashbacks, emotional detachment or numbing of feelings, insomnia, fear, avoidance of reminders, distress when exposed to reminders, irritability, hypervigilance, and heightened startle response [4].
Several lines of evidence support neurochemical, functional and structural alterations in the neuroendocrine system in patients with PTSD. The neurochemical alterations range from adrenergic hyperresponsiveness, increased thyroid activity, increased levels of corticotrophin-releasing factor, to low cortisol levels and increased negative feedback sensitivity of the hypothalamo-pituitary-adrenal-axis after the administration of low-dosage dexamethasone [5]. Functional brain imaging studies in patients with PTSD demonstrate increased function in the amygdala and decreased activity in the hippocampus and prefrontal cortex (PFC) [6;7]. These functional alterations are consistent with the structural findings in animal models of PTSD that neurons in the hippocampus and PFC display atrophy while those in amygdala show growth in response to repeated stress [8].
DLPFC including BA46 is one of the three regions of PFC, which regulates working memory and execution of fear responses [9]. This brain region has been correlated with structural and functional alterations, and with the treatment response of patients with PTSD. In children with PTSD symptoms, decreased volume of gray matter in the DLPFC is correlated with increased functional impairment [10]. Adult patients with PTSD core symptoms (i.e. re-experiencing, avoidance) were markedly improved by treatment with 10-Hz repetitive transcranial magnetic stimulation over the right DLPFC [11]. Furthermore, the indicator of neuronal viability, N-acetylaspartate, in DLPFC was correlated positively in healthy people with verbal intelligence [12], and negatively in patients with a generalized anxiety disorder following a previous traumatic event [13].
Although the prefrontal cortex, amygdala and hippocampus are the brain regions considered to be related to PTSD [6;7], the underlying molecular mechanisms are unknown. One of the possibilities is that functional and structural changes in the brain may result from mitochondria-centered responses to repeated or chronic harmful stresses [14;15]. Mitochondria provide the cell with energy in the form of ATP and play fundamental roles in many metabolic pathways, such as β-oxidation, the tricarboxylic acid cycle and urea cycle, the synthesis of steroid hormones and heme, and calcium signaling. In addition, mitochondrial biochemical and molecular pathways are a natural central target of diverse pharmacological agents [16]. However, reactive oxygen species (ROS), an inevitable by-product of mitochondrial oxidative phosphorylation, can impair molecules, contributing to various diseases including neurodegeneration [17]. Thus, homeostasis of these mitochondrial functions is critical in human brain because of its high energy demand.
Mitochondrial dysfunctions are increasingly recognized as key components in stress related mental disorders [14;15]. Although long or repetitive exposure to stress can induce a neurological change resulting in PTSD, the molecular pathology involving in such a neuronal damage remain elusive. Psychological stress was associated with oxidative stress and telomere shortening [18]. Overexpression of antioxidant enzymes (glyoxalase and glutathione reductase 1) in mouse brain was also associated with increase in anxious behavior [19]. In addition, chronic stress of rats inhibits the activities of mitochondrial respiratory complexes (inhibition of 69% in complex I-III and of 67% in complex II-III) [20]. Cortisol has widespread functions including activation of glucocorticoid receptor which bind directly to mitochondrial membrane and regulate apoptosis [15]. The steroidogenic acute regulatory protein is one of the outer mitochondrial membrane protein required for stress responses [21]. Although all these studies indicate mitochondrial dysfunction, they are all focusing on a specific candidate gene(s). The lack of a holistic study of all the mitochondria-related genes hinders the progress of the PTSD research.
To date, methods employing a systems biology approach to identify the mitochondria-focused genes underlying the pathogenesis in PTSD-related brain regions have not been documented, probably due to the lack of human postmortem brain tissue from PTSD patients. The expression signatures in blood cells have been associated with changes in peripheral lymphocytes of subjects with PTSD [22;23], but provide no information for molecular abnormalities in brain tissues. Here we report the identification of expression signatures, canonical pathways, molecular networks and drug targets of dysregulated neuropsychiatric disease-related genes in BA46 of postmortem brain tissue from patients with PTSD using a human mitochondria-focused cDNA microarray (hMitChip3) [24]. Our results indicate mitochondrial dysfunction in the DLPFC BA46 and may prove useful for developing methods for diagnosis and treatment of PTSD.
Materials and Methods
Postmortem Brain Tissue: Postmortem brain tissues were collected by the Traumatic Stress Brain Study Group who diagnosed, collated, planned, coordinated, made decisions on inclusion and exclusion, established procedures of diagnosis and interrogated reliability, and obtained normal brains for controls as well. Medical records were meticulously reviewed and clinical diagnoses were confirmed using DSM-IV criteria [4]. Table 1 lists all available brain tissues employed for this study, with some clinical data including patient's gender and age (≤4-year difference), brain pH (≤0.65 difference) and reasons of death. Eight subjects died from myocardial infarction/heart attack, two from suicide by overdose, one from alcoholism complications and another from pulmonary embolism. The brain pH was determined from homogenized brain occipital cortex tissue. Tissue (0.4-0.8g) was thawed and homogenized in 10 times the tissue volume of deionized distilled H2O and the homogenate pH was measured with the PerpHect pH 370 model meter with a compatible PerpHect sure-flow type probe (ATI Orion, Boston, MA, USA). The pH electrode was washed thoroughly in deionized distilled H2O after each sample and recalibrated at the standard 3 points of pH (4, 7, and 10).
Demographic Data for Autopsy Cases
| Case | Sex | Age (yr) | PMI (hr) | Brain PH | Cause of Death |
|---|---|---|---|---|---|
| PTSD Patients | |||||
| P_1 | F | 39 | 39 | 6.45 | SUIC/OD |
| P_2 | F | 40 | 38 | 6.35 | SUIC/OD |
| P_3 | F | 42 | 36 | 6.58 | CARDIAC |
| P_4 | F | 44 | 46 | 6.56 | CARDIAC |
| P_5 | F | 47 | 46 | 6.77 | ALC/COM |
| P_6 | M | 54 | 86 | 6.77 | CARDIAC |
| Control Subjects | |||||
| N_1 | F | 35 | 40 | 5.80 | PUL/EMB |
| N_2 | F | 39 | 24 | 6.88 | CARDIAC |
| N_3 | F | 44 | 25 | 6.30 | CARDIAC |
| N_4 | F | 44 | 28 | 6.59 | CARDIAC |
| N_5 | F | 44 | 10 | 6.20 | CARDIAC |
| N_6 | F | 56 | 29 | 6.78 | CARDIAC |
PMI: postmortem interval; SUIC/OD: suicide by overdose; CARDIAC: myocardial infarction/heart attack; ALC/COM: alcoholism complications; PUL/EMB: pulmonary embolism.
