Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2008; 4(6):368-378. doi:10.7150/ijbs.4.368 This issue Cite
Research Paper
Biological and functional analysis of statistically significant pathways deregulated in colon cancer by using gene expression profiles
Angela Distaso1, Luca Abatangelo1, Rosalia Maglietta1, Teresa Maria Creanza1, Ada Piepoli2, Massimo Carella3, Annarita D'Addabbo1, Nicola Ancona1 ![]()
1. Istituto di Studi sui Sistemi Intelligenti per l'Automazione, CNR, Via Amendola 122/D-I, 70126 Bari, Italy
2. Unità Operativa di Gastroenterologia, IRCCS, “Casa Sollievo della Sofferenza”-Ospedale, 71013 San Giovanni Rotondo (FG), Italy
3. Servizio di Genetica Medica, IRCCS, “Casa Sollievo della Sofferenza”-Ospedale, 71013 San Giovanni Rotondo (FG), Italy
Received 2008-7-8; Accepted 2008-10-7; Published 2008-10-14
Abstract
Gene expression profiling offers a great opportunity for studying multi-factor diseases and for understanding the key role of genes in mechanisms which drive a normal cell to a cancer state. Single gene analysis is insufficient to describe the complex perturbations responsible for cancer onset, progression and invasion. A deeper understanding of the mechanisms of tumorigenesis can be reached focusing on deregulation of gene sets or pathways rather than on individual genes. We apply two known and statistically well founded methods for finding pathways and biological processes deregulated in pathological conditions by analyzing gene expression profiles. In particular, we measure the amount of deregulation and assess the statistical significance of predefined pathways belonging to a curated collection (Molecular Signature Database) in a colon cancer data set. We find that pathways strongly involved in different tumors are strictly connected with colon cancer. Moreover, our experimental results show that the study of complex diseases through pathway analysis is able to highlight genes weakly connected to the phenotype which may be difficult to detect by using classical univariate statistics. Our study shows the importance of using gene sets rather than single genes for understanding the main biological processes and pathways involved in colorectal cancer. Our analysis evidences that many of the genes involved in these pathways are strongly associated to colorectal tumorigenesis. In this new perspective, the focus shifts from finding differentially expressed genes to identifying biological processes, cellular functions and pathways perturbed in the phenotypic conditions by analyzing genes co-expressed in a given pathway as a whole, taking into account the possible interactions among them and, more importantly, the correlation of their expression with the phenotypical conditions.
Keywords: Microarray, pathway analysis, prediction accuracy, machine learning, gene expression, colon cancer.
Introduction
Gene expression profiling has become a mainstay of the current research in applied genomics [1]. In oncology, in particular, the advent of DNA microarray technology has allowed a deeper understanding of the role that many genes play in onset, progression and treatment of tumors [2,3]. Typically, specimens of tissues in two different phenotypical conditions (e.g diseased patients vs. healthy controls, or patients in two different stages of the same pathology) are collected and genes which are differentially expressed (DE) in the two conditions analyzed are determined. To this end, appropriate univariate statistical tests are applied to the gene expression profiles of the specimens and genes with high statistical significance levels (p-value) are considered associated with the trait of interest. Due to the huge number of hypotheses tested simultaneously, suitable statistical strategies are applied to control false positive findings [4]. Finally, the list of DE genes statistically associated to the phenotype is used to find the main pathways or biological processes involved in the analyzed pathology. Such processes are coded through lists of genes defined on the basis of a-priori biological knowledge or experimentally. In the first case, such lists are composed of those genes which cooperate or are co-expressed in a particular cellular mechanism or function [5-7]. In the second case, the gene set represents the signature (response) of cells (system) to a given stimulus [8]. Many tools have been proposed for measuring deregulation of pathways and for assessing their statistical significance [9].
This approach has a few major limitations. a) The results obtained with this method are not always confirmed by studies carried out on independent cohorts of subjects [10]. This means that studies performed by different groups on the same trait or pathology may produce lists of DE genes with little overlap. b) The information embedded in genes weakly connected with the phenotype may be lost due to both the statistic adopted and the correction for multiple hypothesis testing. c) Single gene analysis provides a limited view of the phenomena under examination since it does not take into account interactions among genes and is unable to uncover the correlation between groups of genes and phenotype. Many different genes contribute to a given disorder, with no particular gene having a remarkably large effect [11]. Thus, a specific phenotype may result from the combination of effects by a large number of moderately contributing genes.
To overcome these drawbacks, a new approach is emerging in genomics research in which instead of inferring pathways involved in a given disorder starting from the analysis of DE genes, it aims to measure pathway deregulation by considering simultaneously all the genes co-operating in the pathway [12-14]. In this new perspective, the focus shifts from finding DE genes to identifying biological processes, cellular functions and pathways perturbed in the phenotypic conditions by analyzing genes co-expressed in a given pathway as a whole, taking into account the possible interactions among them and, more importantly, the correlation of their expression with the phenotypical conditions [8,15].
In this paper we describe the results obtained by applying this new approach to a data set composed of gene expression profiles relative to patients affected by colon cancer, collected in Casa Sollievo della Sofferenza Hospital, Foggia -Italy [16]. Two well known methods recently proposed for finding deregulated pathways were applied. GSEA (Gene Set Enrichment Analysis) [12] finds perturbed pathways comparing the rank distribution of genes belonging to a given gene set with the rank distribution of the remaining genes. To this end a Kolmogorov-Smirnov like statistic is used for assessing the statistical significance of the deregulation. GLAPA (Gene List Analysis with Prediction Accuracy) [14] uses the prediction accuracy of the phenotypic status of specimens to find the pathways involved in the pathology. Both use non parametric permutation tests [17] and false discovery rate (FDR) [4] for assessing the statistical significance of the estimates. The database of gene sets we use in this study is the Molecular Signatures Database (MSigDB) [12]. This is a collection of 1687 curated gene sets with sizes ranging from 2 to 1594 genes, obtained from online pathway databases, publications in PubMed and expert knowledge.
Our study highlights two relevant and different aspects of the application of pathway analysis in oncology. In fact it shows that specific pathways deregulated in different types of tumors are found perturbed in colon cancer with high statistical significance. Moreover, such an approach provides a more complete portrait of complex diseases like tumors because it points out genes moderately associated to the trait which would not be detected by using classical univariate statistics.
