Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2008; 4(5):270-278. doi:10.7150/ijbs.4.270 This issue Cite
Research Paper
Preconditioning of Carbon Monoxide Releasing Molecule-derived CO Attenuates LPS-induced Activation of HUVEC
Bingwei Sun ![]() , Xiangqian Zou, Yueling Chen, Ping Zhang, Gengsheng Shi
, Xiangqian Zou, Yueling Chen, Ping Zhang, Gengsheng Shi
Department of Burn and Plastic Surgery, Affiliated Hospital, Jiangsu University, Zhenjiang 212001, Jiangsu Province, PR China
Received 2008-6-23; Accepted 2008-8-20; Published 2008-8-22
Abstract
Objective: To investigate the effects and potential mechanisms of preconditioning of tricarbonyldichlororuthenium (III) dimer (CORM-2)-liberated CO on LPS-induced activation of endothelial cells (HUVEC).
Methods: HUVEC were pretreated with CORM-2 at the concentration of 50 or 100μM for 2 hrs, washed and stimulated with LPS (10μg/ml) for additional 4 hrs. Activation (oxidative stress) of HUVEC was assessed by measuring intracellular oxidation of DHR 123 or nitration of DAF-FM, specific H2O2 and NO fluorochromes, respectively. The expression of HO-1, iNOS (Western blot) and ICAM-1 (cell ELISA) proteins and activation of inflammation-relevant transcription factor, NF-κB (EMSA) were assessed. In addition, PMN adhesion to HUVEC was also assessed.
Results: The obtained data indicate that pretreatment of HUVEC with CORM-2 results in: 1) decrease of LPS-induced production of ROS and NO; 2) up-regulation of HO-1 but decrease in iNOS at the protein levels; 3) inhibition of LPS-induced activation of NF-κB; and 4) downregulation of expression of ICAM-1, and this was accompanied by a decrease of PMN adhesion to LPS-stimulated HUVEC.
Conclusions: Preconditioning of CO liberated by CORM-2 elicited its anti-inflammatory effects by interfering with the induction of intracellular oxidative stress. In addition, it also supports the notion that CO is a potent inhibitor of iNOS and NF-κB.
Keywords: carbon monoxide, preconditioning, oxidative stress, NF-κB, ICAM-1
Introduction
Endothelial cells (ECs) control many aspects of haemostasis, immunity and inflammatory reactions in addition to its function as a selective barrier between the blood and the tissues. In the inflammation process, the activation of ECs is recognized as an early step that involves the expression of several adhesion molecules [1]. Also, one of the primary cellular targets of the inflammatory cytokines and their modulatory factors is the endothelial cell, whose activation is crucial during inflammatory processes [2]. This activation triggers a change from a non-thrombogenic surface to one in which circulating cells adhere to the apical membrane of endothelial cells [3]. Increased expression of cell adhesion molecules such as ICAM-1, VCAM-1 and E-selectin are characteristic of the altered gene expression pattern of the activated endothelial cell [4,5]. This process in particular leads to thrombogenesis and extravasation of leukocytes into the tissues [6]. It has been shown that endothelial cell activation, at the molecular level, is characterized by the translocation of the transcription factor NF- κB into the nucleus and an immediate NF- κB-dependent gene activation [7,8].
Bacterial LPS is known to be an in vivo trigger for EC activation, and it has been shown to induce functional alterations in ECs in in vitro culture, including the synthesis and release of IL-6 and IL-8 and alterations in the expression of cell surface structures involved in adhesion [9-11]. LPS is an integral component of the outer membrane of all Gram-negative bacteria. It is released from their surface during both in vitro and in vivo growth or may originate from the lysis of the bacterial cell following exposure to antibiotics or human serum [12,13]. LPS can initiate a potent inflammatory response when large amounts enter circulation, such as during endotoxemia [14]. In the present study, LPS was used to stimulate HUVECs, which were then activated to mimic the inflammation process.
Carbon monoxide (CO) has long been known in biology and medicine as a toxic compound, due to its ability to bind hemoglobin with a much higher affinity than oxygen [15]. Evidence accumulated to date suggests that endogenous carbon monoxide (CO), a bi-product of inducible heme oxygenase (HO-1) can modulate inflammation, inhibits lipopolysaccharide (LPS)-induced production of cytokines both in vivo and in vitro, and consequently exhibits important cytoprotective function and anti-inflammatory properties that are beneficial for the resolution of acute inflammation [16-18]. Inhaled CO at concentrations of 250–500 parts per million (ppm) has also been shown to be beneficial in a number of lung injury models, including hyperoxic injury allergen-induced inflammation [19].
Recently, transitional metal carbonyls have been identified as potential CO-releasing molecules (CO-RMs) with the potential to facilitate the pharmaceutical use of CO by delivering it to tissues and organs [20]. CO-RMs have been shown to act pharmacologically in rat aortic and cardiac tissue where liberation of CO produced vasorelaxant effects [21-24] and decreased myocardial ischemia–reperfusion damage [25,26] in the absence of dramatic changes in blood carboxy-hemoglobin (COHb) levels.