RNA Preparation: DLPFC BA46 tissues were dissected from postmortem brains and the samples contained primarily gray matter with small but random amount of white matter because the dissections were done from frozen tissue blocks but not from cryostat sections. Total RNA was isolated using TRIZOL reagents (Invitrogen Inc. Carlsbad, CA, USA) and then purified with RNeasy kit (Qiagen, Valencia, CA), following the manufacture's instructions.
hMitChip3 Gene Chip and Microarray Data Analysis: A third-generation human mitochondria-focused cDNA microarray (hMitChip3) containing 37 mitochondrial DNA-encoded genes, 1,098 nuclear DNA-encoded and mitochondria-related genes and 225 controls was printed as described previously [24]. A total of 1135 mitochondria-related genes include 946 genes associated with 645 molecular functions, 930 genes with 612 biological processes, 476 genes with biological chemistry pathways, 227 genes with 23 reactome events, 237 genes with 320 genetic disorders, and 55 genes with 87 drugs targets[24]. For examples, 89 genes are related to oxidative phosphorylation, 20 genes to apoptosis, and 14 genes to neurotransmitter (tyrosine, L-dopa and dopamine) metabolism. Five μg of RNA per sample were used for microarray labeling and hybridization as previously described [24]. Slides were scanned using the ScanArray Express Microarray Scanner (PerkinElmer, Boston, MA, USA) as described previously [24]. Microarray database construction, data filtering and normalization were performed as described previously [24;25]. The normalized data were used to cluster and visualize expression levels of genes in these brain tissue specimens by using Eisen's Cluster software [26]. Heat map was visualized by using Eisen's Maple Tree software (http://rana.lbl.gov/EisenSoftware.htm).
Gene Information Analysis: Gene ID, symbols and names were derived from human UniGene Build 204 (ftp://ftp.ncbi.nih.gov/repository/UniGene/) based on human cDNA I.M.A.G.E. clone ID (http://image.llnl.gov/). Ontology, pathways and phenotypes of genes were compiled from Entrez (ftp://ftp.ncbi.nlm.nih.gov/gene) and DAVID Bioinformatics Resources 2008 (http://david.abcc.ncifcrf.gov/). Ingenuity Pathway Analysis software IPA version 6.0 (Ingenuity Systems Inc., Redwood City , CA, USA) was used to map canonical pathways, diseases, disorders, molecular networks and drug targets and to calculate the percentages and p-values (Fisher exact test). The reference gene set in canonical pathway was within the software.
Quantitative RT-PCR (qRT-PCR): Two μg of total RNA was reverse-transcribed into cDNA by using SuperScript TM First-Strand Synthesis System (Invitrogen). 30 ng cDNA was used for qPCR reactions with the Universal PCR Master Mix (No AmpErase UNG) on an Applied Biosystems 7300 Real Time PCR System (Foster City, CA, USA), following the manufacturer's instructions. After 40 cycles, data reduction was performed with the 7300 PCR Software. Triplicate qRT-PCR experiments were performed for each gene. Relative RNA concentrations were calculated using the published methods[27;28]. TaqMan probes and primers for qPCR were purchased from Applied Biosystems and include those of glyceraldehyde-3-phosphate dehydrogenase (GAPDH, 4352934E), DnaJ (Hsp40) homolog subfamily C member 19 (DNAJC19, Hs00829488_s1), solute carrier family 1 (high affinity aspartate/glutamate transporter) member 6 (SLC1A6, Hs00192604_m1), solute carrier family 9 (sodium/hydrogen exchanger) member 6 (SLC9A6, Hs00234723_m1), amyloid beta (A4) precursor protein (APP, Hs00169098_m1), and estrogen receptor 2 (ER beta) (ESR2, Hs00230957_m1).
Statistics: Statistical calculations were performed on triplicate array experiments using XLSTAT 2006 (XLSTAT, New York, NY, USA). Differentially expressed genes were identified arbitrarily by ≥1.25-fold change in the average expression of the background-subtracted mean intensity ratios of a gene between PTSD and control, with p-value <0.05. Student t-test was used to calculate p-values for gene expression, while Fisher exact test in Ingenuity Pathway Analysis software was used to calculate p-values for pathways and diseases. The level of statistical significance was set at a p-value <0.05.
RESULTS
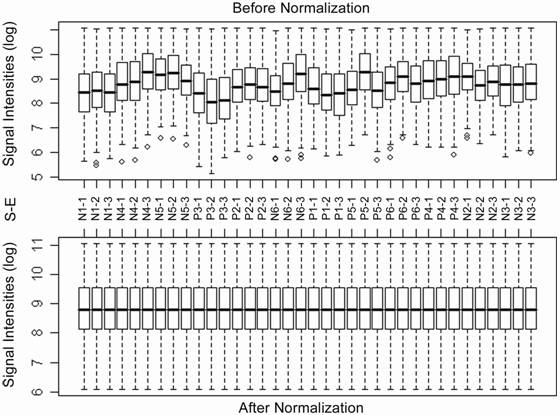
Expression clusters of mitochondria-focused genes distinguished the PTSD and control brains. Table 1 lists demographic data for autopsy cases used for this study. Total RNA samples were extracted from gray matter that was dissected from postmortem brain DLPFC BA46 of PTSD brains (n=6) and unaffected controls (n=6) and labeled for triplicate microarray experiments using our recently developed third generation human mitochondria-focused cDNA microarray hMitChip3. Because use of 3 replicates in cDNA microarrays analysis of even a single specimen greatly reduces misclassification rates [29], the hMitChip3 genes were all measured 9 times (3 identical probes per microarray and 3 microarray experiments per specimen), which generated reliable expression data for further analysis. The microarray data of 4,080 spots across all 36 gene chips used for 12 RNA samples were filtered by uniform statistic and bioinformatic criteria as described previously [24;30], which generated 800 genes with informative expression profiles. The supplemental table 1 reports the expression data of the selected 800 genes in each of the triplicate experiments before and after normalization as the raw and processed data, respectively; while Figure 1 shows the box plots of mRNA levels of 800 genes before and after the data normalization. The normalized data were used for unsupervised clustering analysis and visualization. The resultant dendrograms for all of these 800 genes and 12 brain DLPFC BA46 specimens classified P1, P2, P3, P5, P6 and N2 in one group and N1, N3, N4 N5, N6 and P4 in another (Fig. 2A and Supplemental Figure 1); these analysis separated the PTSD brain from the control except for N2 and P4 (opposite from their clinical diagnosis).
Box plots of expression data before and after normalization. The quantile normalization algorithms[25] were used to adjust the values of the background-subtracted mean pixel intensities of each and every set of 800 genes that were selected from the hMitChip3 triplicate microarray experiments hybridized with PTSD and control prefrontal cortex RNA samples as described previously[24]. In contrast to the pre-normalization boxplots (top panel), the post-normalized boxplots distribute in the same intervals with the same density center, indicating successful adjustment of data. The post-normalized data were used for clustering analysis. S-E: Sample and independent microarray experiment.
