Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2009; 5(2):135-146. doi:10.7150/ijbs.5.135 This issue Cite
Research Paper
Antitumor Activity of T Cells Generated from Lymph Nodes Draining the SEA-expressing Murine B16 Melanoma and Secondarily Activated with Dendritic Cells
Jiyun Yu1*, Rong Tian1*, Bingshui Xiu1*, Jinqi Yan1, Rui Jia1, Liang Zhang1, Alfred E. Chang2, Hongbin Song3 ![]() , Qiao Li1, 2
, Qiao Li1, 2 ![]()
1. Institute of Basic Medical Sciences, Academy of Military Medical Sciences, Beijing, China
2. Division of Surgical Oncology, University of Michigan, Ann Arbor, Michigan, USA
3. Institute of Disease Control and Prevention, Academy of Military Medical Sciences, Beijing, China
* These authors contributed equally to the work
Received 2009-1-13; Accepted 2009-1-19; Published 2009-1-20
Abstract
The successful use of tumor-draining lymph nodes (TDLN) as a source of effector cells for cancer immunotherapy depends largely on the immunogenicity of the tumor drained by the lymph nodes as well as the methods for secondary in vitro T cell activation and expansion. We transferred the bacterial superantigen staphylococcal enterotoxin A (SEA) gene into B16 murine melanoma tumor cells, and used them to induce TDLN (SEA TDLN) in syngeneic hosts. Wild-type (wt) TDLN induced by parental B16 tumor was used as a control. In vitro, SEA TDLN cells proliferated more vigorously, produced more IFNγ and demonstrated higher CTL activity than wt TDLN cells when activated with anti-CD3/anti-CD28/IL-2. In vivo, SEA TDLN cells mediated tumor eradication more effectively than similarly activated wt TDLN cells (p<0.01). Furthermore, use of dendritic cells (DC) plus tumor antigen in vitro in addition to anti-CD3/anti-CD28/IL-2 stimulation further amplified the immune function and therapeutic efficacy of SEA TDLN cells. DC-stimulated SEA TDLN cells eliminated nearly 90% of the pulmonary metastasis in mice bearing established B16 melanoma micrometastases. These results indicate that enforced expression of superantigen SEA in poorly immunogenic tumor cells can enhance their immunogenicity as a vaccine in vivo. The combined use of genetically modified tumor cells as vaccine to induce TDLN followed by secondary stimulation using antigen-presenting cells and tumor antigen in a sequential immunization/activation procedure may represent a unique method to generate more potent effector T cells for adoptive immunotherapy of cancer.
Keywords: Staphylococcal enterotoxin A (SEA), B16 melanoma, dendritic cells (DC), T lymphocyte, Adoptive therapy
Introduction
Adoptive T cell therapy has been used with success for the treatment of malignant diseases [1-4]. In addition to peripheral blood and tumor-infiltrating lymphocytes (TIL) [1-2], tumor-draining lymph nodes (TDLN) have been used successfully to provide “pre-effectors” for the generation of antitumor “effector” T cells for adoptive transfer [3-8]. Tumor cells are typically poorly immunogenic. The same mechanisms that developed to avoid the induction of immune responses against self tissues, e.g. autoimmunity, have to be overcome during the immunotherapy of cancer. Since the majority of malignancies arise in immunocompetent hosts, it is suggested that tumor cells escape normal host surveillance and defenses. Therefore, appropriate immune modification and stimulation may grant therapeutic benefits.
To modulate tumor immunogenicity, multiple strategies have been attempted. Genetic modification by introducing genes encoding immunomodulating molecules into tumor cells represents one approach. To this end, retroviruses encoding MHC II molecules were introduced directly into mammary carcinoma and ocular melanoma cells to facilitate the presentation of MHC class II-restricted tumor peptides to CD4 T cells for tumor rejection [9]. Genetically modified tumors expressing costimulatory molecule CD40L elicited profoundly expanded tumor immune responses [10-11]. B7-dependent costimulatory signals play a central role in T cell activation. It has been proposed that the lack of immunogenicity of many tumor types could be due to the lack of B7 expression. Indeed, it was proved that transfection of B7 genes greatly augmented the immunogenicity of EL-4 lymphoma cells [12], leukemia cells [10], and sarcoma cells by inducing significant T cell activation, proliferation and production of Th1 cytokines, e.g. IL-2 and IFNγ [13]. Moreover, C26 carcinoma cells transduced with OX40 ligand (OX40L) together with GM-CSF, which is known to activate antigen-presenting cells (APC), mounted a superior antitumor response by interacting directly with both APC and T cells in vivo [14]. It was likewise reported that vaccination with rat 9L gliosarcoma cells expressing IL-4 induced specific protective immune response against challenge with parental 9L tumors by promoting both cellular and humoral responses [15]. In addition to mono- or bi-gene transfer, combinatorial costimulator protein transfer onto tumor cell surfaces was also evaluated for the induction of tumor immunogenicity. In highly aggressive L5178Y-R lymphomas, for example, a "tetra-costimulator" combination of B7-1, 4-1BBL, CD48, and CD40L induced complete tumor regression in 45% of treated mice and systemic antitumor immunity [16]. Overall, these achievements have made gene modification an attractive approach to enhance tumor immunogenicity. In addition, the role of BCG as an adjuvant in autologous tumor vaccines has been examined [17]. In patients with renal cell cancer, we inoculated irradiated tumor cells alone (wild-type, WT) or tumor cells with BCG intradermally into contralateral thighs to induce draining vaccine-primed lymph nodes (WT-VPLN and BCG-VPLN). BCG increased the number of harvested VPLN cells by 10-fold. BCG-VPLN had significantly greater percentages of CD3+ and CD4+ T cells compared to WT-VPLN. Anti-CD3/CD28-activated BCG-VPLN cells were found to have a greater release of IFNγ compared with WT-VPLN cells. Collectively, these data demonstrated the immunological effects of BCG and thus support its use as an adjuvant in autologous tumor vaccines.