Material and methods
Data set description
Study population
Twenty-five patients (14 males; mean age: 60 ± 14 years), who underwent colonic resection for colorectal cancer (CRC) at CSS hospital, were prospectively recruited into this study. Two samples from each patient were available, one from colon cancer tissue and one from normal colonic mucosa tissue. The samples had been obtained during the surgery, immediately frozen in liquid nitrogen and then stored at −80°C. All of them were reviewed by the same experienced pathologist to confirm the histological diagnosis. None of the patients suffered from hereditary CRC or had received preoperative chemo-radiotherapy. Informed consent to take part in this study was obtained from all the patients. The study was approved by the Hospital's Ethics Committee.
RNA extraction from fresh frozen tissue
Total RNA from 150-200 mg of fresh frozen tissue was isolated by phenol-chloroform extraction (TRIzol Reagent, Invitrogen, Carlsbad, CA) and subsequently purified through column chromatography (RNeasy Mini Kit, Qiagen, Valencia, CA) according to the manufacturer's instructions. RNA integrity was monitored using denaturing agarose gel electrophoresis in 1X MOPS. Three neoplastic samples were discarded from the final analysis since their RNA preparation was suboptimal.
Microarray assays
Biotinylated target cRNA was generated from 12 mg as described by the Affymetrix Expression Analysis GeneChip Technical Manual (Affymetrix, Santa Clara, California). Briefly, double-stranded cDNA was synthesized from total RNA using the SuperScript Choice System (Invitrogen, Carlsbad, California), a primer containing poly(dT) and a T7 RNA polymerase promoter sequence. In vitro transcription using double-stranded cDNA as a template in the presence of biotinylated UTP and CTP was carried out using BioArray High Yield RNA Transcript Labeling Kit (Enzo Diagnostics, Farmingdale, New York). The resulting biotynilated-cRNA ”target” was purified and quantified. Fifteen micrograms of biotinylated cRNA were randomly fragmented to an average size of 50 nucleotides by incubating in 40mM TRIS-acetate, pH 8.1, 100 mM potassium acetate, and 30 mM magnesium acetate at 94°C for 35 minutes. The fragmented cRNA was hybridized for 16 hours at 45°C on Human Genome U133A GeneChips containing a total of 22,283 probe sets and after stained in a Fluidics station with streptavidin/phycoerythrin, followed by staining through a streptavidin antibody and streptavidin/phycoerythrin. Arrays were scanned on a Genearray scanner G2500A by using standard Affymetrix protocols. Absolute data analysis was performed using the Affymetrix Microarray Suite 5.0 software.
GSEA
This method provides an enrichment score of pre-defined gene sets whose magnitude is proportional to the association of the gene set to the particular phenotype [12]. Given a gene expression dataset, the genes are ordered in a ranked list S according to their differential expression between the two classes. GSEA provides a score which measures the degree of enrichment of a given gene set L at the extremes (top or bottom) of the rank-ordered list S. The method is based on a maximum deviation statistic of two distribution functions, similarly to the Kolmogorov-Smirnov test that is used to estimate the difference between two distributions. In fact, the score is calculated by walking down the list S, increasing a running-sum statistic when a gene in the gene set is encountered, and decreasing it when genes not belonging to the gene set are encountered. The magnitude of the increment depends on the correlation of the gene with the phenotype. The enrichment score (ES) is the maximum deviation from zero in the walk. These ES are then normalized to take into account the size of the gene sets resulting in a normalized enrichment score (NES). The gene sets related to the phenotypic distinction will tend to show high values of NES. The significance of NES is assessed by permutation testing: the observed NES is compared with the distribution of enrichment scores under the null hypothesis that the gene expression levels and the phenotype are independent random variables. The nominal p-value (py) is given by the percentage of random normalized enrichment scores greater than the observed value of NES.
GLAPA
This method uses an estimate of the generalization error of predictors trained by using raw expression levels of the genes belonging to a gene set L as a measure of enrichment of L [14]. The rationale is that a functional category coded through a list of genes is perturbed in a particular disease if it is possible to correctly predict the occurrence of the pathology in new subjects on the basis of the expression levels of those genes only. In other words, a functional category is informative for or is deregulated in a disease if the expression levels of the genes involved in the category are useful for training classifiers able to generalize, that is, able to correctly predict the status of new subjects [18]. So, the generalization ability of predictors trained by using the expression levels of the genes co-operating in a given cellular mechanism or function can be seen as a measure of the relevance of the function in the pathology at hand. The phenotype is predicted through regularized least squares (RLS) classifiers with linear kernel [16, 19-21]. The error rate of the phenotype eL is estimated by using a multiple random validation strategy which provides a statistically significant estimate of the generalization error of outcome cancer predictors [22, 23]. The statistical significance of the measured accuracy is assessed against a couple of null hypothesis by using two independent permutation tests [17]. The first one (T1) controls for how likely the error rate eL was due to chance. The statistical significance py, power πy and false discovery rate FDRy were estimated by phenotypic permutations. The second one (T2) controls for the effect of the gene set size in the error rate eL. The statistical significance pn, power πn and false discovery rate FDRn were estimated by randomly selecting gene sets of the same size as L.
Results and discussion
Statistical analysis
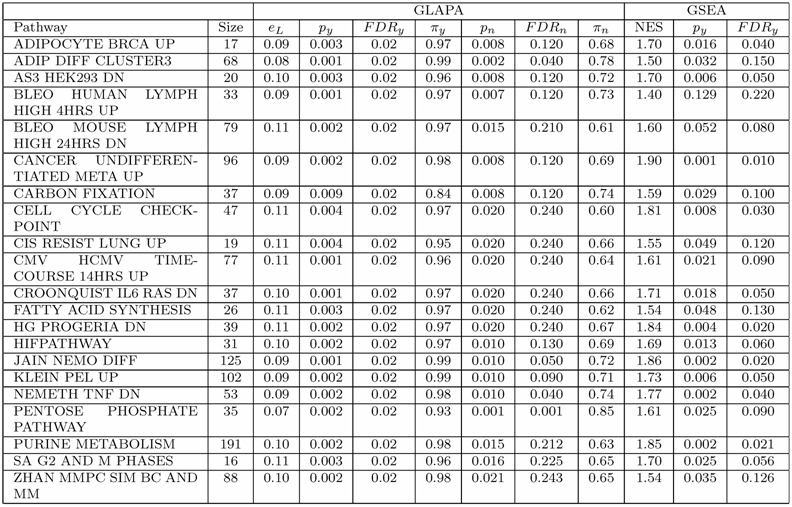
The deregulation of the whole collection of gene sets belonging to MSigDB was measured applying GSEA and GLAPA tools on our colon cancer data set independently. The GSEA software parameters were set to their default values. The statistical significance of the normalized enrichment score (NES) associated to each gene set was assessed through a non parametric permutation test in which 1000 random permutations of the phenotypic labels were carried out. GSEA found 915 gene sets up-regulated in tumor and 769 up-regulated in normal specimens. Among these, only 399 gene sets up-regulated in tumor and 3 up-regulated in normal were found statistically significant with FDRy ≤ 25%. For measuring the deregulation of each gene set L with GLAPA, we measured the prediction error of the phenotype eL associated to L. To this end, for each gene set, 1000 cross validations of the data set were carried out. In each cross validation, we used 30 examples for training and the remaining 17 for testing RLS classifiers with linear kernel. We found 1381 pathways with an error rate eL ≤ 25%. To assess the statistical significance of eL, 1000 random permutations of the phenotypic labels were performed. This permutation test revealed 690 statistically significant gene sets (py ≤ 0.01, FDRy ≤ 0.024) having error rates eL ≤ 17%. In order to determine if the deregulation of a particular pathway was due to the identity of the genes cooperating in the given pathway, or simply to the number of genes present in the gene set, a second permutation test was carried out. Specifically, indicated with n the size of a gene set L, 1000 gene sets were generated composed of n probes randomly drawn from the ones available on the microarray. The error rate associated to each random gene set was evaluated performing 200 cross validations and compared with the error rate eL. Such analysis revealed 58 pathways (pn ≤ 0.02, FDRn ≤ 0.25) having an error rate eL ≤ 11% (py ≤ 0.010, FDRy ≤ 0.024). Table 1 shows the 21 (P − value =0.021 Fisher's exact test) statistically significant pathways found deregulated by GSEA and GLAPA methods simultaneously.