Dendrograms and heat maps of gene expression in human brain DLPFC BA46 tissue samples. (A) Dendrogram of unsupervised cluster of the PTSD and control DLPFC BA46 tissue samples based on expression similarities of 800 human mitochondria-focused genes (Supplemental Table 1); (B) Dendrograms and heat maps of 119 (±≥1.25, p<0.05) and (C) 42 (±≥1.60, p<0.05) dysregulated genes across all of these 12 BA46 samples. The results revealed the PTSD (P) and non-PTSD (N) groups. The control case N2 is clustered in the PTSD group, while the PTSD case P4 in the control group. Green: down-regulation; red: up-regulation; black: no change. The case numbers and gene symbols are indicated. The color map indicates degrees of up- and down-regulation of gene expression.
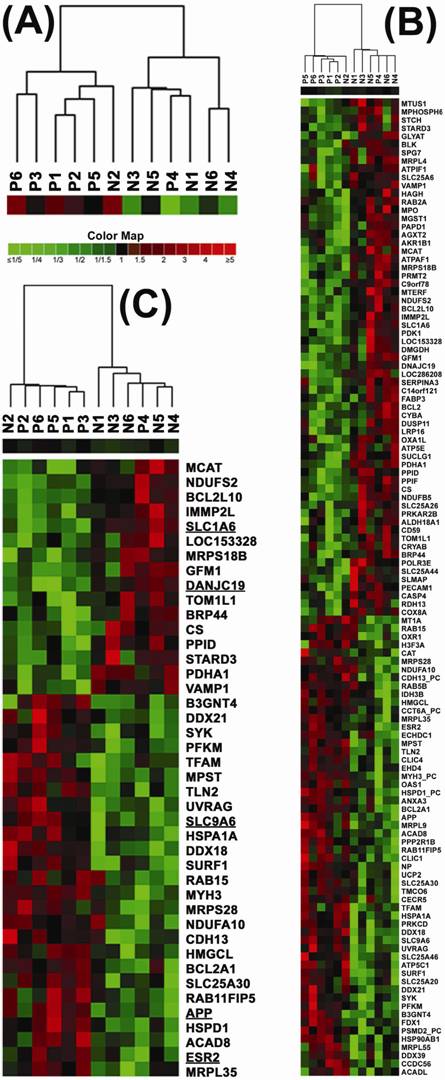
Based on the unsupervised cluster results, we calculated an average expression level of each gene in the PTSD group (P1, P2, P3, P5, P6 and N2) and in the non-PTSD control group (N1, N3, N4 N5, N6 and P4). The ratios and p-values (t-test) were calculated between the PTSD and control specimens, resulting in the identification of 119 genes (p<0.05, ±≥1.25) and 42 genes (p<0.05, ±≥1.60) whose expression were dysregulated in the PTSD DLPFC BA46 as compared to the controls (Supplemental Table 2). Clustering analysis of these 119 and 42 genes across all 12 brain samples generated double dendrograms and heat maps that clearly distinguish the PTSD DLPFC BA46 specimens from the controls (Fig. 2B and 2C). The identification of these dysregulated genes provides candidates with biological clues for validation of mRNA expression and future functional studies.
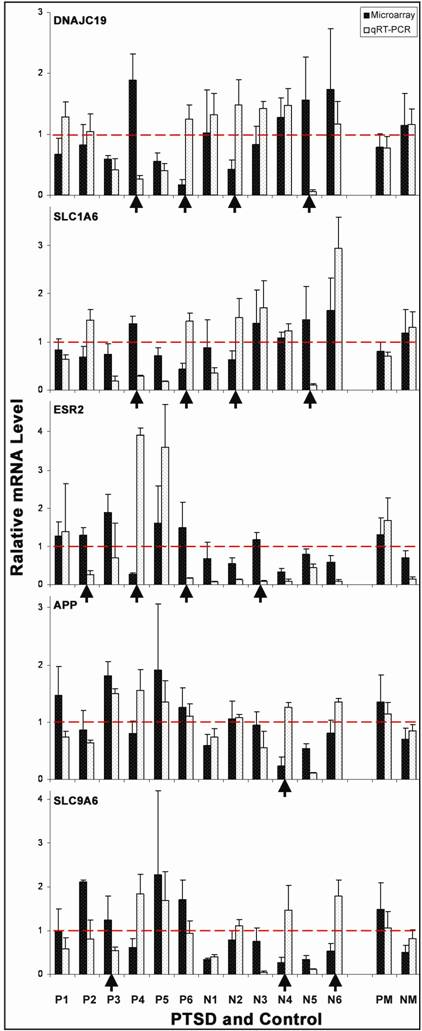
qRT-PCR confirmation of microarray results. To validate the microarray results, we conducted qRT-PCR analysis on 5 genes including APP, DNAJC19, ESR2, SLC1A6 and SLC9A6 because of the highest (SLC9A6) and lowest (DANJC19) expression levels in PTSD BA46 or because of their known relevance to neuropsychiatric disorders (APP, EST2 and SLC1A6). The results showed that the relative mRNA levels of these genes measured by qRT-PCR were essentially in agreement with the data detected by microarray experiments (Fig. 3). Specifically, 11 of 12 (92%) of expression changes were consistent between these two methods for APP, 9 of 12 (75%) for SLC9A6, and 8 of 12 (67%) for DNAJC19, SLC1A6 and ESR2. The discrepant data were randomly distributed among the genes and samples tested (Fig. 3), indicating the absence of a systematic error in these two methods. Thus, we subjected all the 119 dysregulated genes to analysis of a systems biology including the pathway, ontology and network.
The mitochondrial dysfunction and oxidative phosphorylation pathways contained the highest number of dysregulated genes. To see functional relationships, we conducted the canonical pathway analysis on 119 dysregulated genes. The results showed that out of 94 canonical pathways, 16 (17.0%) contained a significant number of dysregulated genes per pathway, of which 10 were listed in Figure 4A. The mitochondrial dysfunction pathway contained 8 (4.8%) dysregulated genes (p=6.61x10-6) including upregulated APP, CAT, NDUFA10 and UCP2 and downregulated COX8A, NDUFB5, NDUFS2 and PDHA1. The oxidative phosphorylation pathway had 6 (3.8%) dysregulated genes (p=9.04x10-4) including upregulated ATP5C1 and NDUFA10 and downregulated ATP5E, COX8A, NDUFB5 and NDUFS2. The citrate cycle pathway had 3 (5.1%) dysregulated genes (p=1.71x10-3) including upregulated IDH3B and downregulated CS and SUCLG1. The methane metabolism pathway contained 2 (3.1%) dysregulated genes (p=5.38x10-3) including upregulated CAT and downregulated MPO. The ubiquinone biosynthesis pathway had 3 (2.9%) dysregulated genes (p=1.11x10-2) including upregulated NDUFA10 and downregulated NDUFB5 and NDUFS2. The ERK/MARK signaling pathway included upregulated H3F3A, PPP2R1B, PRKCD and TLN2, and downregulated PRKAR2B (n=5 [2.8%], p=1.18x10-2). The propanoate metabolism pathway had upregulated ACAD8 and ACADL and downregulated SUCLG1 (n=3 [2.4%], p=1.21x10-2). The xenobiotic metabolism pathway contained upregulated CAT, HSP90AB1, PPP2R1B and PRKCD and downregulated ALDH18A1 and MGST1 (n=6 [2.4%], p=1.28x10-2). The valine, leucine and isoleucine degradation pathway contained upregulated ACAD8, ACADL and HMGCL (n=3 [2.8%], p=1.38x10-2). The pyruvate metabolism pathway had downregulated AKR1B1, HAGH and PDHA1 (n=3 [2.1%], p=2.05x10-2) (Fig. 4A).