Bacterial superantigen staphylococcal enterotoxin A (SEA) is an extremely potent activator of T lymphocytes when presented on MHC II molecules. SEA and SEB interact directly with the antigen-binding groove of MHCⅡ molecules and ligate T cell receptor on its Vβ region without the need of APC or antigen processing [18]. This leads to powerful activation of T cells, determined by TCR Vβ repertoire and MHC haplotype [19]. In one study, a tumor cell vaccine modified with transmembrane-anchored SEA (TM-SEA) using a protein transfer technique provoked a strong antitumor effect both in vitro and in vivo [20]. An alternative approach used SEA conjugated to tumor-specific mAb to deliver SEA to specific antigen-expressing tumor cells. The surface-bound SEA directed T cells to lyse the tumor cells in the absence of MHC II via low affinity TCR-SEA interaction [21]. These reports thus raised the prospect that genetic modification of tumor cells with the SEA gene may lead to the development of a novel therapeutic strategy to increase tumor immunogenicity.
Dendritic cells (DC) have been used in vivo either as a vaccine to induce antitumor pre-effector T cells in vaccine-primed lymph nodes (VPLN) or to boost adoptively transferred T cells. We have used un-pulsed DC in a couple of previous studies [22, 23]. DCs were injected intratumorally (i.t). In this way, the un-pulsed DC will present tumor antigen obtained from the tumor, which was proven to be the case in our subsequent studies [24]. We also reported that administration of DC pulsed with tumor lysate resulted in an approximately eightfold hypertrophy of the VPLN, with an increased influx of DC and B cells in the lymph nodes. The adoptive transfer of DC-induced VPLN cells secondarily activated with anti-CD3/IL-2 resulted in regression of established 3-day pulmonary metastases [25]. When tumor-bearing mice were treated with cultured T cells, concurrent vaccination using peptide-pulsed DC during adoptive T cell transfer led to a more robust antitumor response than the use of each treatment individually [26]. DC-mediated potent antitumor immune responses in vivo correlated with DC-modulated vigorous antitumor immunity in vitro [27]. This led to the prediction that DC may function as a powerful stimulator during T cell culture in vitro to generate effector cells for adoptive transfer. We have used polyclonal T cell specific antibodies anti-CD3, anti-CD28 and T cell growth factor IL-2 to generate tumor-reactive T cells for cancer adoptive immunotherapy [7-8, 28-30]. We postulated that use of anti-CD3/anti-CD28/IL-2 in concert with DC plus tumor antigen in culture might generate large numbers of effector T cells from TDLN with augmented antitumor reactivity.
The murine B16 melanoma cell line is poorly immunogenic and highly aggressive. In this study, we transfected SEA gene into B16 melanoma tumor cells by means of liposome-mediated transfection. We explored whether this strategy would increase the immunogenicity of the B16 tumor by examining the induction of tumor-reactive T cells within the lymph nodes draining the genetically modified tumor. Furthermore, we examined the potential of using DC and tumor antigen in addition to anti-CD3/anti-CD28/IL-2 in vitro to generate more potent antitumor effector T cells from these lymph node cells previously sensitized in vivo by the SEA-expressing B16 tumor vaccine.
Materials and methods
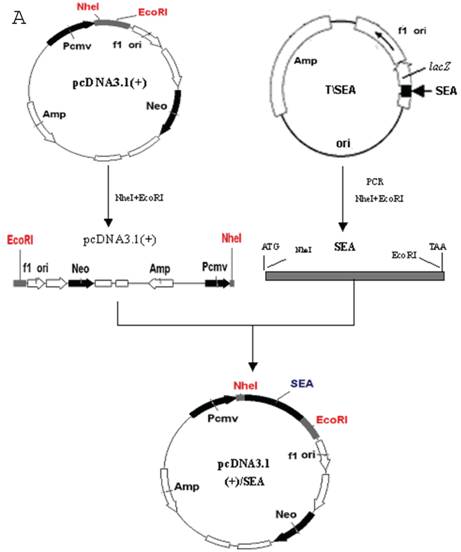
1. The construction of recombinant pcDNA3.1 (+)/SEA
The SEA gene was amplified by PCR using the plasmid pGEM-Teasy-SEA as a template. The sequence was amplified with sense (F1; 5'-CTCAGCTAGCCACCATGAAAAAAACAGCATTTACATTA-3') and antisense (R1; 5'-CCGGAATTCACTTGTATATAAATAT-3') primers. The PCR product was digested with Nhe I and EcoR I and ligated with similarly digested pcDNA3.1 (+). The recombinant pcDNA3.1 (+)/SEA was analyzed by restriction enzyme digestion and the correct plamids were identified by DNA sequencing.
