Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2008; 4(5):259-269. doi:10.7150/ijbs.4.259 This issue Cite
Research Paper
Aberrant c-erbB2 expression in cell clusters overlying focally disrupted breast myoepithelial cell layers: a trigger or sign for emergence of more aggressive cell clones?
Xichen Zhang1, Shahreyar Shar Hashemi2, Morvarid Yousefi2, Jinsong Ni3, Qiuyue Wang1, Ling Gao1, Pengtao Gong1, Chunling Gao4, Joy Sheng5, Jeffrey Mason6, Yan-gao Man6 ![]()
1. College of Animal Science and Veterinary Medicine, Jilin University, Changchun, Jilin, China
2. Departments of Surgery and Internal Medicine, Staten Island University Hospital, NY, USA
3. Norman Bethune College of Medical Science, Jilin University, Changchun, Jilin, China
4. Division of Monoclonal Antibodies, Food and Drug Administration, Bethesda, MD, USA
5. Real-time PCR Technical Support Department, Applied Biosystems, Foster City, CA, USA
6. Armed Forces Institute of Pathology and American Registry of Pathology, Washington, DC, USA
Received 2008-7-9; Accepted 2008-8-7; Published 2008-8-16
Abstract
Our recent studies revealed that cell clusters overlying focal myoepithelial cell layer disruption (FMCLD) had a significantly higher frequency of genetic instabilities and expression of invasion-related genes than their adjacent counterparts within the same duct. Our current study attempted to assess whether these cell clusters would also have elevated c-erbB2 expression. Human breast tumors (n=50) with a high frequency of FMCLD were analyzed with double immunohistochemistry, real-time RT-PCR, and chromogenic in situ hybridization for c-erbB2 protein and gene expression. Of 448 FMCLD detected, 404 (90.2%) were associated with cell clusters that had intense c-erbB2 immunoreactivities primarily in their cytoplasm, in contrast to their adjacent counterparts within the same duct, which had no or barely detectable c-erbB2 expression. These c-erbB2 positive cells were arranged as tongue-like projections, “puncturing” into the stroma, and about 20% of them were in direct continuity with tube-like structures that resembled blood vessels. Aberrant c-erbB2 expression was also seen in clusters of architecturally normal-appearing ducts that had distinct cytological abnormalities in both ME and epithelial cells, whereas not in their clear-cut normal counterparts. Molecular assays detected markedly higher c-erbB2 mRNA and gene amplification in cell clusters associated with FMCLD than in those associated with non-disrupted ME cell layers. Our findings suggest that cell clusters overlying FMCLD may represent the precursors of pending invasive lesions, and that aberrant cerbB2 expression may trigger or signify the emergence of biologically more aggressive cell clones.
Keywords: Tumor invasion, myoepithelial cells, c-erbB2, tumor stem cells, monoclonal proliferation
Introduction
The epithelium of normal and pre-invasive breast tumors is physically separated from the stroma by both the myoepithelial (ME) cells and the basement membrane. ME cells are joined by intercellular junctions and adhesion molecules, constituting a largely continuous sheet encircling the epithelium [1]. The basement membrane is composed of mainly type IV collagen and laminins, forming a continuous lining surrounding and attaching to the ME cell layer [2]. Because of these structural relationships, degradation of both the ME cell layer and the basement membrane is a pre-requisite for tumor invasion.
Breast carcinogenesis is believed to be a multistage process, progressing sequentially from normal to hyperplastic, to in situ, and to invasive stages [3]. Progression from an in situ to the invasive stage is believed to be triggered primarily by the overproduction of proteolytic enzymes by cancer cells, which cause degradation of the basement membrane [4]. These theories are consistent with data derived from studies in cell cultures or animal models [5], whereas they are hard to reconcile with three facts. First, a subset of morphologically normal breast tissues shares a similar immunohistochemical or genetic profile with their malignant counterparts [6-9]. Second, a vast majority of in situ tumors express high levels of proteolytic enzymes, but only 10-30% of the untreated in situ tumors progress to invasive lesions during patients' lifetime [10,11]. Third, results from recent worldwide clinical trials of treating and preventing cancer invasion with a wide variety of proteolytic enzyme inhibitors have been very disappointing [12].
These facts argue that alternative mechanisms may exist for breast tumor invasion in some cases. As the diagnosis of in situ breast tumors has increased by almost 10-fold in the US and Europe during the last 20 years, and over 95% of cancer-related death result from invasion-related illness [13,14], there is an urgent need to identify the intrinsic mechanism of invasion and the specific precursor of invasive lesions.
Promoted by the fact that the ME cell layer is the sole source of several tumor suppressors [15-17], and that ME cell degradation is the most distinct morphological sign of invasive breast lesions [1,2], our recent studies have attempted to identify the early signs of ME cell degradation.
Our initial studies examined the physical integrity of ME cell layers in 220 patients with estrogen receptor (ER) positive ductal carcinoma in situ (DCIS). Of 5,698 ducts examined, 405 were found to harbor focal disruptions (the absence of ME cells resulting in a gap greater than the combined size of at least 3-epithelial cells) in surrounding ME cell layers. Compared to adjacent counterparts within the same duct, but distant from the disruption, cell clusters overlying focal ME cell layer disruptions (FMCLD) had a significantly higher frequency of ER negativity, proliferation, genetic instabilities, expression of tumor invasion-related genes, and aberrant expression of cellular adhesion molecules [18-26].
Together, these findings suggest that FMCLD may represent an early sign of ME cell degradation, and that cell clusters overlying FMCLD may represent a biologically more aggressive clone, which is at higher risk for invasion. Since c-erbB2 is a well documented onco-protein closely associated with tumor aggressiveness and invasiveness [27-29], our current study attempted to assess whether c-erbB2 would be exclusively or preferentially expressed in cell clusters overlying FMCLD.
Materials and Methods
Formalin-fixed, paraffin-embedded breast tissue blocks from 40 females with co-existing normal, hyperplastic, and DCIS components in all cases, and 10 females with additional infiltrating ductal carcinoma (IDC) were used in this study. All the malignant components were grade I or II, and stage I or II lesions. These cases were selected from our previous studies of over 500 DCIS and IDC cases [18-26]. All the selected cases harbored a high frequency of FMCLD, based on our previous immunohistochemical studies [18-26].