The consistency in RNA levels measured by microarray and qRT-PCR. Relative RNA levels of 5 genes were measured and analyzed by both microarray and qRT-PCR experiments. The arrows indicate obvious inconsistent results between these two experiments. Out of these 5 genes in 12 tissues compared, 8 of 12 (66.7%) are consistent for DNAJC19, SLC1A6 and ESR2, 11 of 12 (92%) for APP, and 9 of 12 (75%) for SLC9A6. PM: mean and SD of PTSD cases; NM: mean and SD of non-PTSD controls.
The bar graphs indicate canonical pathways and neuropsychiatric and systemic disorders associated with dysregulated genes in the PTSD DLPFC BA46. (A) 10 canonical biochemical and molecular biology pathways with significantly (p<0.05, Fisher's Exact Test) dysregulated genes, in the PTSD DLPFC BA46 group (P1, P2, P3, P5, P6 and N2) in comparison to the non-PTSD controls (N1, N3, N4, N5, N6 and P4). The ratio is calculated as the number of genes in a given pathway divided by a total number of genes that make up that pathway. The mitochondrial dysfunction pathway contains 8 (4.8%) dysregulated genes (p=6.61x10-6) including upregulated APP, CAT, NDUFA10 and UCP2 and downregulated COX8A, NDUFB5, NDUFS2 and PDHA1. (B) Six categories of abnormalities in which the significant (p<0.05, Fisher's Exact Test) number of the dysregulated genes in the PTSD DLPFC BA46 are known to involve, by searching the literature in the NCBI PUBMED, DAVID, OMIM and Ingenuity databases. Red: upregulation; green downregulation.
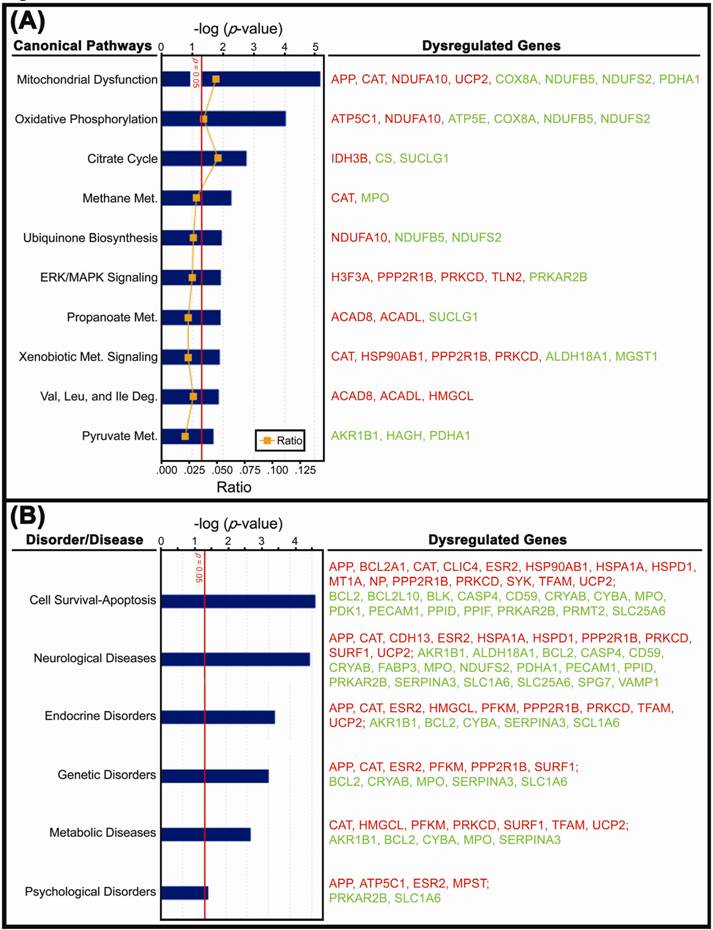
46 dysregulated genes were mapped to neurological diseases and other systemic disorders. To see phenotypes involved, we performed mapping of all 119 dysregulated genes to known human diseases and disorders. The results revealed that 35 dysregulated genes had known roles in a variety of neurological diseases (28 genes) and other systematic disorders including endocrine (14 genes), genetics (11 genes), metabolism (12 genes) and psychology (6 genes). The number of these dysregulated genes were all statistically significant (p<0.05). In addition, 17 (61%) of the 28 genes that were mapped to neurological diseases were known to play a role in survival and apoptosis of a cell. Moreover, a total of 30 dysregulated genes were mapped to the cell survival-apoptosis category (Fig. 4B). Many of these genes are involved in multiple abnormalities, suggesting that PTSD is a neurological and systemic disorder.
30 dysregulated genes have known roles in neurological and psychiatric diseases. Table 2 lists 30 PTSD-dysregulated genes that have been described to play a pathogenic role in various neurological diseases and psychiatric disorders. Upregulated APP induces changes in nitric oxide production and mitochondrial activity, leading to apoptosis [31], which have been implicated in amyloidosis [32;33], neurodegeneration [34], Alzheimer's disease [35;36] and Down syndrome [37]. Considering the role of myeloperoxidase (MPO) in modulating vascular inflammatory responses and transporting molecules as well as its incapability of clearing up amyloid when mutated [38-40], MPO downregulation may lead to regional accumulation of amyloid, resulting in mitochondrial dysfunction and cell death. Peptidylprolyl isomerase D (PPID) suppresses apoptosis via a mitochondrial hexokinase II-dependent mechanism and its decreased expression has been associated with corticobasal degeneration characterized by nerve cell loss and atrophy [41]. Dysregulation of genes with known functions as neurotransmitters in synaptic vesicles (SLC1A6 and VAMP1) and cell adhesion (PECAM1, CDH13 and SPG7) are related to diseases in the central nervous system. Dysregulation of these genes might reduce synaptic communication, and jeopardize cell-to-cell interactions and/or cell-to-matrix adhesion. These are but a few examples (Table 2).