Pathways of MSigDB database deregulated in our colon cancer gene espression data set. For each pathway we report the name, the number of probes (size) and the most relevant statistical parameters as measured by GLAPA and GSEA tools.
Biological and functional analysis
We analyzed in depth some gene sets found deregulated with high statistical significance in the current experimental conditions for finding biological confirmations of their involvement in the pathology. In particular, we studied 2 pathways found perturbed by both methods: ADIPOCYTE BRCA UP and CELL CYCLE CHECKPOINT. The former was analyzed because this pathway seemed at first glance to be not correlated with colon cancer. In fact this gene set was found to be deregulated in breast cancer [24]. Moreover, both methods indicated a strong and statistically significant deregulation of this pathway in the current data set (see Table 1). The latter was analyzed because this pathway is not cancer specific. In fact, cell cycle has been identified as one of the hallmarks of cancer [25]. Moreover, we studied the biological relevance of a third gene set, HDACI COLON SUL12HRS UP, which is known to be correlated to colon cancer [26], and found statistically deregulated by GLAPA only. In fact this method showed a prediction error of 13% (py =0.006, FDRy =0.024,pn =0.074, FDRn =0.439) in classifying the phenotypic status of specimens by using the expression levels of the genes belonging to this gene set.
ADIPOCYTE BRCA UP
This gene set, composed of ten genes, was experimentally determined and found upregulated in breast cancer cells (MCF-7) treated with adipocyte-conditioned growth media [24]. However, an analysis in detail of the genes co-expressed shows a strong correlation of this pathway with colon cancer. ATF3 (activating transcription factor 3; Location: 1q32.3), a member of the ATF/CREB family, is a eukaryotic transcription factor that is upregulated transcriptionally during the cellular response to a variety of stresses, in particular the DNA damage [27]. Dysfunction of ATF3 impairs the p53-mediated cellular response to DNA damage, allowing cells to be readily transformed by oncogenes. Consistent with this notion is the observation of downregulated ATF3 expression in most human cancers [28,29]. Furthermore, ATF3 may play a pivotal role in DIM(3,3'-diindolylmethane)-induced NAG-1(Nonsteroidal anti-inflammatory drug-activated gene-1) expression in human colorectal cancer cells [30]. The second gene analyzed was IGF2 (insulin-like growth factor 2; Location: 11p15.5) that plays a critical role in the regulation of cell growth and transformation. IGF-I and IGF-II inhibit apoptosis, promote tumor growth, and induce transformation and metastasis in many types of malignancies. The gastrointestinal system may be one of the major targets of the IGF action and there is increasing evidence that alterations in IGF signaling are involved in the neoplastic transformation and progression of the colorectal carcinoma. A significant overexpression of IGF-II mRNA and protein levels have been reported in 30-40% of colorectal carcinoma patients [31]. It has also been suggested that IGF-II plays a role in the development of liver metastasis from colorectal cancer and that 44% of colorectal cancer patients showed loss of imprinting of IGF-II [32]. For this reason, it may be a valuable predictive marker of colorectal cancer [33]. Another gene analyzed was MMP1 (matrix metallopeptidase 1; Location: 11q22.3). It is a component of matrix metalloproteinases (MMPs) that collectively degrade most of the components of the extracellular matrix (ECM), contributing to the proliferation, invasion and metastasis of tumor cells by eliminating the surrounding ECM barrier [34,35]. Numerous MMPs, including MMP1, MMP3 and MMP7, have been associated also with the development of colorectal cancer [35, 36]. Another gene belonging to ADIPOCYTE BRCA UP gene set was NFkB (nuclear factor of kappa light polypeptide gene enhancer in B-cells 1; Location: 4q24) that is a generic name for a transcription-factor system that is involved in the regulation of cell proliferation, development, and apoptosis. The analysis of the expression of NFkB in various colorectal carcinoma cell lines shows that the inactive cytoplasmic NFkB form is evidently up-regulated in the tumor epithelium, especially in the metastatic cases, as compared to normal tissue. The transcription factor SOX9 (sex determining region Y-box 9; Location: 17q24.3-q25.1) is another gene co-expressed in this pathway. A study carried out on a human colon carcinoma cell line showed that this gene down-regulates the human carcinoembryonic antigen (CEA) gene expression which contributes to the carcinogenesis, and induces apoptosis [37]. Altered patterns of STC1 (stanniocalcin 1; Location: 8p21-p11.2) expression have a role in human cancer development. Hypoxia can stimulate STC1 gene expression in various human cancer cell lines, including those derived from colon carcinomas [38].