On the basis of these data, the present study was, therefore, designed as a prospective laboratory experiment to investigate the effects of preconditioning of tricarbonyldichlororuthenium (Ⅱ) dimer (CORM-2) [27], one of the novel group of CO-RMs, on decrease of inflammatory responses and oxidative stress in LPS-stimulated HUVECs (human umbilical vein endothelial cells), and discussed the possible molecular mechanisms. Our results show that preconditioning of endothelial cells (HUVEC) by CO liberated from CORM-2 elicits anti-inflammatory effects by interfering with redox-sensitive cell signaling and subsequent inhibition of pro-adhesive phenotype. In addition, it also supports the notion that CO is a potent inhibitor of iNOS.
Material and methods
Materials
Medium 199 (M199), fetal calf serum (FCS), penicillin, and streptomycin were purchased from GIBCO BRL (Gland Island, NY). Tricarbonyldichlororuthenium(II) dimer (CORM-2) was obtained from Sigma Aldrich and solubilized in dimethyl sulfoxide (DMSO) to obtain a 10mM stock. LPS (Escherichia coli serotype 055:B5) was purchased from Sigma. Anti-ICAM-1, HO-1 and iNOS polyclonal antibodies were purchased from Transduction Laboratories (Lexington, KY). Anti-mouse IgG conjugated to horseradish peroxidase was purchased from Kirkegaard and Perry Laboratories (Gaithersburg , MD).
Isolation and culture of human umbilical vein endothelial cells (HUVEC)
Human umbilical vein endothelial cells (HUVEC) were harvested from the fresh human umbilical vein of newborns by collagenase treatment (Worthington Biochem, Freehold, NJ) as previously described [28,29]. The cells were grown in medium 199(M199; GIBCO, Burlington, Canada) supplemented with 10% heat-inactivated FCS (Intergen, Purchase, NY), 2.4mg/l thymidine (Sigma Chemical, Oakville, Canada), 10IU/ml heparin sodium, antibiotics (100U/ml penicillin and 100µg/ml streptomycin; GIBCO), 1.5µg/ml fungizone (GIBCO), and 80µg/ml endothelial mitogen (Biomedical chnologies, Stoughten, MA). The cell cultures were incubated in room air with 5% CO2 at 37°C and 95% humidity and were expanded by brief trypsinization with 0.25% trypsin in PBS containing 0.025% EDTA. The experiments were conducted on passage 3-5 HUVECs. After 18h, the medium was changed to 500μl of fresh complete M199. Cells were pre-treated with CORM-2 (50 and 100μM) for 2h followed by stimulating with LPS (10μg/ml) for additional 4h. Cells co-incubated with CORM-2 (50 and 100μM) were as the controls. After stimulation for 4 h, the cells and medium were harvested separately.
Oxidant production
Oxidant production within HUVEC was assessed by measuring the oxidation of intracellular dihydrorhodamine 123 (DHR 123; Molecular Probes, Inc.), an oxidant-sensitive fluorochrome, as described previously [30]. Briefly, the cells were treated with DHR 123 (5mM) for 1h before being subjected to LPS stimulation. After LPS stimulation the cells were washed with PBS, lysed, and DHR 123 oxidation was assessed spectrofluorometrically at excitation and emission wavelengths of 502 and 523 nm, respectively.
Nitric oxide production
NO production by HUVEC was assessed by measuring the fluorescence of 4-amino-5-methylamino-2´, 7´-difluorofluorescein diacetate (DAF-FM diacetate), a specific NO probe (Molecular Probes, Inc.)[31]. Briefly, DAF-FM diacetate (10mM) in M199 was added to the HUVEC 1h before the LPS stimulation. After LPS stimulation, the HUVEC and supernatants were collected and analyzed spectrofluorometrically at excitation and emission wavelengths of 495 and 515 nm, respectively.
Cell ELISA
For assessment of ICAM-1 surface expression level, an ELISA was performed [32] on HUVEC grown in 96-well cell culture plates (Corning). HUVEC were fixed in 4% paraformaldehyde at 4°C for 30min. The cells were then washed two times with cold PBS and were incubated with the mouse primary antibody against human ICAM-1 at a concentration of 10µg/ml for 1h at room temperature. After this treatment, immunocytochemical staining of HUVEC monolayers was performed using an avidin-biotin-conjugated peroxidase mouse IgG staining kit, and MAb binding was subsequently quantified with a microplate reader (model 3550-UV; Bio- Rad) at 450-nm wavelength.
SDS-polyacrylamide gel electrophoresis and Western blotting
SDS-polyacrylamide gel electrophoresis and Western blotting were performed as described previously [33]. Samples (3μg of protein) were subjected to electrophoresis on 7% (for iNOS) or 12% (for HO-1) SDS-polyacrylamide gels, with the use of the discontinuous system and transferred onto nitrocellulose membranes. The membranes were probed with anti-iNOS polyclonal antibody (1:2000), and anti-HO-1 polyclonal antibody (1:2000). Anti-mouse IgG conjugated to horseradish peroxidase (1:2500) was used as a secondary antibody. The bands were visualized by the use of ECL reagent and Hyperfilm ECL (Amersham, Arlington Heights, IL) as described by the manufacturer. Films were scanned using a flatbed scanner and the bands were quantified using Basic Quantifier software (Bio Image, Ann Arbor, MI), an image analysis program, on a Macintosh computer.