2. Cell lines and transfections
B16 tumor cells were maintained in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 15% fetal bovine serum, 100U of penicillin and 100 μg of streptomycin/ml. Cells were transfected with 10 μg of plasmid by Lipofectamine 2000 following the manufacturer's instructions (Invitrogen, Carlsbad, CA). The stably transfected B16 cells were seeded into selection medium containing 400 μg/ml of G418 to obtain G418-resistant clones. The G418-resistant clones were then maintained in G418 at a concentration of 200 μg/ml. Total RNA was isolated and cDNA was synthesized by AMV reverse transcriptase (Promega, Madison, WI). SEA gene was amplified by PCR with F1 and R1. The products were then identified by DNA agarose gel electrophoresis.
3. Animals and tumors
B16 melanoma was from Cell Centre of Shanghai, China. The syngeneic mice, C57BL/6 (B6) mice, female, were purchased from the Animal Center of Academy of Military Medical Sciences (Beijing, China) and were used at 8 weeks or older.
4. Generation of DC
Mice were injected via tail vein with cyclophosphamide at 200 mg/kg to induce splenomegaly. Eight days later, spleens were harvested. Splenocyte suspension was prepared and depleted of erythrocytes. The resultant cells were suspended at 1×106 /ml in complete medium (CM). One liter CM is composed of 1, 000 ml RPMI 1640 medium (Gibco, Grand Tsland, NY) supplemented with 10% fetal calf serum (FCS), 2.60g Hepes (Huamei, , Beijing, China), 2g NaHCO3 and penicillin/streptomycin (Sigma, St.Louis, MO) at a final concentration of 100U/mL. Cells were cultured in flask at 37o with 5% CO2. Three days later, culture medium and the non-adherent cells were removed and discarded. Adherent cells were then cultured with fresh CM containing GM-CSF and IL-4. Recombinant mouse (rm) GM-CSF (20 ng/ml Gibco) and rmIL-4 (10 ng/ml Gibco) were added to CM for DC culture. Half of the culture medium was replaced with fresh medium every 2 days. Five days later, TNFα (1 ng/ml) was added. After culture for 2 more days, cells were collected for morphological observation under microscopes, and DC phenotype profile was confirmed by FACS staining.
5. Morphological observation of DCs
Phase-contrast microscope (Carl Zeiss, Oberkochen, Germany) was used for DC morphological observation. For use of scanning electron microscopy, cultured DC cells were put on glass cover slips. The slips were washed with PBS and then fixed in 4% precooled glutaraldehyde for 2 hour at 4oC. After washing 2 times with PBS, the cells were fixed again in 1% osmic acid for 1 hour at 4oC. Cells were washed with PBS twice followed by dehydration through a graded series of ethanol and were dried at CO2 critical-point. Specimens were then observed in a scanning electron microscope S-4500 (Hitachi, Ibaraki, Japan) at an accelerating voltage of 3kV. For use of transmission electron microscopy, fixed cells were embedded in Epon-812. Thin sections were then cut, stained with uracyl acetate and lead nitrate, and then examined in a transmission electron microscope TEM-600 (Hitachi, Ibaraki, Japan) at an accelerating voltage of 50kV.
6.Phenotype analysis of DC and intracellular staining of cytokines secreted by stimulated TDLN cells
DC cultures were sampled at day 3, 5, and day 7 respectively and stained with FITC conjugated anti-CD11c, anti-I-Ab, and anti-CD86 (all from Diaclone, Besancon, France). After activation, TDLN cells were permeablized and stained with FITC conjugated anti-IFN-γ or PE conjugated anti-IL-10 (both from Diaclone). Isotype-matching controls were used. FACS analysis was performed using a FACScan flow microfluorometer (Coulter, Fullerton, CA).
7. Induction of TDLN
To induce TDLN, 2×106 irradiated (25Gy) parental B16 cells or SEA gene-modified B16 cells plus 20 μl BCG (Sigma) in 0.1 ml PBS was inoculated subcutaneously (s.c) into the lower flanks of the B6 mice. The draining inguinal lymph nodes were collected 10 days later, and were designated as wt TDLN and SEA TDLN respectively.
8. Preparation of tumor lysate
B16 cells were suspended in CM (1×108/ml) and subjected to three times of rapid freeze-thaw exposures in liquid N2 and water bath, then spun at 12, 000rpm for 20min to remove cellular debris. The supernatant was saved and kept at -20oC for future use. Wild-type B16 tumor lysate was used in all of the experiments involving DC stimulation of either wt TDLN or SEA TDLN during the culture of cells.
9. Stimulation of TDLN T cells in vitro
Multiple inguinal TDLN were pooled from groups of mice and were processed using mechanical dissociation for lymphoid cell suspension. T cells were purified using a nylon wool column. Purified T cells suspended in CM were then stimulated in 96-well culture plate or in culture flask under the following conditions: (1) No stimulation (medium only); (2) Anti-CD3 (1 μg/ml) + anti-CD28 (1 μg/ml) + IL-2 (1000IU/ml) (all from Diaclone); (3) Anti-CD3 (1 μg/ml) + anti-CD28 (1 μg/ml) + IL-2 (1000IU/ml) + DC + tumor lysate. For each 1×105 T cells, 2×104 DCs were used along with tumor lysates equivalent to 1×105 tumor cells. Stimulation lasted for 72 hours at 37oC with 5% CO2.