CELL CYCLE CHECKPOINT
This gene set belongs to the Gene Ontology (GO) data base and is composed of twenty six genes. Cell cycle checkpoints are essential in eukaryotes for ensuring high fidelity transmission of genetic information from one generation to the next. They include DNA damage checkpoints, DNA replication checkpoints, spindle assembly checkpoints, and cytokinesis checkpoints. Also in this case we give a short description of the single genes belonging to this gene set, underlining their importance in oncogenesis. ABL1 (v-abl Abelson murine leukemia viral oncogene homolog 1; Location:9q34.1) proto-oncogene has been implicated in processes of cell differentiation, cell division, cell adhesion, death, and stress response. Several findings suggest that the 9q34 region was altered in some cases of sporadic colorectal carcinomas [39]. The protein encoded by ATM (ataxia telangiectasia mutated; Location:11q22-q23) gene is an important cell cycle checkpoint kinase which functions as a regulator of a wide variety of downstream proteins. ATM and the closely related kinase ATR are thought to be master controllers of cell cycle checkpoint signaling pathways which are involved in the cell response to DNA damage and for genome stability. The ATM gene could be valuable in the cancer's gene therapy. Frequent allelic imbalances at the ATM locus have been reported in colorectal cancer and some findings led us to hypothesize that loss of expression of this gene may have a role in the early stage of colorectal cancer development and it may be related to advanced tumor stage and poorer patient survival [40]. Another interesting gene analyzed was BRCA1 (breast cancer 1; Location: 17q21) which plays a role in maintaining genomic stability and acts as a tumor suppressor.
Defects in BRCA1 are a cause of genetic susceptibility to breast cancer, and BRCA1 mutation carriers have a 4-fold increased risk of colon cancer. Recent evidence shows that the expression of ATM and BRCA1 is a prognostic marker in colorectal cancer [40]. CHEK1 (checkpoint homolog; Location: 11q24-q24) gene is essential in human cells for cell cycle arrest in response to DNA damage, and has been shown to play an important role in the G2/M checkpoint. Some results suggest that the CHEK1 gene is a target of genomic instability in microsatellite instability (MSI)-positive colorectal cancers and that mutations of this gene might be involved in colorectal tumorigenesis [41]. Also CHEK2 (checkpoint homolog; Location: 22q12.1) is a cell cycle checkpoint regulator and it was considered a putative tumor suppressor. Recently, a functionally defective CHEK2 variant I157T has been proposed to associate with an increased risk of colorectal cancer in a large population based study including a significant number of familial and sporadic colorectal cancer cases [42]. Also analysis of GADD45 (growth arrest and DNA-damage-inducible, alpha; Location: 1p31.2-p31.1) was interesting. It is a growth arrest-associated gene that is induced in response to DNA damage. This gene is a target for coordinated regulation by both ZBRK1 and BRCA1. Analyzing the relationships between GADD45, ZBRK1, and BRCA1 expression in colon carcinomas, it was reported that this pathway is deregulated in colon carcinomas [43].
Another important finding was MAD2L1 gene (MAD2 mitotic arrest deficient-like 1; Location: 4q27) which expression was found higher in colorectal cancer than in the corresponding normal tissue. The expression of Mad2 in colorectal cancer was related with histological differentiation and lymph node metastasis. Overexpression of Mad2 protein in cancer tissue might be a marker for the prognosis of colorectal cancer [44].
MRE11A (MRE11 meiotic recombination 11 homolog A Location: 11q21) encodes a nuclear protein involved in homologous recombination, telomere length maintenance, and DNA double-strand break repair. MRE11 may be considered as a new common target in the mismatch repair deficient tumorigenesis with a role in colorectal carcinogenesis [45]. Also mutations of the mismatch repair gene, MSH3 (mutS homolog 3 ; Location: 5q11-q12) might play a role in the progression of tumors by increasing instability. Common polymorphisms in MSH3 may increase the risk of colorectal cancer, especially proximal colon cancer [46]. NBS1 (nibrin; Location: 8q21) is a member of the MRE11/RAD50/NBN complex which plays a critical role in the cellular response to DNA damage and the maintenance of chromosome integrity. Also NBS1 could be a tumor suppressor gene involved in proximal colorectal cancer [47]. Another important gene analyzed was RAD17 (RAD17 homolog; Location: 5q13) which is a cell cycle checkpoint gene required for cell cycle arrest and DNA damage repair. It is overexpressed in various cancer cell lines and in colon carcinoma. Its chromosomal localization suggests that a variety of human cancers shows a deletion of this gene [48].
RPA1 (replication protein A1; Location: 17p13.3) and RPA2 (replication protein A2; Location: 1p35) are required for the stabilization of single-stranded DNA during the DNA replication. Experimental studies in colon cancer cell lines have shown that the RPA protein may be the target of cytotoxins designed to inhibit cellular proliferation. So RPA1 and RPA2 proteins appear to be useful prognostic indicators in colon cancer patients and attractive therapeutic targets [49].
Finally we have analyzed TP53 (tumor protein p53; Location: 17p13.1) which plays an essential role in the regulation of cell cycle, specifically in the transition from G0 to G1. p53 is a tumor suppressor which activates the expression of downstream genes which inhibit growth and/or invasion, and induces apoptosis. Over 8000 mutations of this gene have been identified, and the spectrum of p53 mutations varies among tumor types. The G→A transition in codon 175 of p53 gene may be useful as a potential marker of colorectal cancer progression and in evaluating the margins of surgical resection [50].
HDACI COLON SUL12HRS UP
This gene set, composed of twenty six genes, seems to be specific for colorectal cancer. It was obtained experimentally by SW620 colonic epithelial cells as described in [26]. This gene set was found deregulated by GLAPA software only. ANXA2 (annexin A2; Location: 15q21-q22) gene encodes a member of the annexin family which plays a role in the regulation of cellular growth and in signal transduction pathways. In addition to these functions, it has been suggested that annexin II is involved in cell proliferation/differentiation and in the pathogenesis of carcinoma. Overexpression of annexin II has been reported in various carcinomas including colon malignant tumors and it may be related to the progression and metastatic spread of colorectal carcinoma [51]. Also up-regulation of ANXA5 (annexin A5; Location: 4q28-q32) is associated with human colon adenocarcinoma cell differentiation [52]. Another gene belonging to this list is CD55 gene (decay accelerating factor (DAF) for complement; Location: 1q32), a membrane glycoprotein which regulates the activation of the complement. The expression of DAF is enhanced in colorectal cancer cells and in the colonic epithelium of ulcerative colitis in relation to the degree of mucosal inflammation [53, 54]. The expression of DAF is detected in stool specimens of patients with colorectal cancer and its presence may be a valuable test for the detection of colorectal cancer [55-57]. The CDH1 (cadherin 1, type 1, E-cadherin (epithelial); Location: 16q22.1) gene belongs to the cadherin superfamily. Mutations in this gene are correlated with gastric, breast, colorectal, thyroid and ovarian cancer. Loss of function is thought to contribute to progression in cancer by increasing proliferation, invasion, and/or metastasis. The examination of E-cadherin expression and distribution in colorectal tumors can be extremely valuable in predicting disease recurrence [58]. Moreover, some findings suggest that E-cadherin may play an important role in tumor metastasis in colorectal cancer [59]. Another important gene analyzed was GSR (glutathione reductase; Location: 8p21.1). The gastrointestinal tract is particularly susceptible to reactive oxygen species attacks which lead to carcinogenesis, and an important role in the defense strategy against these reactive oxygen species is played by antioxidants. The activity of glutathione reductase results increased in colorectal tumor [60]. HSP90AA1 (heat shock protein 90kDa alpha (cytosolic), class A member 1; Location: 14q32.33) gene is a member of molecular chaperones and it was thought to promote tumor cell survival. HSP90 was low or non-detectable in normal colon tissues while high levels of HSP90 expression were observed in human colon cancer tissues, suggesting that HSP90 expression is a promising marker for malignant colon cancer [61]. Furthermore HSP90 are marker genes for the stress signaling pathway, so the induction of these genes in colorectal cancer tissue indicate the activation of stress signaling pathway in cancer tissue [62]. The protein encoded by MYC (v-myc myelocytomatosis viral oncogene homolog (avian); Location: 8q24.21) gene is a multifunctional, nuclear phosphoprotein that plays a role in cell cycle progression, apoptosis and cellular transformation. It functions as a transcription factor that regulates transcription of specific target genes. Some findings indicate that failure of the normal apoptotic process together with de-regulation of c-MYC proto-oncogene might promote the development of colorectal tumors and its overexpression is observed in most colorectal cancers [63, 64].