HUVEC nuclear protein extraction
Nuclear protein was extracted from HUVEC as previously described [34,35]. Cells were grown to confluence in Petri-dish, scraped, washed with cold PBS, and incubated in 150µl of buffer E(+) (0.3% Nonidet P-40, 10mM Tris (pH 8.0), 60mM NaCl, 1mM EDTA, 0.5mM dithiothreitol (DTT), 1µg/ml aprotinin, 1µg/ml leupeptin, and 1mM phenylmethylsulfonyl fluoride ) for 5 min on ice. Samples were centrifuged at 4°C for 5min at 500g. The supernatant was then removed, and the pellets (nuclei) were resuspended in 150µl of buffer E (10mM Tris (pH 8.0), 60mM NaCl, 1mM EDTA, and 0.5mM DTT) and centrifuged at 500 g for 5 min at 4°C. The nuclei were then extracted in 30–50µl of buffer E(c)(20mM HEPES, 0.75mM spermidine, 0.15mM spermine, 0.2mM EDTA, 2mM EGTA, 2mM DTT, 20% glycerol, and 1mM PMSF (4°C) in the presence of 0.4MnaCl) and were incubated on ice for 20min. Finally, the samples were centrifuged for 10min at 500g (4°C), and the supernatants were collected and saved as the nuclear protein fraction.Samples were stored at -80°C.
Electrophoretic mobility shift assay(EMSA)
The double-stranded oligonucleotide containing consensus (58-AGGGACTTCCGCTGGGGACTTTCC-38) binding sites for NF-κB (synthesized on site; Beckman-Oligo 1000M DNA synthesizer) were end labeled with [γ-32P] ATP (Amersham) by using T4-polynucleotide kinase (MBI Fermentas, Flamborough, ON), as described previously [35]. One picomole of the labeled oligonucleotide was incubated with 5µg of nuclear extract protein in the presence or absence of 50X excess of cold oligonucleotide. Samples were incubated for 30 min at room temperature and then run through a 4% nondenaturing polyacrylamide gel at 280V for 45-60 min. The gel was dried and then exposed to X-ray film (Kodak) in cassettes for 2-4 h at -80°C with intensifying screens.
PMN adhesion assays
Human neutrophilic PMN were isolated from the venous blood of healthy adults using standard dextran sedimentation and gradient separation on Histopaque-1077. This procedure yields a PMN population that is 95–98% viable (trypan blue exclusion) and 98% pure (acetic acid-crystal violet staining).
For the static adhesion assay, isolated neutrophils were suspended in PBS buffer and radiolabeled by incubating the cells at 5 × 107 cells/ml with 50µCi Na51CrO4/ml PMN suspension at 37°C for 60min. The cells were then washed with cold PBS to remove unincorporated radioactivity. Radiolabeled PMN (5 × 105/well) were added to HUVEC monolayers grown in 48-well plates (Costar), and 30min later the percentage of added PMN that remained adherent after a wash procedure was quantitated as follows: %PMN adherence = lysate (cpm)/[supernatant (cpm) + wash (cpm) + lysate (cpm)], where cpm is counts per minute.
Statistical Analysis
All of the values are presented as means ± SD. Statistical analysis was performed with the use of ANOVA and Student's t-test for the comparisons. A value of P<0.05 was considered to be statistically significant.
Results
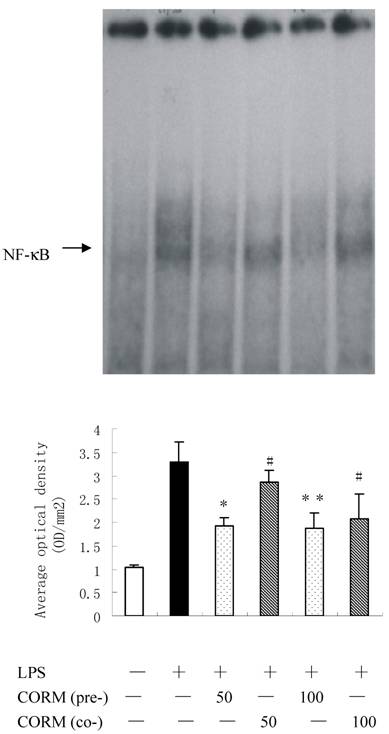
Effects of CORM-2 pretreatment on induction of intracellular oxidative stress (ROS) and NO production in LPS-stimulated HUVEC
As shown in Fig. 1, LPS stimulation resulted in a significant increase in HUVEC oxidant production. Pretreatment of HUVEC with CORM-2 for 2h significantly reduced oxidative stress (A) and NO production (B) in response to LPS stimulation.