10. Proliferation of TDLN T cells in vitro
Purified TDLN T cells were suspended at 5×106/ml in CM. 100μl of cell suspension was added to each well of a 96-well culture plate. The experiment was preformed in triplicate. Stimuli were added as described above at 100 μl/well. The cells were incubated at 37oC with 5% CO2 for 48 hours. 100μl supernatant was removed from each well and 10μl MTT (Sigma, stock 5mg/ml) was added per well. Cells were cultured for another 4 hours before the addition of 100μl 10% SDS and 0.01mol LHC to each well. Cell culture continued overnight. OD values were then measured at wave length 570nm.
11. Adoptive Immunotherapy
Pulmonary metastases were induced by tail vein injection of 1 x 106 of B16 cells in 0.2 ml PBS in B6 mice. Three days after tumor inoculation, the tumor-bearing mice were treated with secondarily activated TDLN cells through tail vein injection (2 x 106 cells/mouse). Approximately 10 days after T cell transfer, all mice were randomized and sacrificed, and lungs were harvested for enumeration of pulmonary metastatic nodules.
12. CTL assay
TDLN T cells were suspended at 1×106/ml in CM and were stimulated as described above. Stimulated T cells were resuspanded at 4×106/ml and mixed with 3H-TdR (Amersham, Little Chalfont, U.K.)-labeled B16 cells at 40: 1, 20: 1, 10: 1 in a U-bottom 96-well plate. The volume was 200 μl/well. After culture at 37 oC with 5% CO2 for 18 hours, cpm was measured by 3H-counter (Backman, Fullerton, CA) and killing (%) was calculated.
13. Statistical Analysis
For comparison of tumor treatment in adoptive immunotherapy, Wilcoxon test was performed. Students t-test was used to analyze cell proliferation and cytokine release. Statistical analysis was performed using SPSS software. Statistical significance was achieved when p<0.05.
Results
1. Construction of recombinant pcDNA3.1 (+)/SEA and the transfection of B16 tumor cells
We constructed a eukaryotic expression vector pcDNA3.1-SEA and transfected it into the B16 murine melanoma tumor cells by means of liposome-mediated transfection.
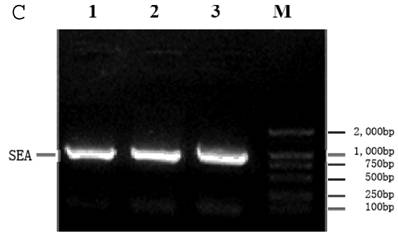
The SEA gene was amplified by PCR using the plasmid pGEM-Teasy-SEA (T/SEA) as a template. The PCR product was digested with Nhe I and EcoR I and ligated with similarly digested pcDNA3.1 (+) using T4 ligase (Fig.1A). The recombinant pcDNA3.1 (+)/SEA was analyzed by restriction enzyme digestion at 37oC for 1 hour. The products were revealed on a 1.5% electrophoresis agarose gel (Fig.1B). It shows that enzyme digestion using NheI and EcoRI resulted in a band of approximately 770bp, consistent with the estimated size of the SEA gene. In addition, the correctly constructed plasmids were confirmed by DNA sequencing (data not shown). B16 cells were transfected with 10 μg of the recombinant plasmid by Lipofectamine 2000. The stably transfected B16 cells were selected with G418 to obtain G418-resistant clones. Total RNA was isolated from the transfected cells and the cDNA was synthesized. The cDNA was then PCR amplified and the products were shown by DNA agarose gel electrophresis (Fig 1C). The bands at approximately 770bp represent the amplified SEA gene from three clones of SEA-transfected B16 tumor cells, thus confirming the successful gene transfer.
A: Scheme for construction of recombinant expression vector pcDNA3.1 (+)/SEA. B: Restriction enzyme digestion of the recombinant expression vector pcDNA3.1-SEA. 1. λDNA/HindIII ladder; 2. pcDNA3.1-SEA/NheI + EcoRI; 3. pcDNA3.1-SEA/EcoRI; M: DL2000 marker. C: Transfected SEA fragment confirmed by RT-PCR product on agarose gel electrophoresis. Lanes 1-3: Three clones of SEA-B16 cells; M: DNA marker (DL2000).