30 Dysregulated Genes in the PTSD DLPFC BA46 and Neuropsychiatric Disorders*
| No. | Gene | Name | Change | Role | Neuropsychiatric Disorder |
|---|---|---|---|---|---|
| 1 | Bcl2 | B-cell CLL/lymphoma 2 | -0.49 (0.005) | Mitochondrial dysfunction and cell death | Neurodegeneration |
| 2 | PPID | Peptidylprolyl isomerase D | -0.94 (0.002) | Neurodegeneration | |
| 3 | APP | Amyloid beta A4 precursor protein | 1.05 (0.002) | Amyloidosis; Neurodegeneration; Alzheimer disease; Down syndrome | |
| 4 | MPO | Myeloperoxidase | -0.36 (0.017) | Alzheimer disease | |
| 5 | CASP4 | Caspase 4, apoptosis-related cysteine peptidase | -0.41 (0.017) | Cell death | Alzheimer disease |
| 6 | CD59 | CD59 molecule, complement regulatory protein | -0.57 (0.041) | Cerebral infarction | |
| 7 | SLC25A6 | Solute carrier family 25 (mitochondrial carrier; adenine nucleotide translocator), member 6 | -0.47 (0.036) | Mitochondrial transporter | Parkinson disease |
| 8 | ALDH18A1 | Aldehyde dehydrogenase 18A1 | -0.57 (0.032) | Mitochondrial enzyme | Neurodegeneration |
| 9 | PDHA1 | Pyruvate dehydrogenase alpha 1 | -0.79 (0.003) | Severe neurological dysfunction and growth retardation | |
| 10 | AKR1B1 | Aldo-keto reductase family 1 member B1 | -0.48 (0.027) | Diabetic retinopathy | |
| 11 | MPST | Mercaptopyruvate sulfurtransferase | 1.02 (0.0003) | Bipolar affective disorder | |
| 12 | NDUFS2 | NADH dehydrogenase (ubiquinone) Fe-S protein 2 | -0.71 (0.004) | Mitochondrial oxidative phosphorylation | Mitochondrial encephalomyopathy |
| 13 | SURF1 | Surfeit 1 | 0.95 (0.004) | COX-deficient Leigh syndrome | |
| 14 | ATP5C1 | ATP synthase, H+ transporting, mitochondrial F1 complex, gamma polypeptide 1 | 0.60 (0.007) | Bipolar affective disorder | |
| 15 | SLC1A6 | Solute carrier family 1 (high affinity aspartate/glutamate transporter), member 6 | -1.06 (0.0002) | Neurotransmitter | Schizophrenia |
| 16 | VAMP1 | Vesicle-associated membrane protein 1 (synaptobrevin 1) | -0.88 (0.010) | Neurological lethal-wasting; Schizophrenia | |
| 17 | PECAM1 | Platelet/endothelial cell adhesion molecule | -0.55 (0.006) | Cell adhesion | Multiple sclerosis, ischaemic stroke; Alzheimer disease |
| 18 | CDH13 | Cadherin 13 | 0.71 (0.011) | Aggregation of brain cancer cell line; inhibition of cancer cell growth; broadly expressed in central nervous system | |
| 19 | SPG7 | Spastic paraplegia 7 | -0.46 (0.042) | Autosomal recessive spastic paraplegia | |
| 20 | CAT | Catalase | 0.61 (0.048) | Stress response and reactive oxygen species | Inhibition of neurite outgrowth |
| 21 | UCP2 | Uncoupling protein 2 | 0.58 (0.041) | Schizophrenia | |
| 22 | HSPA1A | Heat shock 70kDa protein 1A | 0.90 (0.003) | Stress response and protein folding | Neurodegeneration |
| 23 | CRYAB | Crystallin, alpha B | -0.61 (0.046) | Neurodegenerative disease | |
| 24 | HSPD1 | Heat shock 60kDa protein 1 (chaperonin) | 0.89 (0.002) | Spastic paraplegia | |
| 25 | PPP2R1B | Protein phosphatase 2 (formerly 2A), regulatory subunit A, beta isoform | 0.58 (0.004) | Protein phosphatase | Alzheimer disease |
| 26 | PRKAR2B | Protein kinase, cAMP-dependent, regulatory, type II, beta | -0.46 (0.042) | Protein kinase | Cataleptic behavior |
| 27 | PRKCD | Protein kinase C, delta | 0.61 (0.008) | Parkinson disease model and its treatment | |
| 28 | FABP3 | Fatty acid binding protein 3 | -0.55 (0.049) | Fatty acid metabolism | Down syndrome and Alzheimer's disease |
| 29 | SERPINA3 | Serpin peptidase inhibitor clade A member 3 | -0.35 (0.011) | Protease inhibitor | Parkinson disease; Alzheimer disease |
| 30 | ESR2 | Estrogen receptor 2 (ER beta) | 0.98 (0.025) | transcription factor | Alzheimer disease; Anorexia nervosa; Major depressive disorder (MDD) |
* Change: fold changes of the average mRNA levels of a gene in the PTSD BA46 vs. non-PTSD controls; the numbers outside of parentheses indicate the fold changes and those in the parentheses are p-values.
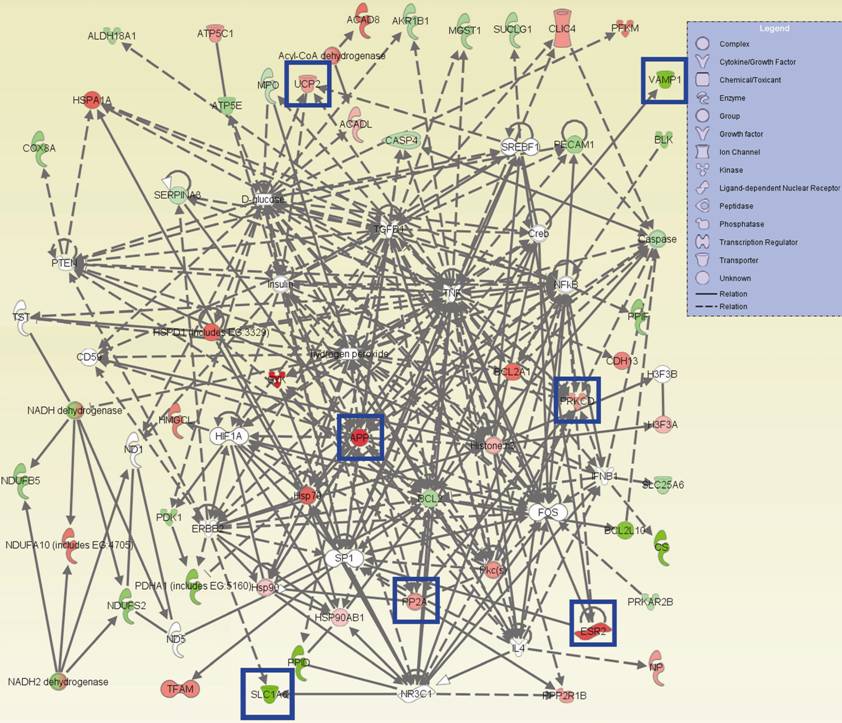
Networks of 54 dysregulated genes involved in neuron function and survival and 7 protein targets for neuropsychiatric drugs: To reveal complex interactions among dysregulated genes and to identify gene targets for the central nervous system drugs, we mapped 54 dysregulated genes to molecular networks. These genes were selected for analysis because of their obvious relevance to neurological diseases, psychological disorders, psychiatric disorders, and stress response in the central nervous system. The results revealed interactive networks with a total of 75 molecular elements that maintain neuronal function and survival. Within these networks, 27 (36%) genes were upregulated; 27 (36%) genes were downregulated; and 21 (28%) elements appear to be unchanged (Fig. 5). These data strongly suggest molecular and cellular abnormalities in neuronal function and survival in PTSD DLPFC BA46.