Effects of CORM-2 pretreatment on induction of intracellular oxidative stress (ROS) and NO production in LPS-stimulated HUVEC. Confluent HUVEC monolayers (48-well cell culture dishes) were pretreated with CORM-2 (50 and 100μM) for 2h. Subsequently, HUVEC were washed and loaded with DHR123 or DAF-FM for additional 1h. Then, HUVEC were stimulated with LPS (10μg/ml) for 4hrs and oxidative stress (DHR123 oxidation) and NO production (DAF-FM nitration) were assessed. In some experiments, naïve (not-pretreated) HUVEC were stimulated with LPS in the presence of CORM-2 (50 and 100μM). Note that pretreatment of HUVEC with CORM-2 reduced oxidative stress (A) and NO production (B) in response to LPS stimulation. All values are expressed as mean ± SD (n = 4). #p <0.05 as compared to LPS; *p <0.05 as compared to co-incubation with 50 μM CORM-2; * *p <0.05 as compared to co-incubation with 100 μM CORM-2.
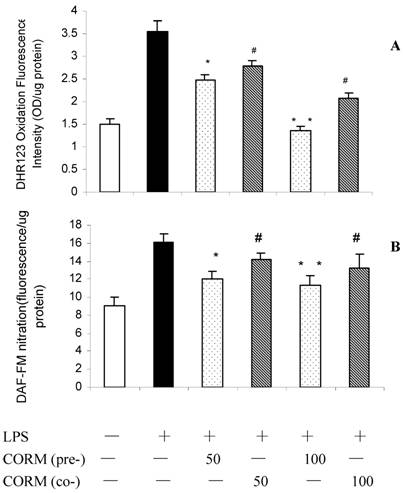
Effect of CORM-2 pretreatment on iNOS and HO-1 expression in LPS-stimulated HUVEC (Western blotting)
At 4h after LPS stimulation, the expression of iNOS (Fig.2 A) and HO-1 (Fig.2 B) in HUVEC significantly increased compared to the control. Both pretreatment and coincubation of CORM-2 in LPS-stimulated HUVECs significantly downregulated the expression of iNOS. However, CO preconditioning was more effective to downregulate the expression of iNOS compare to coincubation group. Interestingly, not only pretreatment, but also coincubation of CORM-2 in LPS-stimulated HUVECs significantly upregulated the expression of HO-1.
Effects of CORM-2 pretreatment on iNOS and HO-1 expression in LPS-stimulated HUVEC. The experimental conditions were the same as described in Fig.1. iNOS and HO-1 expression were determined by Western blot. Note that pretreatment of HUVEC with CORM-2 resulted in more effective inhibition of iNOS protein expression in LPS stimulated HUVEC compare to coincubation group,#p <0.05 as compared to LPS; *p <0.05 as compared to co-incubation with 50 μM CORM-2; * *p <0.05 as compared to co-incubation with 100 μM CORM-2 (A). Both pretreatment and coincubation of CORM-2 significantly upregulated expression of HO-1 in LPS-stimulated HUVECs. Shown is a representative image from three experiments. *p <0.05 as compared to control, # p <0.05 as compared to LPS (B).
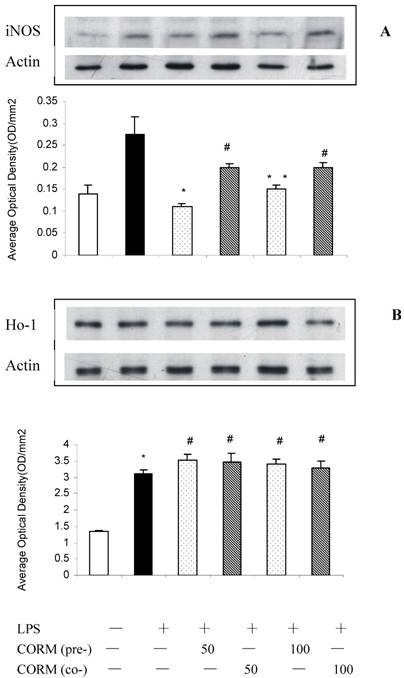
Effect of CORM-2 pretreatment on ICAM-1 expression in LPS-stimulated HUVEC (cell ELISA)
At 4h after LPS stimulation, the expression of ICAM-1 in HUVEC significantly increased compared to the control, while this increase was effectively inhibited after pretreatment or coincubation of CORM-2 in HUVEC with no significant different.
Effects of CORM-2 pretreatment on ICAM-1 surface expression in LPS-stimulated HUVEC. The experimental conditions were the same as described in Fig.2. HUVEC pro-adhesive phenotype was assessed by measuring cell surface levels (cell ELISA) of ICAM-1 protein. Note that LPS-induced up-regulation of ICAM-1 surface levels was significantly reduced in CORM-2-pretreated or coincubated HUVEC. n=4; * P<0.05 compared to LPS. *# P>0.05 compared to those co-incubated with 50 or 100 μM of CORM-2.
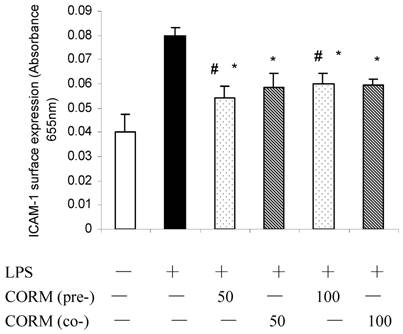
Effect of CORM-2 pretreatment on activities of NF-κB in LPS-stimulated HUVEC
Binding activities of nuclear protein to the radiolabeled consensus binding sequences of NF-κB was assessed by EMSA. At 4h after LPS stimulation, the NF-κB activation in HUVEC was markedly increased and this activity was inhibited by pretreatment of CORM-2 with a concentration-dependent manner (Fig. 4).