Consecutive sections at 5-7μm thickness were cut and placed on positively charged slides. The first and last sections were H&E stained for morphological classification using our published criteria [30]. Sections were subjected to double immunohistochemistry with mouse monoclonal antibodies against the external domain of c-erbB2 (Novocastra, Newcastle, UK) and smooth muscle actin (Vector, Burlingame, CA, USA), a ME cell phenotypic protein using our published protocols [31-32].
To assess the specificity of c-erbB2 immunostaining, three approached were used. First, different controls were included: (1) substitution of the primary antibody with normal serum; (2) omission of the secondary antibody from the immunostaining sequence; (3) serial dilutions of the primary antibody; (4) inclusion of sections from normal lymph-nodes. Second, the same immunostaining protocol was used for the same cases, while substituted with epitopically similar antibodies from different manufacturers (Lab Vision, Fremont, CA, USA and Dako (Glostrup, Denmark). Third, both the avidin-biotin-peroxidase and avidin-biotin-alkaline phosphatase detection kits (Vector, Burlingame, CA, USA) were used to assess the expression of c-erbB2 in consecutive sections of the same cases. Imunostaining was repeated at least twice. Immunostained sections were independently evaluated by at least two investigators, to determine the overall expression status of c-erbB2 in different tissue components, and to count the actual number of c-erbB2 positive cells overlying each FMBLC. The frequency of c-erbB2 expression in morphologically similar ducts (the same histological type and grade, and similar size and shape) with and without FMCLD was statistically compared with the Pearson's Chi-squared test. A cell was considered c-erbB2 positive, if intense (at least ++) immunoreactivities were seen in its membrane or cytoplasm, whereas all the controls were devoid of immunoreactivities.
To correlate c-erbB2 protein expression with c-erbB2 mRNA levels, three cases with multiple large c-erbB2 positive cell clusters (≥100 cells/cluster) overlying FMCLD were selected. A total of 50 consecutive sections at 10-12µm thickness were made from each case. Sections 1, 11, 21, 31, and 41 were subjected to immunostaining as described above. The remaining sections were lightly stained with hematoxylin. Using immunostained sections as references, c-erbB2-positive cells overlying FMCLD and adjacent negative cells within the same duct, but distant from the disruption, were microdissected under a standard microscope, as previously described [21].
Microdissected cells were subjected to RNA extraction with Trizol (Invitrogen, Carlsbad, CA, USA) using the protocol provided by the manufacturer. RNA concentrations were measured with a model 2100 Bioanalyzer (Agilent, Santa Clara, CA, USA) using the manufacturer's Pico-RNA chip. RNA extracts were incubated with DNase for 15 minutes at room temperature before RT-PCR. For each RNA extract, 10 µl of the RNA were reversely transcribed into cDNA using random hexamer primers and omniscript reverse transcriptase (Qiagen Technology, Germantown, MD, USA) in a 20μl volume.
The cDNAs were subjected to real-time PCR. TaqMan MGB probes (5'-FAM dye-labeled) and primers were designed using the Applied Biosystems' Primer Express 3.0 (Foster City, CA, USA). The sequences (Table 1) of primers and probes were checked for specificity by searching the NCBI BLAST database. All primers and probes are obtained from Applied Biosystems. These assays were designed in such a way that the amplified RNA sequences span over exon-exon junctions, consequently the sequence in the genome is undetectable. Real-time PCR was carried out using an ABI 7500 fast Sequence Detection System. Singleplex real-time PCR reactions were performed using 10ng RNA from paired samples, and the TaqMan® Fast Universal PCR Master Mix (Applied Biosystems, Foster City, CA, USA). A thermal cycling profile, consisting of an initial DNA polymerase activation for 20 seconds at 95 ºC followed by 40 cycles of PCR (95 ºC for 3 seconds, 60 ºC for 30 seconds) was used for all analyses. Changes in the gene expression levels were calculated using the ∆∆Ct method [33]. Each datum set was analyzed in triplicate, and was normalized by using beta-2-microglobulin as a run standard.
Primer and probe sequences used in the assays
| Part number | Name | Sequence | Tm (ºC) | GC% |
|---|---|---|---|---|
| 4304970 | HER2-F | CCTACCTGCCCACCAATGC | 59.9 | 63 |
| 4304970 | HER2-R | GTTGTGAGCGATGAGCACGTA | 58.5 | 52 |
| 4316034 | HER2-P | FAM-CCTGCAGGATATCCA-MGB | 69.0 | 53 |
| 4304970 | β2M-246F | TGACTTTGTCACAGCCCAAGATA | 58.4 | 43 |
| 4304970 | β2M-330R | AATCCAAATGCGGCATATTC | 57.2 | 40 |
| 450025 | β2M-275P | VIC-TGATGCTGCTTACATGTCTC GATCCCA-TAMRA | 68.2 | 48 |
To correlate c-erbB2 protein expression with c-erbB2 gene amplification, tissue sections from 10 cases (6 DCIS and 4 IDC) with elevated c-erbB2 protein expression were subjected to chromogenic in situ hybridization (CISH) with a c-erbB2 DNA probe, a tissue pretreatment kit, and a polymer detection kit from Zymed Laboratories, Inc (South San Francisco, CA, USA), following the protocol provided by the manufacturer. The interpretation of c-erbB2 status was based on a previously published criterion [34]. Using immunohistochemically stained sections as references, cell clusters overlying FMCLD and their adjacent counterparts within the same duct were identified in CISH stained sections. Pictures in these locations were taken under high magnification, and enlarged color prints were made. The number of dots in at least 60 nuclei/per location in enlarged color printers were then counted and compared.
Results
Distinct typical membranous c-erbB2 expression was detected in six DCIS and four IDC. The number of c-erbB2 positive cells in these cases accounted for about 30-50% of the cancer cell population. The stromal tissues and negative controls were devoid of c-erbB2 expression. Antibodies from different companies and different detection kits yielded similar results (not shown).