An essential requirement for the development, progression and metastasis of malignant tumors is angiogenesis. VEGF (vascular endothelial growth factor; Location: 6p12), a member of the PDGF/VEGF growth factor family, plays an essential role in the development of angiogenesis of numerous solid malignancies including colon cancer. VEGF is associated with the development and prognosis of colorectal cancer, but its relation with degree of differentiation remains to be studied [65]. Some data suggested that VEGF functions as regulator of colon cancer cell invasion.
A key regulator of the expression of VEGF is the Sodium butyrate (NaB), a short-chain fatty acid naturally present in the human colon. NaB is able to induce cell cycle arrest, differentiation and apoptosis in colon cancer cells suggesting a possible clinical application of this fatty acid as an anti-angiogenic compound in association with conventional chemotherapeutic agents [66]. Other factors which may induce alterations of VEGF expression in colon cancer are hypoxia, mutations of p53 and activation of the Ras/MAPK pathway [67, 68].
Conclusions
In this paper we have described the biological and functional relevance in colon cancer of some pathways found deregulated in a gene expression profile data set composed of normal and tumor specimens of patients affected by this pathology [16]. Other studies have pointed out the fundamental role of pathways in studying onset and progression of tumors. In [8], human primary mammary epithelial cell cultures (HMECs) were used for studying in vitro pathways associated to the activation of Myc, Ras, E2F3, Src and β-catenin oncogenes. To this end, recombinant adenoviruses were used to express the activities of these oncogenes in an otherwise quiescent cell and RNA from multiple independent infections were collected for DNA microarray analysis using Affymetrix Human Genome U133 Plus 2.0 Array. For each oncogene, a microarray data set was used to identify a gene set (signature) associated to the activity of the oncogene. This unique signature was used to assess the activity of the oncogene in lung and breast cancer. In [69], a pathway approach was used to study the genetic perturbations implicated in initiation and progression of prostate cancer and melanoma. In particular, pathways interaction networks were inferred for relevant pathways over the steps in progression.
Finally, in [15] the deregulation of pathways was measured in human prostate cancer by using gene sets experimentally derived from cell types other than prostate. In our study, two well founded statistical methods were applied to measure the deregulation of pathways and to assess their statistical significance. GSEA [12] is an associative method which determines gene sets statistically correlated with the phenotype. GLAPA [14] is a predictive method which classifies the specimens by using the expression levels of the genes belonging to a given pathway. Since it evaluates the prediction error of the phenotypic status of new specimens, it can be used as a prognostic tool. In particular, we measured the deregulation in colon cancer of the whole MSigDB collection, a curated pathway database composed of 1687 gene sets obtained from different sources [12]. In our analysis, we have shown the relevance in colon cancer of two pathways found statistically altered by both methods. The former was a gene set which seemed to be not associated to colon cancer. In fact it was found implicated in breast cancer [24]. The latter was a pathway usually altered in cancer. In fact, cell cycle has been identified as one of the hallmarks of cancer [25]. Moreover, we have studied the biological relevance of a third gene set, which is known to be correlated to colon cancer and that was found implicated in the pathology at hand by GLAPA only.
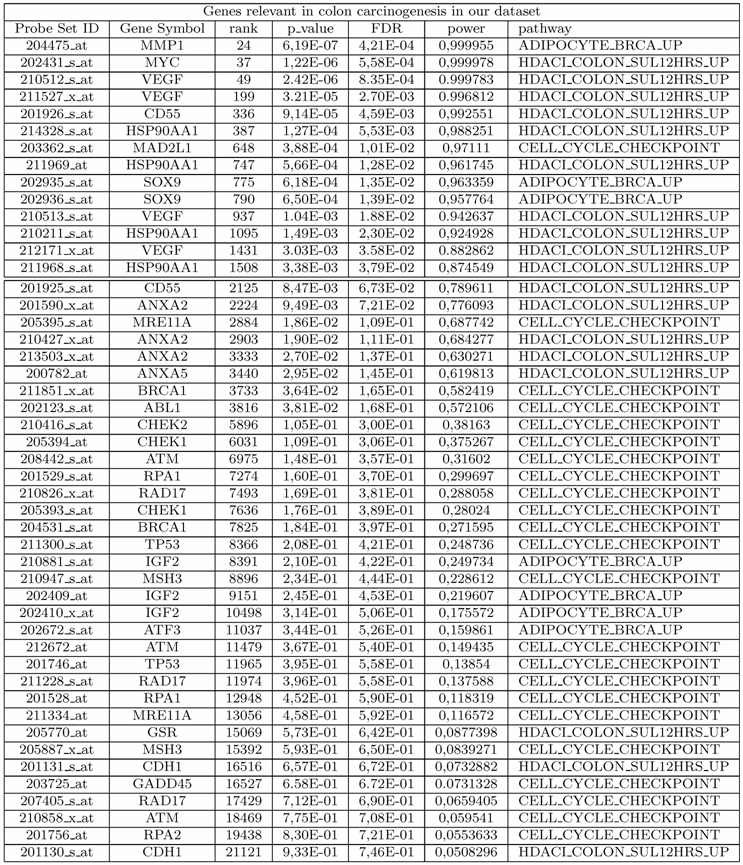
Our study highlights the importance of using gene sets rather than single genes for understanding the main biological processes and pathways involved in colorectal cancer. Our analysis shows that many of the genes involved in pathways found deregulated are strongly associated to colorectal tumorigenesis. Many of the genes that we have discussed in this work, and found involved in the pathology at hand, would not have been detected by the classical single gene approach. In fact, by applying a two-sample t-test statistics to the expression levels of each of 22283 probes in our data set, we found 1743 statistically significant DE probes with FDR ≤ 5%. Among these, only 14 belong to the set of probes determined by using the pathway approach and found strongly implicated in colon cancer as demonstrated by our analysis (see Table 2). Note that, 34 out of 48 probes showed associated to the pathology, having rank in the range [2125,21121], would not have been analyzed by adopting classical single gene approach. This highlights the importance of pathway approach to the study of complex diseases because it allows detection of genes weakly correlated to the phenotype of interest which would be difficult to find using classical univariate statistics. So individual gene analysis could give a limited interpretation of the processes involved in the tumorigenesis and could not be able to provide a full description of the complex interactions among genes. Unlike single gene analysis, pathways analysis allows to get a more complete picture of altered biological processes in cancer pathologies.