Effects of CORM-2 pretreatment on NF-kB activation in LPS-stimulated HUVEC. HUVEC were grown in 60 mm Petri dishes until confluence and pretreated with CORM-2 (50 and 100μM) for 2 h. The control HUVECs were treated with the vehicle only. Subsequently, HUVEC were washed and stimulated with LPS (10 µg/ml) for 4 hrs in the presence (coincubation) or absence of CORM-2 (50-100µM). NF-κB activation was assessed by EMSA. Note that LPS-induced activation of NF-κB was effectively prevented in CORM-2-pretreated HUVEC. Shown is a representative image from four experiments. #p <0.05 as compared to LPS; *p <0.05 as compared to co-incubation with 50 μM CORM-2; * *p <0.05 as compared to co-incubation with 100 μM CORM-2.
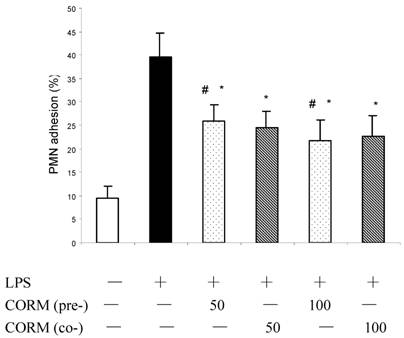
Effect of CORM-2 pretreatment on PMN adhesion to LPS-stimulated HUVEC
As shown in Fig.5, adhesion of PMN to HUVEC is low in control. After monolayer of endothelial cells were stimulated by LPS for 4h, adhesion of PMN to HUVEC significantly increased (P<0.01 compared to control). However, both pretreatment and coincubation with CORM-2 (50 and 100μM) markedly decreased leukocyte–endothelial cells adhesion (P<0.05 compared to LPS group).
Effects of CORM-2 pretreatment on PMN adhesion to LPS-stimulated HUVEC. HUVEC were grown to confluence in 48-well cell culture dishes and pretreated with CORM-2 (50-100µM) for 2 hr. Then HUVEC were washed and stimulated with LPS (10 µg/ml) for additional 4 hrs in the presence (coincubation) or absence of CORM-2. Subsequently, HUVEC were washed and co-incubated with 51Cr-PMN (1×105) for 30 min. Per cent of PMN adhesion to HUVEC was calculated based on the amount of radioactivity remaining in HUVEC monolayers following two subsequent washes. Note that PMN adhesion to LPS-stimulated HUVEC was significantly reduced in CORM-2-pretreated or coincubated HUVEC. All values are expressed as mean ± SE (n = 3). * P<0.01 compared to LPS. *# P>0.05 compared to those coincubated with 50 or 100 μM of CORM-2.
Discussion
The present study investigated the effects of preconditioning of CORM-2 on decrease of inflammatory response and oxidative stress in LPS-stimulated HUVECs and possible molecular mechanisms.
LPS from the bacteria can activate endothelial cells and stimulate their expression of adhesion molecules, elicit the responsiveness of ECs through its ability to affect leukocyte adherence to activate vascular endothelium and the subsequent transendothelial migration. Many experimental studies have highlighted the specific and independent role of exogenous CO in the modulation of inflammation [36,37]. It is well known that heme oxygenase (HO) is a rate-limiting enzyme that is responsible for the catabolism of heme into bilirubin, free iron, and carbon monoxide (CO). Three HO isoforms have been identified; HO-2 and HO-3 isoforms are believed to be constitutive and physiologically expressed, whereas HO-1 isoform is a stress-responsive protein that is induced by various stimuli. The adaptive response of HO-1 to various stimuli suggests that HO-1 may play an important role in protecting against inflammatory response and oxidative injury [38]. Works from other laboratories have shown that the up-regulation of endogenous HO-1 ameliorates inflammatory responses and/or tissue damage [39]. As a new metal carbonyl-based compounds, CO-releasing moleculars (CO-RMs) have the ability to release CO in biological systems. The vasoactive, anti-hypertensive and anti-rejection effects of CO-RMs have been demonstrated to be due to the CO liberated by the compounds. CORM-2, a DMSO- soluble CORM, also has exhibited anti-inflammatory actions in an in vitro model of LPS-stimulated macrophages, and it was reported that the CO derived from 50 μM CORM-2 dissolved in DMSO (generating 50 ppm CO) was not cytotoxic by itself and did not enhance or reduce the cytotoxicity of LPS as was observed by Sawle et al [40]. Therefore, this led us to examine whether preconditioning of CORM-2 could decrease oxidative stress, inhibit LPS-induced HUVEC activation. In this study, we found that HO-1 is significantly up-regulated in HUVECs by LPS stimulation. Interestingly, the expression of HO-1 in LPS-stimulated HUVECs with both preconditioning and coincuation of CORM-2 was more significantly increased compared to LPS group. This result indicated that not only LPS might significantly induce the expression of HO-1, but also the increase of HO-1 expression can be further enhanced by the administration of CORM-2. Through the by-product (CO, and / or biliverdin), the potent cytoprotective and anti-inflammatory functions were ultimately led to exert.