Each of the 50 cases contained multiple (5 to over 20) ducts or acini with FMCLD, and a total of 448 FMCLD were identified. The size of FMCLD varied substantially, ranging from a few cells to over 50% of the entire ME cell layer. The size of the cell clusters overlying FMCLD also varied substantially, ranging from as few as 10 to more than 300 cells. Of the 448 FMCLD, 404 (90.2%) were associated with cell clusters that had intense c-erbB2 immunoreactivities primarily in cytoplasm. Of 448 morphologically similar ducts with non-disrupted ME cell layers, only 38 (0.85%) showed distinct cytoplasmic c-erbB2 expression (Table 2).
Comparison of aberrant c-erbB2 expression in ducts with and without FMCLD
| Duct type | Total number | With c-erbB2 expression | p |
|---|---|---|---|
| With FMCLD | 448 | 404 (90.2%) | < 0.01 |
| W/o FMCLDD | 448 | 38 (0.85%) |
The distribution of c-erbB2 positive malignant or hyperplastic cells associated with FMCLD could be roughly classified into two main categories:
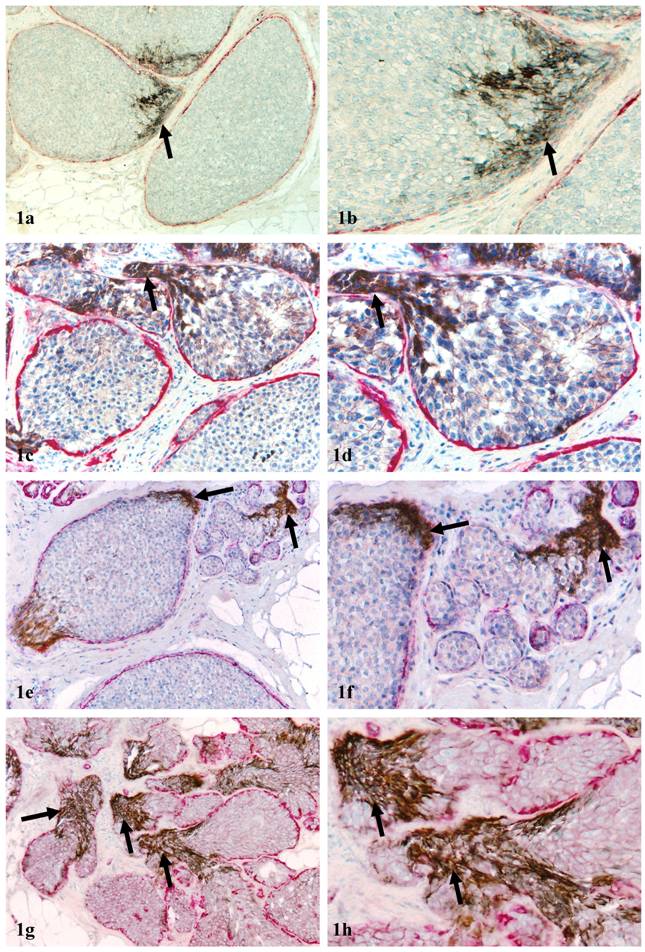
1. Individual duct or small duct clusters: About 70% of c-erbB2 positive cell clusters were distributed in the individual duct or small duct clusters. Under low magnification, a vast majority of c-erbB2 positive clusters were indistinguishable from adjacent counterparts within the same or different ducts. Under high magnification, these c-erbb2 positive cells differed noticeably from their adjacent counterparts within the same duct in size, shape, density, and polarity. A vast majority of the cells overlying FMCLD, however, were morphologically similar with a spindle or elongated shape, and were commonly arranged as tongue-like projections “puncturing” into the stroma (Fig 1).
Aberrant c-erbB2 expression in isolated ducts. Human breast tissue sections were double immunostained for c-erbB2 (brown or black) and smooth muscle actin (red). Arrows identify cell clusters with aberrant c-erbB2 expression overlying FMCLD in DCIS (1a-1f) and hyperplastic ducts (1g-1h). a, c, e, and g: 100X; b, d, f, and h: a higher magnification (300X) of a, c, e, and g, respectively.
2. Tube-like structures that resemble blood vessels: About 20% of c-erbB2 positive cell clusters were in direct continuity with tube-like structures resembling blood vessels (Fig 2). These tube-like structures had a poorly defined “wall” directly connected with the ME cell layer. The “wall”, however, differed from the ME cell layer in four main aspects: (1) it is thinner and elongated; (2) the lumen of these “walls” often contained red blood cells (Figs 2e-2f); [3] it is weakly positive for smooth muscle actin, whereas is totally negative for p63, a nuclear ME cell phenotypic marker (Fig 2g-2h); (4) it often shows distinct immunoreactivities to CD31, a endothelial cell specific marker. The frequency and pattern of CD31 immunoreactivities in these tube-like structures, however, were not consistent (not shown).
Aberrant c-erbB2 expression in vessel-like structures. Sections a-f were double immunostained for c-erbB2 (brown or black) and smooth muscle actin (red). Sections g & h were double immunostained for smooth muscle actin (red) and p63 (nuclear black stain). Circles identify c-erbB2 positive cell clusters and associated vessel-like structures. Thick arrows identify residual ME cell layers. Thin arrow identify ME cells with p63 expression. a, c, e, and g: 100X; b, d, f, and h: a higher magnification (300X) of a, c, e, and g, respectively.