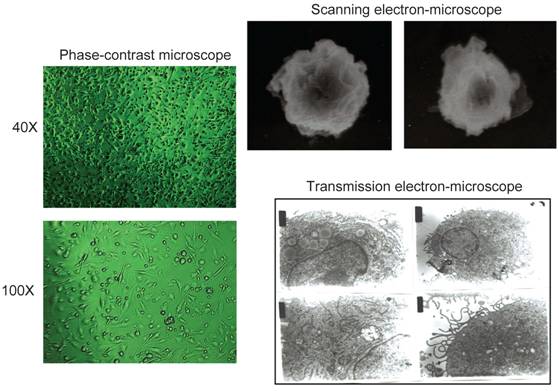
DCs were used in this study during in vitro TDLN cell stimulation. To induce splenomegaly, mice were injected with cyclophosphamide. Eight days later, spleens were harvested. Splenocyte suspension was prepared and depleted of erythrocytes. The resultant cells were suspended at 1×106 /ml in CM. Cells were cultured in a flask at 37oC with 5% CO2. Three days later, culture medium and non-adherent cells were removed and discarded. Adherent cells were then cultured with fresh CM containing GM-CSF (20 ng/ml) and IL-4 (10 ng/ml). Half of the culture medium was replaced by fresh medium every other day. Five days later, TNFα (1ng/ml) was added. After culture for 2 more days, cells were collected for morphological observation under microscope. The GM-CSF and IL-4-cultured cells observed under phase-contrast and electron microscopes demonstrated the typical characteristics of DC morphology (Fig.2). Meanwhile, the phenotype of the cultured DC cells was analyzed by FACS staining. Cell cultures were sampled at day 3, 5, and day 7 without TNFα or day 7 with TNFα respectively and stained with FITC-conjugated anti-CD11c, anti-I-Ab, and anti-CD86. FACS analysis indicated that the expression of CD11c and MHC II (I-Ab) molecules showed an ascending tendency with the time of cell culture (data not shown). On day 7, the CD11c, I-Ab, or CD86 positive cells were 34%, 25%, and 75% respectively. Importantly, with the addition of TNFα, the expression of these markers was enhanced to 78%, 58%, and 81% respectively, signifying the noteworthy role of TNFα in DC maturation.
Morphological observation of DCs under phase contrast microscope, scanning electron microscope, and transmission electron microscope after cell culture for 7 days.
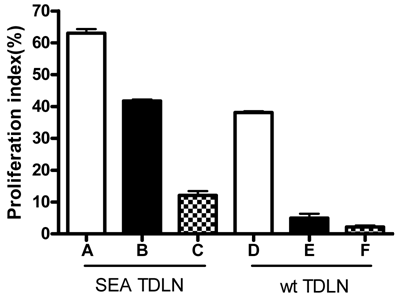
2. Significantly enhanced in vitro proliferation of SEA TDLN cells induced by anti-CD3, anti-CD28 and IL-2 plus DC and tumor antigen
The inguinal lymph nodes draining the parental B16 or SEA-B16 tumor vaccines were collected and were designated as wt TDLN or SEA TDLN respectively. Purified TDLN T cells were activated with T cell specific polyclonal antibody anti-CD3, anti-CD28 mAbs and maintained in culture with exogenous T cell growth factor IL-2 with or without DC plus tumor lysate. MTT analysis was then performed to measure cell proliferation. As shown in Figure 3, group A demonstrated the highest proliferation index (63%), which is significantly higher (p<0.01) than any other groups. The proliferation index of group B (41%) was much lower, but it was significantly higher than control (group C, 12%). These data indicate that anti-CD3, anti-CD28, and IL-2 simulated significant proliferation of SEA TDLN T cells, which could be radically augmented by the simultaneous use of DC and tumor lysates. It is worthy noting, when comparing wt TDLN with SEA TDLN, that SEA TDLN cells proliferated more vigorously than wt TDLN cells in response to the similar activation conditions. For instance, the proliferation indexes of wt TDLN cells after stimulation with anti-CD3, anti-CD28, IL-2 were 38% (group D) and 5% (group E) respectively in the presence and absence of DC plus tumor lysate. These data thus revealed more active proliferation of antibody-activated SEA TDLN cells (group B) compared with antibody-activated wt TDLN cells (group E), and this antibody-activated SEA TDLN cell proliferation can be further increased with DC plus tumor antigen (group A). Collectively, these data suggest the critical role of enforced expression of SEA to enhance the immunogenicity of the tumor vaccine during the in vivo sensitization of TDLN cells, and advocate the usefulness of antigen-presenting cells and tumor antigen during the in vitro activation and expansion of these TDLN cells.
Proliferation of TDLN cells stimulated with different conditions. Multiple inguinal TDLNs were pooled from groups of mice and were processed for lymphoid cell suspension. T cells were purified using the nylon wool column. Purified T cells were stimulated under the following conditions: Group A, D: Anti-CD3 + anti-CD28 + IL-2 plus DC and tumor lysates; Group B, E: Anti-CD3 + anti-CD28 + IL-2; Group C, F: Un-stimulated (medium only). 100μl of cell suspension was added to each well of 96-well culture plate in triplicate. Stimuli described above were added at 100μl/well. After culture for 48 hours, cell proliferation was measured with MTT analysis. P<0.01: A vs. B; A vs. C; A vs. D. B vs. E; C vs. F. D vs. E; D vs. F.
3. Therapeutic efficacy of TDLN cells post antibody, IL-2 and DC stimulation
To test the therapeutic efficacy of T cells generated from the lymph node draining the SEA-expressing tumor and secondarily activated with DC, we performed adoptive transfer experiments with established tumors.