Based on current research findings, oxidative stress is believed to be the major causative agent to damage the organs after injury. Nitrosative stress initiates an inflammatory cascade that includes acute phase protein synthesis, upregulation of inflammatory adhesion molecules, and proinflammatory cytokine release [41,42]. Thus, tissue or organ injury after trauma appears to be mediated both by reactive oxygen species (ROS) and reactive nitrogen species (RNS) such as hydroxyl radical, superoxide anion, hydrogen peroxide, and peroxynitrite. It has been demonstrated that severe injury is associated with lipid peroxidation mediated by ROS and is believed to be an important cause of oxidative damage to cellular membranes and, eventually, cell death [43]. Also, nitric oxide (NO) plays a crucial role in cellular injury after trauma. It has been recognized that NO is produced by cNOS and/or iNOS in mice liver [44,45]. In this study, we found that LPS stimulation produces a significant upregulation of the inducible nitric oxide synthetase (iNOS), iNOS-mediated nitric oxide production then be upregulated. Our studies also demonstrated that administration of CORM-2 markedly down-regulated this increase both preconditioning and coincubation of CORM-2. However, preconditioning of CORM-2 is more effective to decrease the expression of iNOS and then decrease the production of ROS and NO with concentration-dependent manner. These data indicated that preconditioning of exogenous CO was capable of effectively inhibiting inflammatory response and improving oxidative stress in activated HUVEC.
Neutrophil (PMN)–endothelial cell (EC) interactions are supposed to play a central role in the pathogenesis of gut barrier failure following thermal injury and ischemia-reperfusion (I/R) [46]. Intercellular adhesion molecule-1 (ICAM-1), mediating leukocyte adhesion, presence correlates with infiltration of leukocytes into inflammatory lesions [47,48]. Evidences showed that the direct cause of leukocytes sequestration is considered to be the more expression of adhesion molecule (such as ICAM-1). ICAM-1 activates leukocytes and endothelial cells, which in turn prompt the release of various inflammatory mediators, resulting in systemic inflammatory response syndrome (SIRS), acute respiratory distress syndrome (ARDS) and multi-organ dysfunction syndrome (MODS) [49-51]. It was found in the present study that both preconditioning and co-incubation of CORM-2 could down-regulate the expression of ICAM-1 with no significant difference. These results were in accordance with the PMN adhesion experiments (Fig.5) which showed that both preconditioning and co-incubation of CORM-2 could effectively decreased the PMNs adhesion to LPS-stimulated HUVEC.
There is no doubt that the nuclear factor κB (NF-κB) is a ubiquitous, rapidly acting transcription factor involved in immune and inflammatory reactions, it exerts its immune and inflammatory response by regulating expression of cytokines, chemokines, cell adhesion molecules, and growth factors [52,53]. In this study, NF-κB activities in HUVEC were elevated by LPS stimulation while it was markedly inhibited by administration of CORM-2 with preconditioning or coincubation treatment. However, preconditioning of CORM-2 was more effective to inhibit NF-κB activities compared to CORM-2 coincubation group. These data showed that CORM-2 preconditioning plays a pivotal role in inhibition of NF-κΒ activity, subsequently decreased the expression of cellular adhesion molecules (ICAM-1) and LPS-induced pro-inflammatory mediators. Therefore, an effective therapeutic strategy that inhibit this transcription factor would be expected to improve organ functions in SIRS or sepsis.
In summary, the present studies serve to clarify the role of preconditioning of CORM-2, one of the novel CO-releasing molecules, on the mechanisms of attenuation of inflammation and oxidative stress. Preconditioning of CORM-2 on LPS-stimulated HUVECs upregulated the expression of HO-1 while downregulated the expression of iNOS, and subsequently decreased the production of ROS and NO. Also, preconditioning of CORM-2 effectively inhibited activation of NF-κΒ, which was accompanied by a decrease of the expression of ICAM-1. In parallel, PMN adhesion to HUVEC stimulated by LPS were markedly decreased after CORM-2 admiistration both with preconditioning and coincubation treatment. Taken together these findings indicate that preconditioning of CORM-released CO is more effective to decrease the oxidative stress and inflammatory responses by interfering with NF-κΒ activation, protein expression of ICAM-1 and therefore suppressing endothelial cells pro-adhesive phenotype.
Acknoeledgements
We thank professor Gediminas Cepinskas (University of Western Ontario) for the technical assistance. This work was supported by National Natural Science Foundation of China, No.30772256.