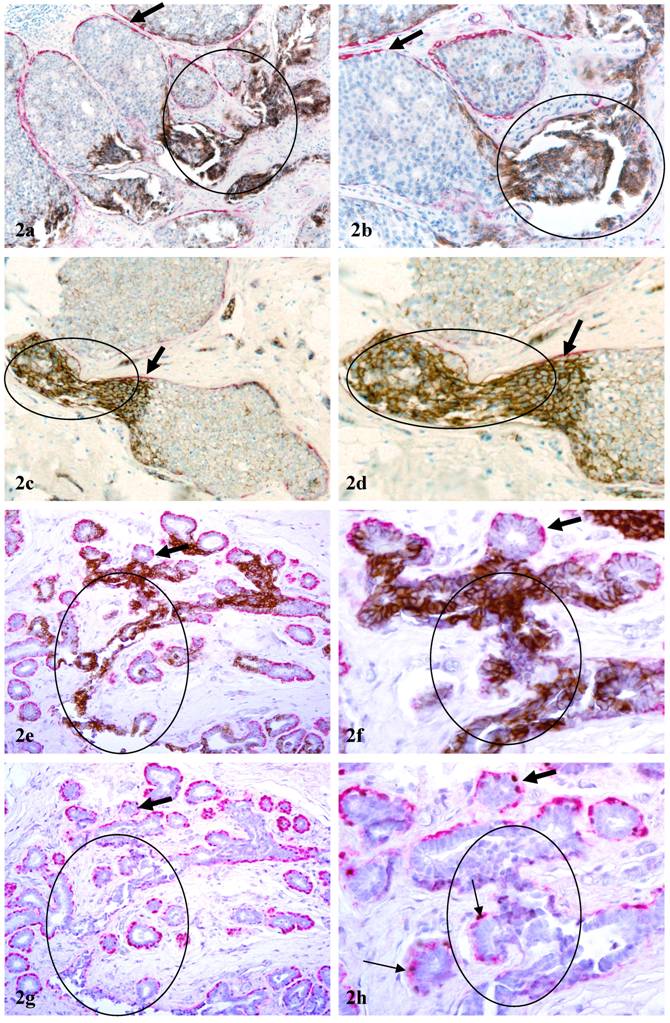
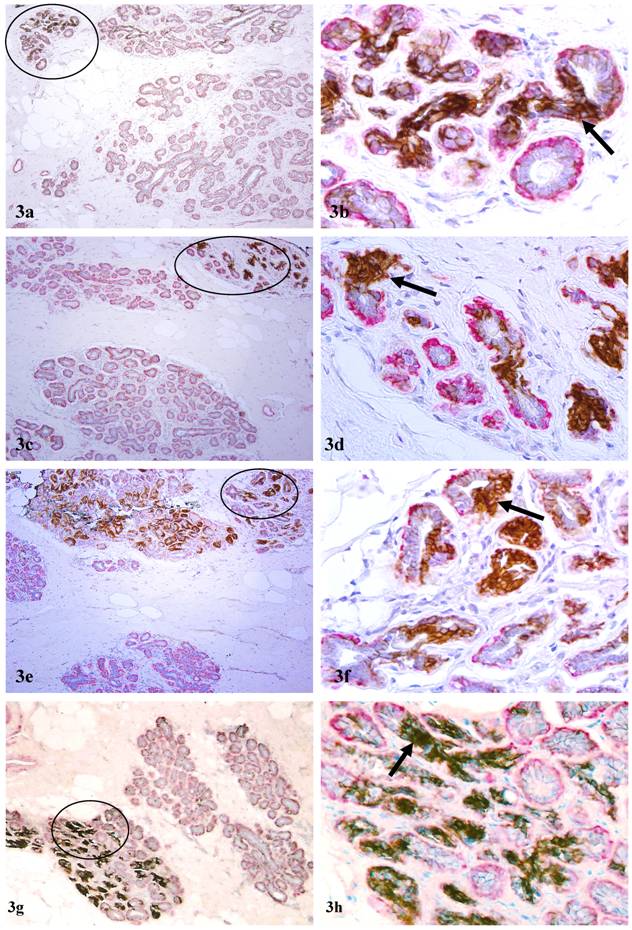
In addition to DCIS and hyperplastic lesions, elevated expression of c-erbB2 was detected in large clusters of morphologically normal-appearing ducts in 7 cases. The numbers of ducts per cluster ranged from 10 to over 50 (Fig 3). Under low magnification, these duct clusters were architecturally comparable to their adjacent normal counterparts. Under high magnification, the ME cell layers in a vast majority of these ducts were attenuated, discontinuous, or totally absent. The epithelial cells of these normal-appearing duct clusters often displayed distinct cytological abnormalities, including enlarged and irregular-shaped nuclei, increased nuclear-cytoplasmic ratio, and substantially enlarged nucleoli, similar to those described in our previous report [8]. Similar to c-erbB2 positive malignant and hyperplastic cells, these c-erbB2 positive normal-appearing cells were also arranged as tongue-like projections “puncturing” deep into the stroma (Fig 3). These normal appearing duct clusters had a distinct boundary with adjacent counterparts that lacked c-erbB2 expression and had non-disrupted ME cell layers (Fig 3).
Aberrant c-erbB2 expression in normal-appearing duct clusters. Human breast tissue sections were double immunostained for c-erbB2 (brown or black) and smooth muscle actin (red). Arrows identify examples of ducts with aberrant c-erbB2 expression. Note that a majority of the ducts in some duct clusters are strongly positive for c-erbB2, whereas all ducts in other duct clusters are devoid of c-erbB2 expression. a, c, e, and g: 100X; b, d, f, and h: a higher magnification (300X) of a, c, e, and g, respectively.
In both pre-invasive and normal appearing ducts, cell clusters overlying FMCLD had a markedly higher expression of c-erbB2, compared to their adjacent counterparts within the same duct, but distant from the disruption. In all cell clusters overlying FMCLD, c-erbB2 expression was predominantly seen in the cytoplasm, despite the presence of typical cell membrane distribution in adjacent cells within the same duct but distant from the disruption (Figs.1c-1d, 2c-2d).