List of genes relevant in colon cancer as measured in our data set and discussed in our analysis. For each probe, we show the probe set ID, the rank, the nominal p-value and power of a two-sample t-test, the FDR and the corresponding pathway The first 14 probes have FDR ≤ 5%.
Conflict of interests
The author(s) declare that they have no competing interests.
Acknowledgements
R.M. and A.D'A. are PhD students of Dipartimento Interateneo di Fisica, Bari, associated to Istituto Nazionale di Fisica Nucleare, sez. di Bari, and to Center of Innovative Technologies for Signal Detection and Processing (TIRES), Università degli Studi di Bari, Italy. This work was supported by grants from Regione Puglia, Progetto Strategico PS 012.
References
1. Schena M, Shalon D, Davis RW, Brown PO. Quantitative monitoring of gene-expression patterns with a complementary-DNA microarray. Science. 1995;270:467-470
2. Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, Coller H, Loh ML, Downing JR, Caligiuri MA, Bloomfield CD, Lander ES. Molecular Classification of Cancer: Class Discovery and Class Prediction by Gene Expression Monitoring. Science. 1999;286:531-537
3. Barrier A, Boelle PY, Roser F, Gregg J, Tse C, Brault D, Lacaine F, Houry S, Huguier M, Franc B, Flahault A, Lemoine A, Dudoit S. Stage II Colon Cancer Prognosis Prediction by Tumor Gene Expression Profiling. J Clin Oncol. 2006;24(29):4685-4691
4. Storey J, Tibshirani R. Statistical significance for genomwide studies. Proc Natl Acad Sci. 2003;100:9440-9445
5. Ashburner M, Ball C, Blake J, Botstein D, Butler H, Cherry J, Davis A, Dolinski K, Dwight S, Eppig J, Harris M, Hill D, Issel-Tarver L, Kasarskis A, Lewis S, Matese J, Richardson J, Ringwald M, Rubin G, Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25-29
6. Kanehisa M, Goto S, Kawashima S, Nakaya A. The KEGG databases at GenomeNet. Nucleic Acids Res. 2002;30:42-46
7. Khatri P, Draghici S, Ostermeier G, Krawetz S. Profiling Gene Expression Using Onto-Express. Genomics. 2002;79(2):266-270
8. Bild A, Yao G, Chang J, Wang Q, Potti A, Chasse D, Joshi M, Harpole D, Lancaster J, Berchuck A, Olson J, Marks J, Dressman H, West J M Nevins. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature. 2006;439(19):353-357
9. Khatri P, Draghici S. Ontological analysis of gene expression data: current tools, limitations, and open problems. Bioinformatics. 2005;21:3587-3595
10. Fortunel NO, Otu HH, Ng HH, Chen J, Mu X, Chevassut T, Li X, Joseph M, Bailey C, Hatzfeld JA. et al. author reply 393. Science. 2003;302:393
11. NJ R. Searching for genetic determinants in the new millennium. Nature. 2000;405:847-856
12. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci. 2005;102:15545-15550
13. Tian L, Greenberg S, Kong S, Altschuler J, Kohane I, Park P. Discovering statistically significant pathways in expression profiling studies. Proc Natl Acad Sci. 2005;102:13544-13549
14. Maglietta R, Piepoli A, Catalano D, Licciulli F, Carella M, Liuni S, Pesole G, Perri F, Ancona N. Statistical assessment of functional categories of genes deregulated in pathological conditions by using microarray data. Bioinformatics. 2007;23(16):2063-2072
15. Creighton CJ. Multiple oncogenis Pathway Signatures Show Coordinate Expression Patterns in Human Prostate Tumors. PLoS ONE. 2008;3(3):e1816
16. Ancona N, Maglietta R, Piepoli A, D'Addabbo A, Cotugno R, Savino M, Liuni S, Carella M, Pesole G, Perri F. On the statistical assessment of classifiers using DNA microarray data. BMC Bioinformatics. 2006;7:387
17. Good P. Permutation tests: a practical guide to resampling methods for testing hypotheses. New York: Springer Verlag, New York, Inc. 1994
18. Vapnik V. The Nature of Statistical Learning Theory. New York: Springer Verlag. 1995
19. Rifkin R, Yeo G, Poggio T. Regularized least squares classification. In: (ed.) Suykens J. et al. Advances in Learning Theory: Methods, Model and Applications, NATO Science Series III: Computer and Systems Sciences. Amsterdam: IOS Press. 2003:131-153
20. Ancona N, Maglietta R, D'Addabbo A, Liuni S, Pesole G. Regularized least squares cancer classifiers from DNA microarray data. BMC-Bioinformatics. 2005;6(Suppl 4):S2
21. Maglietta R, D'Addabbo A, Piepoli A, Perri F, Liuni S, Pesole G, Ancona N. Selection of relevant genes in cancer diagnosis based on their prediction accuracy. Artif Intell Med. 2007;40:29-44
22. Mukherjee S, Tamayo P, Rogers S, Rifkin R, Engle A, Campbell C, Golub TR, Mesirov JP. Estimating dataset size requirements for classifying DNA microarray data. J Comp Biol. 2003;10:119-142
23. Michiels S, Koscielny S, Hill C. Predictor of cancer outcome with microarrays: a multiple random validation strategy. Lancet. 2005;365:488-492
24. Iyengar P, Combs TP, Shah SJ, Gouon-Evans V, Pollard JW, Albanese C, Flanagan L, Tenniswood MP, Guha C, Lisanti MP, Pestell RG, Scherer PE. Adipocyte-secreted factors synergistically promote mammary tumorigenesis through induction of anti-apoptotic transcriptional programs and proto-oncogene stabilization. Oncogene. 2003;22(41):6408-6423
25. Hanahan D, Weinberg RA. The Hallmarks of Cancer. Cell. 2000;100:57-70