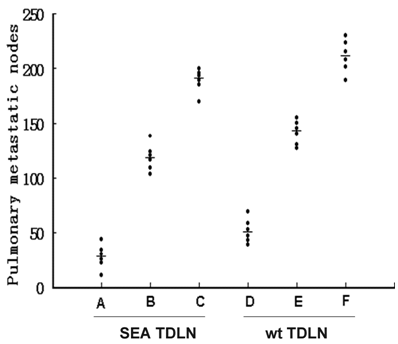
B6 mice bearing three-day wt B16 pulmonary metastases were treated with stimulated SEA TDLN or wt TDLN cells. These TDLN cells were stimulated in vitro using anti-CD3/anti-CD28/IL-2 with or without DC and tumor antigen. Therapeutic efficacy was compared with those using un-stimulated TDLN cells. In control mice that received no treatment, metastatic nodules were more than 250 (data not shown) at the end of the experiments. As shown in Figure 4, unstimulated SEA TDLN and wt TDLN cells mediated marginal tumor regression. The metastatic nodules were around 200, and these T cells inhibited tumor growth by 25% (group C), and 15% (group F) respectively, with SEA TDLN (group C) being more effective. However, anti-CD3/anti-CD28/IL-2- stimulated SEA TDLN and wt TDLN cells mediated significant tumor regression. The metastatic nodules were around 150, and these T cells inhibited tumor growth by 53% (group B), and 43% (group E) respectively, again, with SEA TDLN (group B) being more effective. Importantly, the most potent antitumor effector cells were those generated from SEA TDLN and stimulated in vitro using anti-CD3/anti-CD28/IL-2 plus DC and tumor antigen (group A). These cells inhibited tumor growth by almost 90%. The therapeutic efficacy by these cells was also significantly (p<0.01) higher than that mediated by similarly stimulated wt TDLN cells (group D). Taken together, the therapeutic data confirm that the SEA-expressing B6 tumors are more immunogenic in inducing TDLN cells in vivo, and that the subsequent use of DC to present tumor antigen in vitro in concert with anti-CD3, anti-CD28 and IL-2 can generate more potent antitumor effectors from the TDLN cells.
SEA TDLN cells activated with anti-CD3/anti-CD28/IL-2 plus DC and tumor antigen demonstrated the most potent therapeutic efficacy. In these adoptive transfer experiments, pulmonary metastases were induced by tail vein injection of 1 x 106 of wt B16 cells in 0.2 ml PBS in B6 mice. Three days later, the tumor-bearing mice were treated with variously activated TDLN cells (2 x 106 cells/mouse) by tail vein injection. Approximately 10 days after T cell transfer, all mice were randomized and sacrificed, and lungs were harvested for enumeration of pulmonary metastatic nodules. TDLN cells were activated with anti-CD3/anti-CD28/IL-2 plus DC and tumor antigen (Group A, D); anti-CD3/anti-CD28/IL-2 (Group B, E), or un-stimulated (Group C, F). P<0.01: A vs. B; A vs. C; A vs. D; B vs. E; C vs. F; D vs. E; D vs. F.
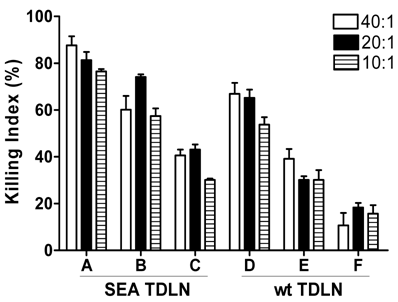
4. Augmented therapeutic efficacy of antibody plus DC-activated SEA TDLN cells correlates with higher CTL activity and increased IFNγ secretion
To appreciate the mechanisms which may be involved in the augmented therapeutic efficacy mediated by antibody plus DC-activated SEA TDLN cells, we examined the immune function of these cells. Killing of wt B16 tumor cells by wt TDLN or SEA TDLN T cells stimulated under various conditions were compared. As illustrated in Figure 5, for SEA TDLN, stimulation with anti-CD3, anti-CD28, and IL-2 plus DC and tumor antigen resulted in a most potent cell killing (group A), followed by stimulation with anti-CD3, anti-CD28 and IL-2 (group B). Wt TDLN also showed superior cell killing when anti-CD3, anti-CD28, IL-2 were used together with DC plus tumor lysate (group D) compared with that when anti-CD3, anti-CD28 and IL-2 were used without DC (group E). These data show that DC and tumor antigen used in vitro boosts CTL activity in TDLN cells induced by antibody and IL-2. On the other hand, SEA TDLN revealed higher tumor cell killing than wt TDLN cells stimulated under the same conditions. For example, the cell killing of SEA TDLN cells after stimulation with antibody and IL-2 in the presence of DC plus tumor lysate (group A) was higher than that of wt TDLN cells similarly stimulated (group D). Also, antibody and IL-2-induced cell killing of SEA TDLN cells in the absence of DC and tumor lysate (group B) was higher than that of wt TDLN cells similarly stimulated (group E). SEA TDLN cells are more cytolytic to tumors after antibody and tumor antigen stimulation compared with wt TDLN cells. These data support the notion that enhanced CTL activity may contribute to the improved therapeutic efficacy of antibody and DC-stimulated SEA TDLN cells.
In vitro CTL activity of TDLN cells stimulated with different conditions. SEA TDLN and wt TDLN T cells were stimulated using anti-CD3 + anti-CD28 + IL-2 with (group A, D) or without (group B, E) DC plus tumor lysate, or left un-stimulated (group C, F), as in Figure 3. Stimulated T cells were washed, re-suspended at 4×106/ml and mixed with 3H-TdR-labeled B16 cells at 40: 1, 20: 1, and 10: 1 in U-bottom 96-well plate. After culture at 37oC with 5% CO2 for 18 hours, cpm was measured and the killing index was calculated.