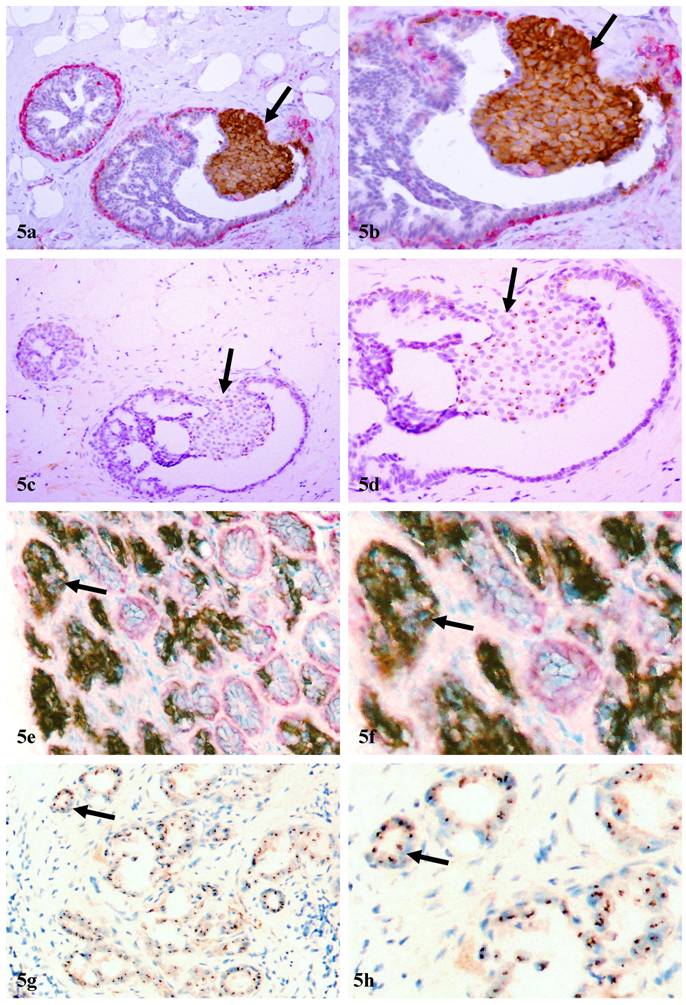
In cases selected for real-time PCR analysis, c-erb2 mRNA expression in cell clusters overlying FMCLD was consistently higher than that of adjacent counterpart within the same duct, but distant from the disruption (Fig.4; Table 3). In 10 cases selected for CISH, a total of 30 small and large cell clusters were found to have elevated atypical c-erbB2 protein expression. Of these, 23 (77%) had high levels of c-erbB2 gene amplification (Figs 5a-5d). High levels of c-erbB2 gene amplification were also seen in normal-appearing ducts (Figs 5e-5h). The c-erbB2 amplification signals were predominantly presented as large dot clusters (Fig 5).
Cell clusters dissected for real-time PCR and the PCR results of a selected case (case 2). Human breast tissue sections were double immunostained for c-erbB2 (brown or black) and smooth muscle actin (red). Thick arrows identify c-erbB2 positive cell clusters overlying FMCLD. Thin arrows identify residual ME cell layers. Curves show c-erbB2 mRNA levels between cell clusters overlying FMCLD (2b) and adjacent counterparts (2a) within the same duct. Each datum set was analyzed in triplicate.
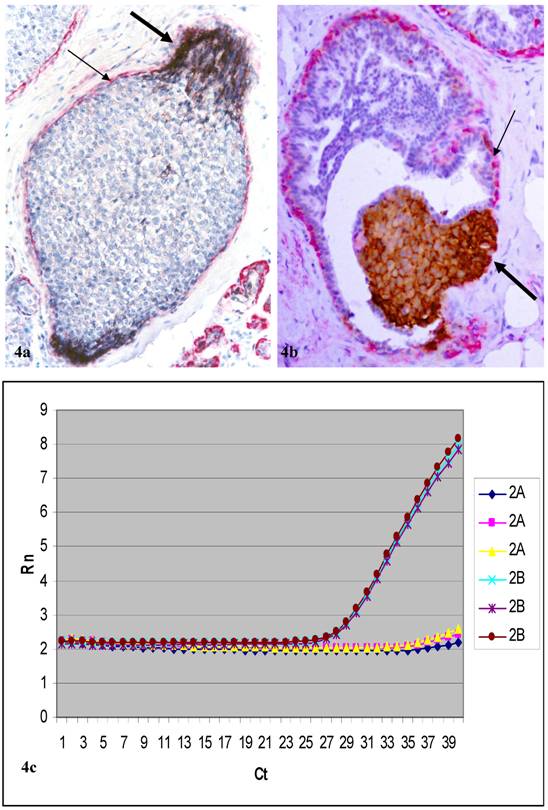
Real-time PCR outcomes
| Sample | Average Ct of target gene | Average Ct of control gene | ∆Ct | ∆∆Ct | Fold of change |
|---|---|---|---|---|---|
| 1A | Undetermined* | 35.78 | |||
| 1B | 31.12 | 35.55 | |||
| 2A | 37.36 | 35.27 | 2.09 | ||
| 2B | 27.56 | 35.03 | -7.74 | -9.83 | 910 |
| 3A | Undetermined* | 36.12 | |||
| 3B | 36.25 | 36.78 |
C-erbB2 amplification in cell clusters overlying FMCLD and in normal appearing ducts. Two sets (a-d and e-h) of adjacent sections were subjected to double immunohistochemical staining (a-b & e-f) and CISH (c-d & g-h). Arrows identify cell clusters with aberrant c-erbB2 protein expression and high level of c-erbB2 gene amplification in DCIS (a-d) and normal appearing ducts (e-h). a, c, e, and g:100X; b, d, f, and h: a higher magnification (300X) of a, c, e, and g, respectively.
Discussion
Our current study detected significantly higher c-erbB2 protein expression and gene amplification in malignant and hyperplastic cell clusters overlying FMCLD than that in adjacent cells within the same duct, but distant from the disruption, and also in morphologically normal-appearing duct clusters with distinct cytological abnormalities in their surrounding ME cell layers and epithelial cells. To our best knowledge, these findings have not been previously reported. The main reasons appear to be: (1) these clusters are indistinguishable from their clear-cut normal counterparts under low magnification of H&E sections, which allow them to escape from detection, (2) the size of these clusters are relatively small and could be elucidated only by double immunohistochemistry, which has not been widely used in clinical samples, and (3) past studies has been focused primarily on the membrane expression of c-erbB2.