Importantly, 7 gene products within the neuronal function-survival networks were identified as known targets of drugs that are used for neurological diseases, psychological and/or behavioral disorders, including APP as a target for Bapineuzumab (AAB-001), VAMP1 for Dysport, SLC1A6 for Riluzole, ESR2 for 17β-estradiol (agonist) and Tamoxifen (antagonist), PRKCD for Tamoxifen and rottlerin, PP2A for Okadaic Acid and Fostriecin, and UCP2 for MPTP (1,2,3,6-methyl-phenyl-tetrahydropyridine).
Discussion
Human brain DLPFC including BA46 is involved in regulation of working memory and preparation and selection of fear responses and has been correlated with structural and functional alterations and treatment response of patients with PTSD [9-13]. However, the underlying cellular and molecular mechanisms are unknown. In this study, we applied human mitochondria-focused cDNA microarrays (hMitChip3) to PTSD brain samples and have successfully identified expression signatures, canonical pathways, molecular networks and drug targets of neurological disease- and psychological/psychiatric disorder-related genes that are dysregulated in the DLPFC BA46. Our results indicate mitochondrial dysfunction involved in neuronal function and survival in the DLPFC BA46 and may prove useful for development of methods for diagnosis, prevention and treatment of PTSD.
Unsupervised clusters of 12 DLPFC BA46 RNA samples based solely on expression similarities of informative 800 mitochondria-focused genes clearly distinguish the PTSD brains from the controls. The identification of 119 (p<0.05, ±≥1.25) and 42 (p<0.05, ±≥1.60) dysregulated genes provides candidates for qRT-PCR validation and for subsequent functional studies. The clustering signatures of these genes may facilitate further development of methods and tools for forensic diagnosis of PTSD. Our approach has generated informative results partially because of our accurate dissection of brain DLPFC BA46 gray matter, and partially because of the utilization of our hypothesis-driven mitochondria-focused cDNA microarrays. However, we cannot explain why the samples P4 and N2 had expression profiles different from their clinical classification. Due to a small sample size, P4 and N2 were kept in the groups based on their gene expression rather than a clinical diagnosis because the former might be more objective than the latter. We would like to repeat the study when more PTSD and control specimens become available in order to improve the molecular classification of PTSD. The subsequent experiments should examine both mRNA and protein levels in the DLPFC BA46, as well as other PTSD-related brain regions such as medial and ventral PFC, hippocampus, and amygdala [6;7;42].
The molecular networks for neuron function and survival. The networks have a total of 75 elements including 27 (36%) upregulated genes, 27 (36%) downregulated genes and 21 (28%) unchanged ones. Twenty-one elements in white color include 7 genes present and 11 genes absent in the hMitChip3 microarray, and 3 small molecules. The symbols in Legend illustrate the networks. The genes in the blue boxes are the known targets for neuropsychiatric drugs. For example: the ESR2 is a target of 17β-estradiol or related ligands. The APP is a target for Bapineuzumab (AAB-001) that is currently used for the Phase 3 clinical trials for the treatment of patients with Alzheimer's disease. The ESR2 is a target for Tamoxifen that is used for the treatment of mania in patients with bipolar disorder. The PRKCD is a target for both Tamoxifen, and rottlerin that has been used in preclinical treatment of Parkinson's disease. The PP2A is a target for Fostriecin that antagonize dopamine activities. The VAMP1 is a target of the neurotoxin protein Dysport that is used in minute doses to treat painful muscle spasms. The SLC1A6 is a target of Riluzole used to treat amyotrophic sclerosis. The UCP2 is a target for protection of dopaminergic neurons from oxidative stress caused by 1,2,3,6-methyl-phenyl-tetrahydropyridine (MPTP) toxicity.
The mitochondria-mediated cellular and molecular response to stress in the central nervous system of patients with PTSD is the key part of our hypothesis because mitochondria are at the center of a cellular response to stress [14;15]. Study of mitochondrial function is inevitable and our application of the mitochondria-focused cDNA microarrays to PTSD is just the beginning. Mitochondrial function in the brain is of particular importance because of its high energy demand. Although human brain represents only 2% of body weight, it receives 15% of the body's cardiac output, and uses 20% of total body oxygen. These high level oxygen and energy requirements are continuous and imply that even brief periods of oxygen or glucose deprivation may impair neuron function and even result in neuron death. Our demonstration that a highly significant number of oxidative phosphorylation genes were dysregulated in the PTSD brain BA46 strongly suggests the presence of at least energy deficiency in this brain region. Our findings support the postulation that mitochondria-focused and stress-responsive genes may play a key role in the pathogenesis of PTSD, although the specific impact of these dysregulated genes and pathways on brain function remains to be determined.
The identification and validation of the dysregulated genes enhances our understanding of the cellular and molecular mechanisms underlying the pathophysiology of PTSD. The genes listed in Table 2 are critical to the BA46 neuronal functions including survival and apoptosis, carbohydrate and lipid metabolism, ATP and ROS production, synthesis and transport of neurotransmitter, mitochondrial and nuclear DNA expression, and protein phosphorylation and dephosphorylation as well as folding and degradation. Because homeostasis of these mitochondrial functions is vital for neuronal function and survival, the presence of a hundred consistently dysregulated genes in the PTSD DLPFC BA46 implies a pathological role. In addition, more genes were downregulated than those upregulated (63 vs. 56 in Supplemental Table 1 and 18 vs. 12 in Table 2). Moreover, some upregulated genes play a degradative or negative role such as proapoptosis (e.g., APP), reduction of ATP synthesis (e.g., UCP2) and protein breakdown (e.g., HSPA1A). Taken together, functional impairment and atrophy in the PTSD DLPFC BA46 might be a consequence of these dysregulated genes. Thus, the neurons in the PTSD DLPFC BA46 may be unable to regulate working memory and preparation and selection of healthy responses to fear or may even send abnormal signals to their downstream executive neurons such as those in the hippocampus and/or amygdala. Our molecular insight in the PTSD DLPFC BA46 pathogenesis is reminiscent of the neurocircuitry model by Bremner [43] and Rauch et al [42] that the fear extinction abnormalities in PTSD are characterized by exaggerated amygdala responses and deficiency in ventral/medial PFC and hippocampus. Therefore, our systems biology results appear not only to extend the neurocircuitry model to brain DLPFC BA46, but they also provide genes and proteins that should be further investigated for better understanding of the molecular pathogenesis of PTSD.