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
1. Pober JS, Cotran RS. The role of endothelial cells in inflammation. Transplantation. 1990;50:537-41
2. Melih K, Bulent U, Betul B. et al. The effect of ethyl pyruvate on oxidative stress in intestine and bacterial translocation after thermal injury. J Surg Res. 2008;144:59-63
3. Ronald JA, Ionescu CV, Rogers KA. et al. Differential regulation of transendothelial migration of THP-1 cells by ICAM-1/LFA-1 and VCAM-1/VLA-4. J Leukoc Biol. 2001;70:601-9
4. Yang Domingos NCK, M WS. Identification of a Novel Family of Cell-surface Proteins Expressed in Human Vascular Endothelium. J Bio Chem. 2002;277:46364-73
5. Madge LA, Pober JS. TNF signaling in vascular endothelial cells. Exp Mol Pathol. 2001;70:317-25
6. Colwell J. Pathogenesis of vascular disease. Diabetes Obes Metab. 2000;2(Suppl 2):S19-24
7. Chen FE, Ghosh G. Regulation of DNA binding by Rel/NF-κB transcription factors: structural views. Oncogene. 1999;18:6845-52
8. Senftleben U, Karin M. The IKK/NF- κB pathway. Crit Care Med. 2002;30:S18-S26
9. Jehle AB, Li Y Stechschulte AC. et al. Endotoxin and mast cell granule proteases synergistically activate human coronary artery endothelial cells to generate interleukin-6 and interleukin-8. J Interferon Cytokine Res. 2000;20:361-8
10. von Asmuth EJ, Leeuwenberg JF, Ceska M. et al. LPS and cytokine-induced endothelial cell IL-6 release and ELAM-1 expression; involvement of serum. Eur Cytokine Netw. 1991;2:291-7
11. Charreau B, Coupel S, Goret F. et al. Association of glucococorticoids and cyclosporin A or rapamycin prevents E-selectin and IL-8 expression during LPS- and TNFα-mediated endothelial cell activation. Transplantation. 2000;69:945-53
12. Dofferhoff AS, Nijland JH, deVries-Hospers HG. et al. Effects of different types and combinations of antimicrobial agents on endotoxin release from gram-negative bacteria: an in vitro and in vivo study. Scand J Infect Dis. 1991;23:745-54
13. Evans ME, Pollack M. Effect of antibiotic class and concentration on the release of lipopolysaccharide from Escherichia coli. J Infect Dis. 1993;167:1336-43
14. Mayeux PR. Pathobiology of lipopolysaccharide. J Toxicol Env Health. 1997;51:415-35
15. Piantadosi CA. Biological chemistry of carbon monoxide. Antioxid Redox Signal. 2002;4:259-70
16. Hayashi S, Takamiya R, Yamaguchi T. et al. Induction of heme oxygenase- 1 suppresses venular leukocyte adhesion elicited by oxidative stress. Role of bilirubin generatedby the enzyme. Circ Res. 1999;85:663-71
17. Lee TS, Chau LY. Heme oxygenase-1 mediates the anti-inflammatory effect of interleukin-10 in mice. Nat Med. 2002;8:240-6
18. Otterbein LE, Soares M, Yamashita K. et al. Heme oxygenase-1: unleashing the protective properties of heme. Trends Immunol. 2003;24:449-55
19. Chapman JT, Otterbein LE, Elias JA. et al. Carbon monoxide attenuates aeroallergen-induced inflammation in mice. Am J Physiol Lung Cell Mol Physiol. 2001;281:L209-L216
20. Motterlini R, Mann BE, Johnson TR. et al. Bioactivity and pharmacological actions of carbon monoxide-releasing molecules. Curr Pharm Des. 2003;9:2525-39
21. Motterlini R, Clark JE, Foresti R. et al. Carbon monoxide-releasing molecules: characterization of biochemical andv ascular activities. Circ Res. 2002;90:E17-24
22. Motterlini R, Sawle P, Bains S. et al. CORM-A1: a new pharmacologically active carbon monoxide-releasing molecule. FASEB J. 2005;19:284-6
23. Johnson TR, Mann BE, Clark JE. et al. Metal carbonyls: a new class of pharmaceuticals? Angew Chem Int Ed Engl. 2003;42:3722-9
24. Foresti R, Hammad J, Clark JE. et al. Vasoactive properties of CORM-3, a novel water-soluble carbon monoxide-releasing molecule. Br J Pharmacol. 2004;142:453-60
25. Clark JE, Naughton P, Shurey S. et al. Cardioprotective actions by a water-soluble carbon monoxidereleasing molecule. Circ Res. 2003;93:2-8
26. Guo Y, Stein AB, Wu WJ. et al. Administration of a Coreleasing molecule at the time of reperfusion reduces infarct size in vivo. Am J Physiol Heart Circ Physiol. 2004;286:H1649-53
27. Bingwei Sun, Zhiwei Sun, Qin Jin. et al. CO-releasing molecules (CORM-2)-liberated CO attenuates leukocytes infiltration in the renal tissue of thermally injured mice. Int J Biol Sci. 2008;4:176-83
28. Yoshida N, Granger DN, Anderson DC. et al. Anoxia/reoxygenation-induced neutrophil adherence to cultured endothelial cells. Am J Physiol Heart Circ Physiol. 1992;262:H1891-8
29. Gimbrone MA, Cotran RS, Folkman J. Human vascular endothelial cells in culture. Growth and DNA synthesis. J Cell Biol. 1974;60:673-84