Aberrant c-erbB2 expression in cell clusters overlying FMCLD is likely to result from or to signify the emergence of biologically more aggressive cell clones for three reasons. First, c-erbB2 expression is absolutely required for breast morphogenesis and angiogenesis [35]. The absence of c-erbB2 expression could result in a number of structural defects, including a significant reduction of the epithelial cells in the terminal duct-lobular units [36]. The c-erbB2 and c-met signaling pathways are also required for the branching and alveolar morphogenesis, by regulating the breakdown of intercellular junctions and shift of the sub-cellular localization of the cell adhesion molecules, so that epithelial cells could migrate to new destinations to form new structures [37-38]. Second, as the epithelium is normally devoid of vascular structures and the ME cell layer is the sole source of several tumor suppressors, a FMCLD could result in several focal alterations: (1) a loss of tumor suppressors and paracrine inhibitory functions, which allow epithelial cells to escape from programmed cell death, (2) alterations in permeability for oxygen or growth factors, which selectively favor the exit of stem cells from quiescence and monoclonal proliferation of the progenitor cells, and (3) direct exposure of the epithelial cells to stromal and immunoreactive cells, which stimulates tumor angiogenesis and epithelial-mesenchymal transition [39-41]. The above alterations may individually or collectively induce the emergence of biologically more aggressive cell clones and aberrant c-erbB2 expression. Consistent with this possibility is the fact that c-erbB2 expression is inducible by a number of factors, including ultraviolet irradiation, hormonal receptors, or developmental stages [43-44]. Third, our recent studies have consistently shown that cell clusters overlying FMCLD are biologically more aggressive, as they have a significantly higher frequency of ER negativity, genetic instabilities, and expression of tumor invasion-related genes than adjacent cells within the same duct [16-26]. Our findings are in agreement with that of a previous study, which shows that microdissected cells from the periphery of DCIS have significantly higher gene expression than their counterparts in the center of the same DCIS, assessed with Atlas human Cancer 1.2 Arrays, which contain 1176 known cancer-related genes [42].
Aberrant c-erbB2 expression in normal appearing duct clusters is likely to result from two factors. First, these clusters may present a population of maturation arrested progenitors derived from monoclonal proliferation of genetically damaged primitive stem cells at the early stages of ductal morphogenesis by trauma, radiation, or other factors [45]. These clusters may retain the potential for unlimited proliferation and multi-lineage differentiation by recapitulation of the process of duct branching and morphogenesis. Since duct branching and tumor invasion share a very similar molecular mechanism [36-38], aberrant c-erbB2 expression in these normal appearing duct clusters is likely to be associated with, or reflect these events. Second, these clusters may represent a previously uncharacterized malignant lesion with genetic defects in the c-erbB2 gene. In agreement with this possibility is the fact that “clinging ductal carcinoma in situ” is morphologically indistinguishable to normal ducts under low magnification of H&E stained sections, but microdissected cells from this lesion share the same genetic instabilities and the same monoclonality with invasive cancers [6, 46]. This possibility is further supported by the fact that recent studies have identified a DNA phenotype, which is identical to that of invasive prostate cancer in certain healthy men and also in morphologically normal tissues adjacent to prostate cancer [47-49]. Previous studies have shown that a point mutation in the transmembrane domain could alter the protein's helical structure and it's insertion into the cell membrane [50]. Mutations also alter the subcellular localization of coded proteins and the biological functions of the c-erbB2 protein [51-52].
The scientific and clinical significance of cytoplasmic c-erbB2 expression has been a subject of debate. Based on data derived from immunohistochemical staining and in situ hybridization, Taylor et al stated that “cytoplasmic staining for c-erbB2 is an immunocytochemical artifact” with no significant implication [53, 54]. In sharp contrast, a number of studies have shown that cytoplasmic expression of c-erbB2 is significantly associated with worse prognosis in breast, endometrial, and other carcinomas [55,56]. Our findings differ from those of the above reports in two main aspects: (1) cytoplasmic c-erbB2 expression is exclusively or preferentially associated with cells overlying FMCLD, and (2) cells with cytoplasmic c-erbB2 expression also show appreciable membranous c-erbB2 expression.
The scientific and clinical significance of our findings cannot be determined at present, since the sample size of our study is small, and the clinical follow-up data are not available. On the other hand, it is clear that cell clusters overlying FMCLD have significantly higher aberrant c-erbB2 expression than their adjacent counterparts within the same duct. Coupled with a significantly higher rate of ER negativity, genetic instabilities, and expression of tumor invasion related genes [18-26], it is very likely that aberrant c-erbB2 expression in these cell clusters is associated with, or signifies the emergence of biologically more aggressive cell clones. Consequently, patients with DCIS that have aberrant c-erbB2 expression in cell clusters overlying FMCLD are very likely to represent the specific individual who will develop, or at greater risk to develop, invasive breast lesions. If confirmed by studies in more cases and by clinical follow-up data, our findings and technical approaches may have significant value in predication of tumor progression or invasion, and in selection of patients for optimal treatment.
Acknowledgements
This study was supported in part by grant 2006CB910505 from the Ministry of Chinese Science and Technology Department to Drs. Xichen Zhang and Yan-gao Man, and also by grants DAMD17-01-1-0129, AMD17-01-1-0130, and PC051308 from the US Congressionally Directed Medical Research Programs, grant BCTR0706983 from The Susan G. Komen Breast Cancer Foundation, and grant 05AA from the AFIP/ARP joint research initiative project to Dr. Yan-gao Man.
The opinions and assertions contained herein represent the personal views of the authors and are not to be construed as official or as representing the views of the Department of the Army or the Department of Defense.