Seven protein targets for neuropsychiatric drugs are present in the neuronal function-survival networks in human brain DLPFC BA46. In addition of dysregulation of 72% (54/75) genes in these networks, many dysregulated genes are associated with multiple neuropsychiatric conditions such as Alzheimer's disease, Parkinson's disease, Schizophrenia, and Major Depressive Disorder (MDD), Down syndrome, attention disorders and mood disorders (Table 2). Obviously, modulations of these genes with drugs would affect neuropsychiatric functions of the neurons in human DLPFC BA46. For example, the APP is a target for the immunotherapeutic drug Bapineuzumab that is currently used for the Phase 3 clinical trials for the treatment of patients with Alzheimer's disease [44]. The ESR2 is a target for 17β-estradiol (agonist) and Tamoxifen (antagonist). Beside the treatment of estrogen-receptor positive breast cancer, Tamoxifen is used to treat mania in patients with bipolar disorder, probably by blocking PRKCD [45]. The PRKCD is also a target for rottlerin used in preclinical study of Parkinson's disease [46]. The PP2A is a target for Fostriecin that antagonizes dopamine activities [47]. The UCP2 is a target for protection of dopaminergic neurons from oxidative stress caused by 1,2,3,6-methyl-phenyl-tetrahydropyridine (MPTP) toxicity [48]. However, whether these drugs and related targets might be useful for prevention and treatment of PTSD is interesting and deserves further studies.
Supplementary Material
Supplemental Table 1a: Microarray Mean Data Before Normalization
Supplemental Table 1b: Microarray Mean Data After Normalization
Supplemental Table 2
Supplemental Figure 1
Acknowledgements
We thank Drs. Ron Duman and John Krystal at Yale University for critical review and helpful discussions of the manuscript. YAS, JW, QZ and BW were supported by the Catherine McCormick Genomics Center, NIH-NIDDK-06-925, the St. Laurant Institution and HU0001-07-M-V451; and LZ, HL and RJU by USUHS; PH by FDA, MJW by Stanley Medical Research Institute; and OMR by NICHD/NIH. DMS is a special volunteer from Walt Whitman High School.
Traumatic Stress Brain Study Group
Traumatic Stress Brain Study Group includes David Benedek and Harry Holloway, Dept of Psychiatry & Center for the Study of Traumatic Stress, USUHS, School of Medicine; Maree J. Webster, Stanley Medical Research Institute; Christopher J. Hough, FDA; Ronald Duman, Abraham Ribicoff Research Facilities Laboratory of Molecular Psychiatry, Yale University School of Medicine; Matthew Friedman, National Center for PTSD, US Dept of Veterans Affairs & Dartmouth Medical School; John Krystal, Clinical Neuroscience Division, VA National Center for PTSD, VA Connecticut Healthcare System, West Haven, CT & Dept of Psychiatry, Yale University School of Medicine; Gregory Leskin, National Center for PTSD, VA Palo Alto Health Care System; James Meyerhoff, Georgetown University School of Medicine & Division of Psychiatry & Neurosciences, Walter Reed Army Institute of Research; Elizabeth Osuch, Dept of Psychiatry, University of Western Ontario, Schulich School of Medicine and Dentistry.
Conflict of Interest
The authors have no conflicts of interest related to this work.
References
1. Kulka RA, Schlenger WE, Fairbank JA, Hough RL, Marmar BK, Weiss DS. Trauma and the Vietnam War Generation: Report of Findings from the National Veitnams Readjustment Study. New York: Brunner/Mazel. 1990
2. Dohrenwend BP, Turner JB, Turse NA, Adams BG, Koenen KC, Marshall R. The psychological risks of Vietnam for U.S. veterans: a revisit with new data and methods. Science. 2006Aug18;313(5789):979-82
3. Breslau N, Davis GC, Andreski P, Peterson EL, Schultz LR. Sex differences in posttraumatic stress disorder. Arch Gen Psychiatry. 1997Nov;54(11):1044-8
4. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition. Washington, DC: American Psychiatric Association. 1994
5. Yehuda R, Giller EL, Southwick SM, Lowy MT, Mason JW. Hypothalamic-pituitary-adrenal dysfunction in posttraumatic stress disorder. Biol Psychiatry. 1991Nov15;30(10):1031-48
6. Shin LM, Rauch SL, Pitman RK. Amygdala, medial prefrontal cortex, and hippocampal function in PTSD. Ann N Y Acad Sci. 2006Jul;1071:67-79
7. Bremner JD. Functional neuroimaging in post-traumatic stress disorder. Expert Rev Neurother. 2007Apr;7(4):393-405
8. McEwen BS. Protection and damage from acute and chronic stress: allostasis and allostatic overload and relevance to the pathophysiology of psychiatric disorders. Ann N Y Acad Sci. 2004Dec;1032:1-7
9. Fales CL, Barch DM, Rundle MM, Mintun MA, Snyder AZ, Cohen JD. et al. Altered Emotional Interference Processing in Affective and Cognitive-Control Brain Circuitry in Major Depression. Biol Psychiatry. 2008;63(4):377-84
10. Richert KA, Carrion VG, Karchemskiy A, Reiss AL. Regional differences of the prefrontal cortex in pediatric PTSD: an MRI study. Depress Anxiety. 2006;23(1):17-25
11. Cohen H, Kaplan Z, Kotler M, Kouperman I, Moisa R, Grisaru N. Repetitive transcranial magnetic stimulation of the right dorsolateral prefrontal cortex in posttraumatic stress disorder: a double-blind, placebo-controlled study. Am J Psychiatry. 2004Mar;161(3):515-24
12. Pfleiderer B, Ohrmann P, Suslow T, Wolgast M, Gerlach AL, Heindel W. et al. N-acetylaspartate levels of left frontal cortex are associated with verbal intelligence in women but not in men: a proton magnetic resonance spectroscopy study. Neuroscience. 2004;123(4):1053-8
13. Mathew SJ, Mao X, Coplan JD, Smith EL, Sackeim HA, Gorman JM. et al. Dorsolateral prefrontal cortical pathology in generalized anxiety disorder: a proton magnetic resonance spectroscopic imaging study. Am J Psychiatry. 2004Jun;161(6):1119-21
14. Zhang L, Zhou R, Li X, Ursano RJ, Li H. Stress-induced change of mitochondria membrane potential regulated by genomic and non-genomic GR signaling: a possible mechanism for hippocampus atrophy in PTSD. Med Hypotheses. 2006;66(6):1205-8
15. Manoli I, Alesci S, Blackman MR, Su YA, Rennert OM, Chrousos GP. Mitochondria as key components of the stress response. Trends Endocrinol Metab. 2007Jul;18(5):190-8
16. Szewczyk A, Wojtczak L. Mitochondria as a pharmacological target. Pharmacol Rev. 2002Mar;54(1):101-27
17. Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006Oct19;443(7113):787-95
18. Epel ES, Blackburn EH, Lin J, Dhabhar FS, Adler NE, Morrow JD. et al. Accelerated telomere shortening in response to life stress. Proc Natl Acad Sci U S A. 2004Dec7;101(49):17312-5