30. Cepinskas G, Lush CW, Kvietys PR. Anoxia/reoxygenation-induced tolerance with respect to polymorphonuclear leukocyte adhesion to cultured endothelial cells: a nuclear factor- kB phenomenon. Circ Res. 1999;84:103-12
31. Parsa CJ, Matsumoto A, Kim J. et al. A novel protective effect of erythropoietin in the infarcted heart. J Clin Invest. 2003;112:999-1007
32. Panes J, Perry MA, Anderson DC. et al. Regional differences in constitutive and induced ICAM-1 expression in vivo. Am J Physiol Heart Circ Physiol. 1995;269:H1955-64
33. Nakatsuka M, Nakatsuka K, Osawa Y. Metabolism-based inactivation of penile nitric oxide synthase activity by guanabenz. Drug Metab Dispos. 1998;26:497-501
34. Cepinskas G, Lush CW, Kvietys PR. Anoxia/reoxygenation-induced tolerance with respect to polymorphonuclearleukocyte adhesion to cultured endothelial cells—a nuclear factor-kB-mediated phenomenon. Circ Res. 1999;84:103-12
35. Kishimoto TK, Warnock RA, Jutila MA. et al. Antibodies against human neutrophil LECAM-1 (LAM-1/Leu-8/DREG-56 antigen) and endothelial cell ELAM-1 inhibit a common CD18-independent adhesion pathway in vitro. Blood. 1991;78:805-11
36. Otterbein LE, Bach FH, Alam J. et al. Carbon monoxide has anti-inflammatory effects involving the mitogen-activatedp rotein kinase pathway. Nat Med. 2000;6:422-8
37. A Moore BA, Murase N Liu F. et al. Immunomodulatory effects of inhaled carbon monoxide on rat syngeneic small bowel graft motility. Gut. 2003;52:1278-85
38. Willis D, Moore AR, Frederick R. et al. Heme oxygenase: a novel target for the modulation of the inflammatory response. Nat Med. 1996;2:87-93
39. Attuwaybi BO, Kozar RA, Moore-Olufemi SD. et al. Heme oxygenase-1 induction by hemin protects against gut ischemia/reperfusion injury. J Surg Res. 2004;118:53-7
40. Sawle P, Foresti R, Mann BE. et al. Carbon monoxide-releasing molecules (CO-RMs) attenuate the inflammatory response elicited by lipopolysaccharide in RAW264.7 murine macrophages. British J Pharmacology. 2005;145:800-10
41. Szabo C, Dawson WL. Role of poly (ADP-ribose) synthetase in inflammation and ischemia-reperfusion. TIPS. 1998;19:287-298
42. Willy C, Dahouk S, Starek C. et al. DNA damage in humane leucocytes after ischemia/reperfusion injury. Free Rad Biol Med. 2000;28:1-12
43. Cetinkale O, Senel O, Bulan R. The effect of antioxidant therapy on cell mediated immunity following burn injury an animal model. Burns. 1999;25:113-118
44. Moriyama A, Tabaru A, Unoki H. et al. Plasma nitrite/nitrate concentrations as a tumor marker for hepatocellular carcinoma. Clinica Chimica Acta. 2000;296:181-191
45. Hara H, Mitani N, Adachi T. Inhibitory effect of nitric oxide on the induction of cytochrome P450 3A4 mRNA by 1, 25-dihydroxyvitamin D3 in Caco-2 cells. Free Rad Res. 2000;33:279-285
46. Oktar B, Coskun T, Bozkurt A. et al. Endothelin-1-induced PMN infiltration and mucosal dysfunction in the rat small intestine. Am J Physiol Gastrointest Liver Physiol. 2000;279:483-491
47. Defazio G, Nico B, Trojano M. et al. Inhibition of protein kinase C counteracts TNF-a-induced ICAM-1expression and fluid phase endocytosis on brain microvascular endothelial cells. Brain Res. 2000;863:245-8
48. Rahman A, True AL, Anwar KN. et al. Gaq and Gbg regulate PAR-1 signaling of thrombin-induced NF-kB activation and ICAM-1 transcription in endothelial cells. Circ Res. 2002;91:398-405
49. Sparkes BG, Gyorkos JW, Gorczynsky RM. et al. Comparison of endotoxins and cutaneous burn toxin as immunosuppressants. Burns. 1990;16:123-7
50. Deveci M, Eski M, Sengezer M. et al. Effects of cerium nitrate bathing and prompt burn wound excision on IL-6 and TNF-α levels in burned rats. Burns. 2000;26:41-5
51. Joseph C, David G, Iris G. Modulation of ndotoxin-induced endothelial activity by microtubule depolymerization. J Trauma. 2003;54:104-13
52. Siebenlist U, Franzoso G, Brown K. Structure, regulation and function of NF-kappa B. Annu Rev Cell Biol. 1994;10:405-55
53. Yin MJ, Yamamoto Y, Gaynor RB. The anti-inflammatory agents aspirin and salicylate inhibit the activity of I (kappa) B kinase-beta. Nature. 1998;396:77-80
Author contact
![]() Correspondence to: Prof. Bingwei Sun, Department of Burn and Plastic Surgery, Affiliated Hospital, Jiangsu University, PR China.Tel.: 0511-85082258; Fax.: 0511-85029089; E-mail address: sunbinwesina.com
Correspondence to: Prof. Bingwei Sun, Department of Burn and Plastic Surgery, Affiliated Hospital, Jiangsu University, PR China.Tel.: 0511-85082258; Fax.: 0511-85029089; E-mail address: sunbinwesina.com