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
1. Tsubura A, Shikata N, Inui T. et al. Immunohistochemical localization of myoepithelial cells and basement membrane in normal, benign and malignant human breast lesions. Virch Arch. 1998;413:133-9
2. Guelstein VI, Tchypysheva TA, Ermilova VD. et al. Myoepithelial and basement membrane antigens in benign and malignant human breast tumors. Int J Cancer. 1993;53:269-77
3. Clarke R, Brunner N, Katzenellenbogen BS. Progression of human breast cancer cells from hormone- dependent to hormone-independent growth both in vitro and in vivo. Proc Natl Acad Sci USA. 1989;86:3649-53
4. Goldfarb RH, Liotta LA. Proteolytic enzymes in cancer invasion and metastasis. Semin Thromb Hemost. 1986;12:294-307
5. Doffy MJ, Maguire TM, Hill A. et al. Metalloproteinases: roles in breast carcinogenesis and metastasis. Breast Cancer Res. 2000:252-7
6. Moinfar F, Man YG, Bratthauer GL. et al. Genetic abnormalities in mammary ductal intraepithelial neoplasia-flat type (Clinging ductal carcinoma in situ)-A simulator of normal mammary epithelium. Cancer. 2000;88:2072-81
7. Deng G, Liu Y, Ziotnikov G. et al. Loss of heterogosity in normal tissue adjacent to breast carcinomas. Science. 1996;274(5295):2057-9
8. Man YG, Nieburgs HE. A subset of cell clusters with malignant features in morphologically normal and hyperplastic breast tissues. Cancer Detect Prev. 2006;30(3):239-47
9. Man YG. Bad seeds produce bad crops:a single step-process of breast carcinogenesis and progression. Bioscience Hypotheses. In press
10. Page DL, Duppont WD, Rogers LW, Landenberger M. Intraductal carcinoma of the breast: follow up after biopsy only. Cancer. 1982;49:751-8
11. Page DL, Dupont WD, Rogers LW, Jensen RA, Schuyler PA. Continues local recurrence of carcinoma 15-25 years after a diagnosis of low grade ductal carcinoma in situ of the breast treated only by biopsy. Cancer. 1995;76:1197-2000
12. Coussens LM, Fingleton B, Matrisian LM. Matrix metalloproteinase inhibitors and cancer: trial and tribulations. Science. 2002;295(5564):2387-92
13. Cancer Trends Progress Report - 2005 Update. National Cancer Institute, NIH, DHHS: Bethesda, MD. http://progressreport.cancer.gov
14. Leonard GD, Swain SM. Ductal carcinoma in situ, complexities and challenges. J Nat Cancer Inst. 2004;96:906-20
15. Zou Z, Anisowicz A, Hendrix MJ. et al. Maspin, a serpin with tumor-suppressing activity in human mammary epithelial cells. Science. 1994;263:526-9
16. Barbareschi M, Pecciarini L, Cangi MG. et al. p63, a p53 homologue, is a selective nuclear marker of myoepithelial cells of the human breast. Am J Surg Pathol. 2001;25:1954-60
17. Sternlight MD, Barsky SH. The myoepithelial defense: a host defense against cancer. Med Hypotheses. 1997;48:37- 46
18. Zhang R, Man YG, Vang RS. et al. A subset of morphologically distinct mammary myoepithelial cells lacks corresponding immunophenotypic markers. Breast Cancer Res. 2003;5:R151-6
19. Man YG, Tai L, Barner R, Vang R. et al. Cell clusters overlying focally disrupted mammary myoepithelial cell layers and adjacent cells within the same duct display different immunohistochemical and genetic features: implications for tumor progression and invasion. Breast Cancer Res. 2003;5:R231-41
20. Man YG, Sang QXA. The significance of focal myoepitehlial cell layer disruptions in breast tumor invasion: a paradigm shift from the “protease-centered” hypothesis. Exp Cell Res. 2004;301:103-18
21. Man YG, Zhang Y, Shen T. et al. cDNA expression profiling identifies elevated expressions of tumor progression and invasion related genes in cell clusters of in situ breast tumors. Breast Cancer Res Treat. 2005;89:199-208
22. Yousefi M, Mattu R, Gao C, Man YG. Mammary ducts with and without focal myoepithelial cell layer disruptions show a different frequency of white blood cell infiltration and growth pattern: Implications for tumor progression and invasion. Appl Immunohistochem Mol Morphol. 2005;13:30-7
23. Man YG, Shen T, Weisz J. et al. A subset of in situ breast tumor cell clusters lacks expression of proliferation and progression related markers but shows signs of stromal and vascular invasion. Cancer Detect Prev. 2005;29:323-31
24. Man YG, Zhao CQ, Wang J. Breast tumor cell clusters and their budding derivatives show different immunohistochemical profiles during stromal invasion: implications for hormonal and drug therapies. Cancer Therapy. 2006;4:193-204
25. Man YG. Focal degeneration of aged or injured myoepithelial cells and the resultant auto- immunoreactions are trigger factors for breast tumor invasion. Medical Hypotheses. 2007;69(6):1340-57
26. Zhang XC, Hashemi SS, Yousefi M. et al. Atypical expression of e-cadherin in cell clusters overlying focally disrupted mammary myoepithelial cell layers: implications for tumor cell motility and invasion. Cancer Biomarkers. Pathol-Res Pract, conditionally. accepted
27. Yarden Y. Biology of HER2 and its importance in breast cancer. Oncology. 2001;61(Suppl 2):1-13
28. Cooke T, Reeves J, Lanigan A, Stanton O. HER2 as a prognostic and predicative marker for breast cancer. Ann Oncol. 2001;12(Suppl 2):S23-8
29. Menard S, Casalini P, Campiglio M. et al. HER2 overexpression in various tumor types, focusing on its relationship to the development of invasive breast cancer. Ann Oncol. 2001;12(Suppl 1):S15-9
30. Tavassoli FA, Man YG. Morphofunctional features of intraductal hyperplasia, atypical hyperplasia, and various grades of intraductal carcinoma. The Breast J. 1995;1(3):155-62