26. Mariadason JM, Corner GA, Augenlicht LH. Genetic reprogramming in pathways of colonic cell maturation induced by short chain fatty acids: comparison with trichostatin A, sulindac, and curcumin and implications for chemoprevention of colon cancer. Cancer Res. 2000;60(16):4561-4572
27. Wang A, Arantes S, Conti C, McArthur M, Aldaz CM, MacLeod MC. Epidermal hyperplasia and oral carcinoma in mice overexpressing the transcription factor ATF3 in basal epithelial cells. Md Carcinog. 2007;46(6):476-487
28. Yan C, Boyd DD. ATF3 regulates the stability of p53: a link to cancer. Cell Cycle. 2006;5(9):926-929
29. Zhang C, Gao C, Kawauchi J, Hashimoto Y, Tsuchida N, Kitajima S. Transcriptional activation of the human stress-inducible transcriptional repressor ATF3 gene promoter by p53. Biochemical and Biophysical Research Communications. 2002;297:1302-1310
30. Lee SH, Kim JS, Yamaguchi K, Eling TE, Baek SJ. Indole-3-carbinol and 3,3'-diindolylmethane induce expression of NAG-1 in a p53-independent manner. Biochem Biophys Res Commun. 2005;328(1):63-69
31. Weber MM, Fottner C, Liu SB, Jung MC, Engelhardt D, Baretton GB. Overexpression of the Insulin-Like Growth Factor I Receptor in Human Colon Carcinomas. CANCER. 2002;95(10):2086-2095
32. Nosho K, Yamamoto H, Taniguchi H, Adachi Y, Yoshida Y, Arimura Y, Endo T, Hinoda Y, Imai K. Interplay of Insulin-Like Growth Factor-II, Insulin-Like Growth Factor-I, Insulin-Like Growth Factor-I Receptor, COX-2, and Matrix Metalloproteinase-7, Play Key Roles in the Early Stage of Colorectal Carcinogenesis. Clinical Cancer Research. 2004;10:7950-7957
33. Woodson W, Flood A, Green L, Tangrea JA, Hanson J, Cash B, Schatzkin A, Schoenfeld P. Loss of Insulin-Like Growth Factor-II Imprinting and the Presence of Screen-Detected Colorectal Adenomas in Women. Journal of the National Cancer Institute. 2004;96(5):407-410
34. Seiki M. Membrane-type 1 matrix metalloproteinase: a key enzyme for tumor invasion. Cancer Letters. 2003;194:1-11
35. Ye S. Polymorphism in matrix metalloproteinase gene promoters: implication in regulation of gene expression and susceptibility of various diseases. Matrix Biol. 2000;19:623-629
36. Hewitt RE, Leach IH, Powe DG, Clark IM, Cawston TE, Turner DR. Distribution of collagenase and tissue inhibitor of metalloproteinases (TIMP) in colorectal tumours. Int J Cancer. 1991;49:666-672
37. Jay P, Berta P, Blache P. Expression of the Carcinoembryonic Antigen Gene Is Inhibited by SOX9 in Human Colon Carcinoma Cells. Cancer Res. 2005;65(6):2193-2198
38. Yeung HY, Lai KP, Chan HY, Mak NK, Wagner GF, Wong CKC. Hypoxia-Inducible Factor-1-Mediated Activation of Stanniocalcin-1 in Human Cancer Cells. Endocrinology. 2005;146(11):4951-4960
39. Bartos JD, Gaile DP, McQuaidb DE, Conroy JM, Darbary H, Nowak NJ, Block A, Petrelli NJ, Mittelman A, Stoler DL, Anderson GR. aCGH local copy number aberrations associated with overall copy number genomic instability in colorectal cancer: Coordinate involvement of the regions including BCR and ABL. Mutation Research. 2007;615:1-11
40. Grabsch H, Dattani M, Barker L, Maughan N, Maude K, Hansen O, Gabbert HE, Quirke P, Mueller W. Expression of DNA Double-Strand Break Repair Proteins ATM and BRCA1 Predicts Survival in Colorectal Cancer. Clin Cancer Res. 2006;12(5):1494-1500
41. Kim CJ, Lee JH, Song JW, Cho YG, Kim SY, Nam SW, Yoo NJ, Park WS, Lee JY. Chk1 frameshift mutation in sporadic and hereditary non-polyposis colorectal cancers with microsatellite instability. EJSO. 2007;33:580-585
42. Kilpivaara O, Alhopuro P, Vahteristo P, Aaltonen LA, Nevanlinna HJ. CHEK2 I157T associates with familial and sporadic colorectal cancer. Med Genet. 2006;43(7):e34
43. Garcia V, Garcia JM, Pena C, Silva J, Dominguez G, Rodriguez R, Maximiano C, Espinosa R, Espana P. The GADD45, ZBRK1 and BRCA1 pathway: quantitative analysis of mRNA expression in colon carcinomas. J Pathol. 2005;206(1):92-99
44. Li GQ, Zhang HF. Mad2 and p27 expression profiles in colorectal cancer and its clinical significance. World J Gastroenterol. 2004;10(21):3218-3220
45. Giannini G, Rinaldi C, Ristori E, Ambrosini MI, Cerignoli F, Viel A, Bidoli E, Berni S, D'Amati G, Scambia G, Frati L, Screpanti I, Gulino A. Mutations of an intronic repeat induce impaired MRE11 expression in primary human cancer with microsatellite instability. Oncogene. 2004;23(15):2640-2647
46. Berndt SI, Platz EA, Fallin MD, Thuita LW, Hoffman SC, Helzlsouer KJ. Mismatch repair polymorphisms and the risk of colorectal cancer. Int J Cancer. 2007;120(7):1548-1554
47. Uhrhammer N, Bay JO, Gosse-Brun S, Kwiatkowski F, Rio P, Daver A, Bignon YJ. Allelic imbalance at NBS1 is frequent in both proximal and distal colorectal carcinoma. Oncol Rep. 2000;7(2):427-431
48. Bao S, Chang MS, Auclair D, Sun Y, Wang Y, Wong WK, Zhang J, Liu Y, Qian X, Sutherland R, Magi-Galluzi C, Weisberg E, Cheng EYS, Hao L, Sasaki H, Campbell MS, Kraeft SK, Loda M, Lo KM, Chen LB. HRad17, a Human Homologue of the Schizosaccharomyces pombe Checkpoint Gene rad17, Is Overexpressed in Colon Carcinoma. CANCER RESEARCH. 1999;59:2023-2028