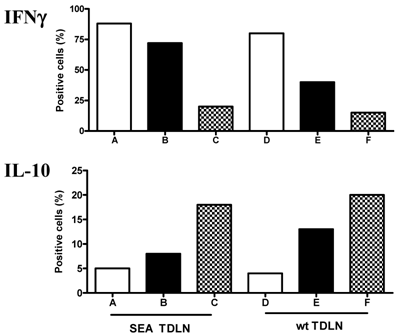
Another critical machinery involved in tumor rejection is the elaboration of cytokines by the effector cells [31, 32]. In parallel with the CTL analysis, we examined the cytokine secretion of SEA TDLN cells. Comparison was made between the wt TDLN and SEA TDLN cells stimulated using anti-CD3 + anti-CD28 + IL-2 with or without the use of DC and tumor lysate. Cytokine secretion was examined at the end of cell stimulation by intracellular staining. IFNγ and IL-10 production are shown in Figure 6. For SEA TDLN, the percentage of IFNγ secreting cells was approximately 70% (group B) and 90% (group A) respectively after T cell stimulation using anti-CD3 + anti-CD28 + IL-2 without or with DC and tumor antigen. Similarly, the use of DC increased the number of IFNγ secreting wt TDLN T cells from 40% (group E) to 80% (group D) after anti-CD3 + anti-CD28 + IL-2 stimulation. In contrast, when IL-10 was examined, an opposed effect of DC was found. In SEA TDLN, the percentage of IL-10-secreting cells was 8% (group B) and 5% (group A) respectively after T cells were stimulated using anti-CD3 + anti-CD28 + IL-2 without or with DC plus tumor antigen. Similarly, the use of DC decreased the number of IL-10-secreting wt TDLN T cells from 13% (group E) to 4% (group D) with anti-CD3+ anti-CD28 + IL-2 stimulation. In line with the proliferation and CTL analyses, these data suggest that DC, when used in vitro to present tumor antigen in concert with anti-CD3/anti-CD28/IL-2, could modulate the immune function of TDLN cells, namely by enhancing the production of type 1 cytokine, e.g. IFNγ and simultaneously inhabiting the secretion of type 2 cytokine, e.g. IL-10.
IFNγ and IL-10 expression on activated TDLN cells. SEA TDLN and wt TDLN T cells were stimulated using anti-CD3 + anti-CD28 + IL-2 with (groups A, D) or without (groups B, E) DC plus tumor lysate, or left un-stimulated (groups C, F), as in Figure 3. After activation for 48 hours, TDLN T cells were permeablized and stained with FITC-conjugated anti-IFN-γ or PE-conjugated anti-IL-10. FACS analysis was then performed.
Discussion
We report in this study that immunization of mice with B16 tumors genetically engineered to express SEA induces the antitumor immune response of T cells in the lymph nodes draining the SEA-expressing tumor. Secondary activation of these TDLN cells with DC plus tumor antigen in the presence of anti-CD3/anti-CD28/IL-2 amplifies their antitumor activity. As a result, adoptive transfer of these T cells mediates more effective tumor regression in the mice bearing the wild type B16 tumor compared with activated wt TDLN cells. These data present evidence that in vivo sensitization of TDLN draining a tumor with modulated immunogenicity followed by in vitro stimulation with combined use of antigen-presenting cells and T cell activation antibodies can profoundly alter the therapeutic efficacy of the TDLN cells. The sequential use of SEA-B16 vaccine in vivo and the use of anti-CD3/CD28 plus DC and tumor antigen in vitro to generator antitumor effector cells for adoptive immunotherapy represents a novel procedure described in this study.
Poorly immunogenic B16 melanoma cells have been genetically engineered to improve their immunogenicity by forced expression of several immune modulator molecules [33-36]. We examined the effects of in vivo transfection of an allogeneic MHC class I gene (H-2Ks) into the B16 melanoma of H-2b origin [35], and the host immune response to B16 melanoma transduced to secrete granulocyte-macrophage colony-stimulating factor (GM-CSF) [34]. Likewise, genetic modification of human melanoma tumor cells has resulted in clinical benefit. Adoptive transfer of GM-CSF-secreting vaccine-primed lymph node cells resulted in a durable complete remission of metastatic tumor in melanoma patients [37], although the clinical response was confined to a limited population of patients. In addition, gene transfer of a foreign MHC class I protein, HLA-B7, into HLA-B7-negative patients with stage IV melanoma stimulated local antitumor immune responses that facilitated the generation of effector cells for immunotherapy of cancer [38]. In addition, we evaluated the in vivo response to B16 melanoma genetically altered to secrete IL-2, IL-4 and IFNγ. Secretion of IL-2 abrogated the tumorigenicity of B16, while IFNγ and IL-4 partially reduced tumorigenicity. However, neither IL-2 nor IFNγ secretion resulted in the induction of immune T cells in the TDLN. These studies demonstrated that reduced tumorigenicity did not correlate with increased immunogenicity [36]. In this study, we found that SEA expression conveyed super antigenicity to tumor cells, which evoked T cells in the draining lymph nodes to suppress tumor growth upon adoptive transfer.