19. Hovatta I, Tennant RS, Helton R, Marr RA, Singer O, Redwine JM. et al. Glyoxalase 1 and glutathione reductase 1 regulate anxiety in mice. Nature. 2005Dec1;438(7068):662-6
20. Madrigal JL, Olivenza R, Moro MA, Lizasoain I, Lorenzo P, Rodrigo J. et al. Glutathione depletion, lipid peroxidation and mitochondrial dysfunction are induced by chronic stress in rat brain. Neuropsychopharmacology. 2001Apr;24(4):420-9
21. Bose H, Lingappa VR, Miller WL. Rapid regulation of steroidogenesis by mitochondrial protein import. Nature. 2002May2;417(6884):87-91
22. Segman RH, Shefi N, Goltser-Dubner T, Friedman N, Kaminski N, Shalev AY. Peripheral blood mononuclear cell gene expression profiles identify emergent post-traumatic stress disorder among trauma survivors. Mol Psychiatry. 2005May;10(5):500-13
23. Zieker J, Zieker D, Jatzko A, Dietzsch J, Nieselt K, Schmitt A. et al. Differential gene expression in peripheral blood of patients suffering from post-traumatic stress disorder. Mol Psychiatry. 2007Feb;12(2):116-8
24. Bai X, Wu J, Zhang Q, Alesci S, Manoli I, Blackman MR. et al. Third-generation human mitochondria-focused cDNA microarray and its bioinformatic tools for analysis of gene expression. Biotechniques. 2007Mar;42(3):365-75
25. Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003Jan22;19(2):185-93
26. Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A. 1998Dec8;95(25):14863-8
27. Manoli I, Le H, Alesci S, McFann KK, Su YA, Kino T. et al. Monoamine oxidase-A is a major target gene for glucocorticoids in human skeletal muscle cells. FASEB J. 2005Aug;19(10):1359-61
28. Holland PM, Abramson RD, Watson R, Gelfand DH. Detection of specific polymerase chain reaction product by utilizing the 5'----3' exonuclease activity of Thermus aquaticus DNA polymerase. Proc Natl Acad Sci U S A. 1991Aug15;88(16):7276-80
29. Lee ML, Kuo FC, Whitmore GA, Sklar J. Importance of replication in microarray gene expression studies: statistical methods and evidence from repetitive cDNA hybridizations. Proc Natl Acad Sci U S A. 2000Aug29;97(18):9834-9
30. Zhang Q, Wu J, Nguyen A, Wang BD, He P, Laurent GS. et al. Molecular mechanism underlying differential apoptosis between human melanoma cell lines UACC903 and UACC903(+6) revealed by mitochondria-focused cDNA microarrays. Apoptosis. 2008Aug;13(8):993-1004
31. Keil U, Bonert A, Marques CA, Scherping I, Weyermann J, Strosznajder JB. et al. Amyloid beta-induced changes in nitric oxide production and mitochondrial activity lead to apoptosis. J Biol Chem. 2004Nov26;279(48):50310-20
32. Van BC, Haan J, Bakker E, Hardy JA, van HW, Wehnert A. et al. Amyloid beta protein precursor gene and hereditary cerebral hemorrhage with amyloidosis (Dutch). Science. 1990Jun1;248(4959):1120-2
33. De JC, Zehr C, Yager D, Prada CM, Younkin S, Hendriks L. et al. Flemish and Dutch mutations in amyloid beta precursor protein have different effects on amyloid beta secretion. Neurobiol Dis. 1998Oct;5(4):281-6
34. Gralle M, Ferreira ST. Structure and functions of the human amyloid precursor protein: the whole is more than the sum of its parts. Prog Neurobiol. 2007May;82(1):11-32
35. Ancolio K, Dumanchin C, Barelli H, Warter JM, Brice A, Campion D. et al. Unusual phenotypic alteration of beta amyloid precursor protein (betaAPP) maturation by a new Val-715 --> Met betaAPP-770 mutation responsible for probable early-onset Alzheimer's disease. Proc Natl Acad Sci U S A. 1999Mar30;96(7):4119-24
36. Chartier-Harlin MC, Crawford F, Houlden H, Warren A, Hughes D, Fidani L. et al. Early-onset Alzheimer's disease caused by mutations at codon 717 of the beta-amyloid precursor protein gene. Nature. 1991Oct31;353(6347):844-6
37. van Leeuwen FW, de Kleijn DP, van den Hurk HH, Neubauer A, Sonnemans MA, Sluijs JA. et al. Frameshift mutants of beta amyloid precursor protein and ubiquitin-B in Alzheimer's and Down patients. Science. 1998Jan9;279(5348):242-7
38. Eiserich JP, Baldus S, Brennan ML, Ma W, Zhang C, Tousson A. et al. Myeloperoxidase, a leukocyte-derived vascular NO oxidase. Science. 2002Jun28;296(5577):2391-4
39. Van Allen MW, Frohlich JA, Davis JR. Inherited predisposition to generalized amyloidosis. Clinical and pathological study of a family with neuropathy, nephropathy, and peptic ulcer. Neurology. 1969Jan;19(1):10-25
40. Reynolds WF, Hiltunen M, Pirskanen M, Mannermaa A, Helisalmi S, Lehtovirta M. et al. MPO and APOEepsilon4 polymorphisms interact to increase risk for AD in Finnish males. Neurology. 2000Nov14;55(9):1284-90
41. Chen W, Ji J, Ru B. Proteomic analysis of corticobasal degeneration: a case study of corticobasal degeneration at the proteome level. J Neuropsychiatry Clin Neurosci. 2005;17(3):364-71
42. Rauch SL, Shin LM, Phelps EA. Neurocircuitry models of posttraumatic stress disorder and extinction: human neuroimaging research--past, present, and future. Biol Psychiatry. 2006Aug15;60(4):376-82
43. Bremner JD. Functional neuroanatomical correlates of traumatic stress revisited 7 years later, this time with data. Psychopharmacol Bull. 2003;37(2):6-25
44. Zimmermann M, Gardoni F, Di LM. Molecular rationale for the pharmacological treatment of Alzheimer's disease. Drugs Aging. 2005;22(Suppl 1):27-37
45. Yildiz A, Guleryuz S, Ankerst DP, Ongur D, Renshaw PF. Protein kinase C inhibition in the treatment of mania: a double-blind, placebo-controlled trial of tamoxifen. Arch Gen Psychiatry. 2008Mar;65(3):255-63
46. Latchoumycandane C, Anantharam V, Kitazawa M, Yang Y, Kanthasamy A, Kanthasamy AG. Protein kinase Cdelta is a key downstream mediator of manganese-induced apoptosis in dopaminergic neuronal cells. J Pharmacol Exp Ther. 2005Apr;313(1):46-55
47. Beaulieu JM, Sotnikova TD, Marion S, Lefkowitz RJ, Gainetdinov RR, Caron MG. An Akt/beta-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell. 2005Jul29;122(2):261-73
48. Conti B, Sugama S, Lucero J, Winsky-Sommerer R, Wirz SA, Maher P. et al. Uncoupling protein 2 protects dopaminergic neurons from acute 1,2,3,6-methyl-phenyl-tetrahydropyridine toxicity. J Neurochem. 2005Apr;93(2):493-501
Author contact
![]() Correspondence to: Yan A. Su, MD and PhD, Ross Hall, Room 555, 2300 I St., NW, Washington, DC 20037; Tel: 202-994-1891; Fax: 202-994-8974; Email: bcmyasedu
Correspondence to: Yan A. Su, MD and PhD, Ross Hall, Room 555, 2300 I St., NW, Washington, DC 20037; Tel: 202-994-1891; Fax: 202-994-8974; Email: bcmyasedu