49. Givalos N, Gakiopoulou H, Skliri M, Bousboukea K, Konstantinidou AE, Korkolopoulou P, Lelouda M, Kouraklis G, Patsouris E, Karatzas G. Replication protein A is an independent prognostic indicator with potential therapeutic implications in colon cancer. Modern Pathology. 2007;20:159-166
50. Krajewska WM, Stawinska M, Brys M, Mlynarski W, Witas H, Okruszek A, Kilianska ZM. Genotyping of p53 codon 175 in colorectal cancer. Med Sci Monit. 2003;9(5):228-231
51. Emoto K, Yamada Y, Sawada H, Fujimoto H, Ueno M, Takayama T, Kamada K, Naito A, Hirao S, Nakajima S. Annexin II Overexpression Correlates with Stromal Tenascin-C Overexpression. A Prognostic Marker in Colorectal Carcinoma. Cancer. 2001;92:1419-1426
52. Guzman-Aranguez A, Olmo N, Turnay J, Lecona E, Perez-Ramos P, Lopez de Silanes I, Lizarbe MA. Differentiation of human colon adenocarcinoma cells alters the expression and intracellular localization of annexins A1, A2, and A5. J Cell Biochem. 2005;94(1):178-193
53. OKAZAKI H, MIZUNO M, NASU J, MAKIDONO C, HIRAOKA S, YAMAMOTO K, OKADA H, FUJITA T, TSUJI T, SHIRATORI Y. Difference in Ulex europaeus agglutinin I-binding activity of decay-accelerating factor detected in the stools of patients with colorectal cancer and ulcerative colitis. J Lab Clin Med. 2004;143(3):169-174
54. Durrant LG, Chapman MA, Buckley DJ, Spendlove I, Robins RA, Armitage NC. Enhanced expression of the complement regulatory protein CD55 predicts a poor prognosis in colorectal cancer patients. Cancer Immunol Immunother. 2003;52:638-642
55. Iwagaki N, Mizuno M, Nasu J, Mizuno M, Okazaki H, Hori S, Yamamoto K, Okada H, Tsuji T, Fujita T, Shiratori Y. Advances in the development of a reliable assay for the measurement of stool decay-accelerating factor in the detection of colorectal cancer. J Immunoassay Immunochem. 2002;23(4):497-507
56. Mizuno M, Mizuno M, Iwagaki N, Nasu J, Okazaki H, Yamamoto K, Okada H, Tsuji T, Fujita T, Shiratori Y. Testing of multiple samples increases the sensitivity of stool decay-accelerating factor test for the detection of colorectal cancer. Am J Gastroenterol. 2003;98(11):2550-2555
57. Nakagawa M, Mizuno M, Kawada M, Uesu T, Nasu J, Takeuchi K, Okada H, Endo Y, Fujita T, Tsuji T. Polymorphic expression of decay-accelerating factor in human colorectal cancer. J Gastroenterol Hepatol. 2001;16(2):184-189
58. Elzagheid A, Algars A, Bendardaf R, Lamlum H, Ristamaki R, Collan Y, Syrjanen K, Pyrhonen S. E-cadherin expression pattern in primary colorectal carcinomas and their metastases reflects disease outcome. World J Gastroenterol. 2006;12(27):4304-4309
59. Furuta K, Yoshioka S, Okabe S, Ikeda M, Oginosawa M, Ikeda S, Nakayama Y, Kikuchi M, Hamilton SR. Expressions of two adenomatous polyposis coli and E-cadherin proteins on human colorectal cancers. Virchows Arch. 2003;442(3):266-270
60. Skrzydlewska E, Kozuszko B, Sulkowska M, Bogdan Z, Kozlowski M, Snarska J, Puchalski Z, Sulkowski S, Skrzydlewski Z. Antioxidant potential in esophageal, stomach and colorectal cancers. Hepatogastroenterology. 2003;50(49):126-131
61. Park KA, Byun HS, Won M, Yang KJ, Shin S, Piao L, Kim JM, Yoon WH, Junn E, Park J, H SJ, Hur GM. Sustained activation of protein kinase C downregulates nuclear factor-kB signaling by dissociation of IKK-g and Hsp90 complex in human colonic epithelial cells. Carcinogenesis. 2007;28(1):71-80
62. Cen H, Zheng S, Fang YM, Tang XP, Dong Q. Induction of HSF1 expression is associated with sporadic colorectal cancer. World J Gastroenterol. 2004;10(21):3122-3126
63. Greco C, Alvino S, Buglioni S, Assisi D, Lapenta R, Grassi A, Stigliano V, Mottolese M, Casale V. Activation of c-MYC and c-MYB proto-oncogenes is associated with decreased apoptosis in tumor colon progression. Anticancer Res. 2001;21(5):3185-3192
64. Seidler HBK, Utsuyama M, Nagaoka S, Takemura T, Kitagawa M, Hirokawa K. Expression level of Wnt signaling components possibly influences the biological behavior of colorectal cancer in different age groups. Experimental and Molecular Pathology. 2004;76:224-233
65. Han J, Xia C, Gao J, Xing C, Yang X, Tang X, Qiu F, Du Y. Expression of vascular endothelial growth factor in colorectal cancer and its clinical significance. Zhonghua Yi Xue Za Zhi. 2002;82(7):481-483
66. Pellizzaro C, Coradini D, Dandone MG. Modulation of angiogenesis-related proteins synthesis by sodium butyrate in colon cancer cell line HT29. Carcinogenesis. 2002;23(5):735-740
67. Faviana P, Boldrini L, Spisni R, Berti P, Galleri D, Biondi R, Camacci T, Materazzi G, Pingitore R, Miccoli P, Fontanini G. Neoangiogenesis in colon cancer: correlation between vascular density, vascular endothelial growth factor (VEGF) and p53 protein expression. Oncol Rep. 2002;9(3):617-620
68. Cassano A, Bagala C, Battelli C, Schinzari G, Quirino M, Ratto C, Landriscina M, Barone C. Expression of vascular endothelial growth factor, mitogen-activated protein kinase and p53 in human colorectal cancer. Anticancer Res. 2002;22(4):2179-2184
69. Edelman E, Guinney J, Chi J, Febbo P, S M. Modeling cancer progression via pathway dependencies. PLoS Comput Biol. 2008;4(2):e28
Author contact
![]() Correspondence to: Nicola Ancona - anconaissia.cnr.it
Correspondence to: Nicola Ancona - anconaissia.cnr.it