31. Man YG, Tavassoli FA. A simple epitope retrieval method without the use of microwave oven or enzyme digestion. Appl Immunohistochem. 1996;4(2):139-41
32. Man YG, Burgar A. An antigen unmasking protocol that satisfies both immunohistochemical and subsequent molecular biological assessments. Pathol-Res Pract. 2003;199:815-25
33. Bustin SA, Nolan T. Data analysis and interpretation. Busti SA (ed) A-Z of quantitative PCR, pp441-492. International University Line, La Jolla, CA, USA. 2004
34. Tanner M, Gancberg D, Di Leo A. et al. Chromogenic in situ hybridization: a pratical alternative for fluorescence in situ hybridization to detect HER-s/neu oncogene amplification in achival breast cancer samples. Am J Pathol. 2000;157:1467-72
35. Klos KS, Wyszomierski SL, Sun M. et al. c-erbB2 increases vascular endothelial growth factor protein synthesis via activation of mammalian target of rapamycin/p70S6K leading to increased angiogenesis and spontaneous metastasis of human breast cancer cells. Cancer Res. 2006;64:2028-37
36. Jackson-Fisher AJ, Bellinger G, Ramabhadran R. et al. ErbB2 is required for ductal morphogenesis of the mammary gland. Proc Natl Acad Sci USA. 2004;101(49):17138-43
37. Niemann C, Brinkmann V, Spitzer E. et al. Reconstitution of mammary gland development in vitro: requirement of c-met and c-erbB2 signaling for branching and alveolar morphogenesis. J Cell Biol. 1998;143(2):533-45
38. Shyamala G, Chou YC, Cardiff RD, Vargis E. Effect of c-neu/ErbB2 expression levels on estrogen receptor alpha-dependent proliferation in mammary epithelial cells: Implications for breast cancer biology. Cancer Res. 2006;66(21):10391-8
39. Oliveira AM, Ross JS, Fletcher JA. Tumor suppressor genes in breast cancer: the gatekeepers and the caretakers. Am J Clin Pathol. 2005;124:S16-28
40. Chakravarthy MV, Spangenhurg EE, Booth FW. Culture in low levels of oxygen enhances in vitro proliferation potential of satellite cells from old skeletal muscles. Cell Mol Life Sci. 2001;58:1150-8
41. Hendrix MJ, Seftor EA, Hess AR, Seftor RE. Vasculogenic mimicry and tumor-cell plasticity: lessons from melanoma. Nat Rev Cancer. 2003;3(6):411-21
42. Zhu G, Reynolds L, Crnogorac-Jurceic T. et al. Combination of microdissection and microarray analysis to identify gene expression changes between differentially located tumour cells of breast cancer. Oncogene. 2003;22:3742-48
43. Madson JG, Lynch DT, Tinkum KL. et al. Erbb2 regulates inflammation and proliferation in the skin after ultraviolet irradiation. Am J Pathol. 2006;169(4):1402-14
44. Darcy KM, Zangani D, Wohlbueter AL. et al. Changes in ErbB2 (her-2/neu), Erbb3, and ErbB4 during growth, differentiation, and apoptosis of normal rat mammary epithelial cells. J Histochem Cytochem. 2000;48(1):63-8
45. Sell S, Pierce GB. Maturation arrest of stem cell differentiation is a common pathway for the cellular origin of teratocarcinomas and epithelial cancers. Lab Invest. 1994;70:6-22
46. Azzopardi JG. Problems in breast pathology. Landon: WB Saunders. 1979
47. Malins DC, Johnson PM, Barker EA. Cancer-related changes in prostate DNA as men age and early identification of metastasis in prostate tumors. Proc Natl Acad Sci USA. 2003;100:5401-6
48. Malins DC, Anderson KM, Gilman NK. Development of a cancer DNA phenotype prior to tumor formation. Proc Natl Acad Sci Sci USA. 2004;101:10721-5
49. Malins DC, Gilman NK, Green VM. A DNA phenotype in healthy prostates, conserved in tumors and adjacent normal cells, implies a relationship to carcinogenesis. Proc Natl Acad Sci USA. 2005;102:19093-6
50. Soumana SO, Aller P, Garnier N, Genest M. Transmembrane peptides from tyrosine kinase receptor. Mutation-related behavior in a lipid bilayer investigated by molecular dynamics simulations. J Biomol Struct Dyn. 2005;23(1):91-100
51. Garnier N, Genest D, Hebert E, Genest M. Influence of a mutation in the transmembranr domain of the p185c-erbB2 oncogene-encoded protein studied by molecular dynamics simulations. J Biomol Struct Dyn. 1994;11(5):983-1002
52. Weiner DB, Liu J, Cohen JA. et al. A point mutation in the neu oncogene mimics ligand induction of receptor aggregation. Nature. 1989;339(6221):230-1
53. Taylor SL, Platt-Higgins A, Rudland PS, Winstanley JH, Barraclough R. Cytoplasmic staining of c- erbB2 is not associated with the presence of detectable c-erbB2 mRNA in breast specimens. Int J Cancer. 1998;76(4):459-63
54. Taylor SL, Rudland PS, Barraclough R. C-erbN-2 mRNA in breast cancer specimens that exhibit membrane or cytoplasmic immunoreactivity for c-erbB-2. Oncol Res. 1999;11(7):311-7
55. Keshgegian AA, Cnaan A. erbB-2 oncoprotein expression in breast carcinoma. Poor prognosis associated with high degree of cytoplasmic positivity using CB-11 antibody. Am J Clin Pathol. 1997;108(4):456-63
56. Lambropoulou M, Stefanou D, Alexiadis G, Tamiolakis D, Tripsisnis G, Chatzaki E. et al. Cytoplasmic expression of c-erb-B2 in endometrial carcinomas. Onkologie. 2007;30(10):495-500
Author contact
![]() Correspondence to: Yan-gao Man, MD., PhD., Director of Gynecologic and Breast Research Laboratory, Department of Gynecologic and Breast Pathology, Armed Forces Institute of Pathology and American Registry of Pathology, 6825 16th Street, NW, Washington DC 20306-6000. Phone: 202-782-1612; Fax: 202-782-3939; E-mail: manosd.mil
Correspondence to: Yan-gao Man, MD., PhD., Director of Gynecologic and Breast Research Laboratory, Department of Gynecologic and Breast Pathology, Armed Forces Institute of Pathology and American Registry of Pathology, 6825 16th Street, NW, Washington DC 20306-6000. Phone: 202-782-1612; Fax: 202-782-3939; E-mail: manosd.mil