SEA has been previously expressed on tumor cells to increase their immunogenicity and to serve as a target for T cells [20, 21, 39]. This was done either by modifying the tumor with transmembrane-anchored SEA (TM-SEA) using the protein transfer technique [20], or by antibody targeting using a recombinant protein of SEA and the Fab-fragment of a tumor-reactive antibody [21, 39]. In this report, we modified B16 by SEA gene transfer. This strategy altered the immunogenicity of the genetically modified tumor when used as vaccine, and induced more potent tumor-reactive TDLN cells. This was demonstrated by more vigorous cell proliferation, higher CTL activity, and significantly augmented therapeutic efficacy of the SEA TDLN cells compared with similarly in vitro simulated TDLN cells draining the parental tumor.
It was shown that genetic modification of tumors could be made more efficient in tumor immunotherapy by concomitant DC administration [11]. On the other hand, Ag-specific in vitro DC-driven proliferation and differentiation of T cells from HIV-1-infected patients were documented [40]. Antigen specific T cells were also generated from peripheral blood [41] or from naive spleen-derived T cells [27] by in vitro stimulation with DC and the relevant antigen. DCs have also been used in vitro to generate human antitumor effectors. For example, cancer patients' memory T cells from bone marrow could be specifically activated to become cytotoxic effector cells by short-term culture with autologous DC pre-pulsed with tumor lysates [42]. Both human CD4+ and CD8+ renal cell carcinoma-reactive T cells were generated using a completely autologous system of DCs presenting whole ultraviolet-irradiated apoptotic tumor cells [43]. The function of CD4 cells relevant to CD8 cells generated from TDLN has also been documented in the adoptive immunotherapy models [30, 44, 45]. In this report, we used DC and tumor antigen in vitro to stimulate TDLN cells isolated from the hosts vaccinated with a genetically modified tumor, and the in vitro stimulation was performed in the presence of T cell specific antibodies, anti-CD3 and anti-CD28 plus the T cell growth factor IL-2. The secondary stimulation in vitro with the use of DC amplifies the antitumor activity induced by the genetically modified tumor as vaccine in vivo. Consequently, DC-stimulated SEA TDLN cells eliminated nearly 90% of the pulmonary metastasis in mice bearing established B16 melanoma micrometastases. The sequential use of genetically modified tumor as vaccine to induce TDLN followed by secondary stimulation using APCs and tumor antigen represents a unique combination method to generate tumor reactive T cells.
We found in this study that addition of DC and tumor lysate to T cell culture with anti-CD3/anti-CD28/IL-2 increases the elaboration of IFNγ but diminishes the secretion of IL-10 by TDLN cells. This cytokine profile correlated with a superior therapeutic efficacy. Several groups, including our own, have reported previously that the cytokine profile secreted by lymphoid cells correlates with the outcome of antitumor activity in cancer immunotherapy [8, 28, 29, 31]. In a tumor-reactive antibody-targeted immunotherapy using the SEA-mAb complex, IFNγ was found to be suppressive to tumor, while in contrast high level of IL-10 was demonstrated to inhibit Fab-SEA tumor therapy, since the therapeutic efficacy was significantly higher in IL-10 KO mice [39]. Moreover, incubation of T cells with autologous ovarian tumor cell lysate-pulsed DC stimulated proliferation of T cells and production of Th1 cytokines [46]. Collectively, these data support the efforts to promote the expression of type1 cytokines in order to develop more effective immunotherapy strategies [7-8, 28]. In this regard, DC may play an important role even though the molecular mechanisms remain to be defined to further elucidate these observations.
With the use of T cells generated from lymph nodes draining an SEA-expressing murine B16 melanoma and secondarily activated with DC plus B16 tumor antigen in this study, significant remission of established solid tumors was achieved. It would be interesting to see what will happen when B16 tumor cells are inoculated into the mice that have been previously immunized with SEA-expressing B16 cells. Normal mice injected with SEA-B16 followed by inoculation of wt-B16 tumor cells to see if wt-B16 will grow or the growth will be retarded should allow us to test the protective immunity which may be induced by SEA-B16 as a vaccine.
While the lung metastatic model has been used extensively in studies of adoptive transfer therapy including TDLN-related approaches as we described in this study, the use of subcutaneous (sc) tumor model [22, 23, 45] would provide additional data to support the use of SEA-B16 vaccine in vivo and anti-CD3/CD28 plus DC and tumor antigen in vitro to generator antitumor effector cells for adoptive immunotherapy. Nevertheless, the preclinical studies reported herein provide compelling evidence that genetic introduction of bacterial superantigen SEA into poorly immunogenic tumors appears to be a promising strategy for rendering tumors more immunogenic. SEA-modified tumor vaccine conferred the induction of more potent tumor-reactive T cells within the lymph nodes draining the vaccine. Subsequent in vitro stimulation of these lymph node cells using DC and tumor antigen amplifies the antitumor activity and results in more potent therapeutic effector T cells. These findings have shown the feasibility to combine the use of a genetically modified tumor vaccine with a secondary in vitro activation procedure involving antigen presenting cells in future clinical trials, and may lead to the development of novel protocols for cancer immunotherapy.
Acknowledgements
The authors thank Ms. Kerry Odneal and Ms. Ele Kotowski for their excellent assistance in the preparation of this manuscript.
This work was supported in part by National 863 Project (No.2006AA02A237; No.2007AA02Z451), P.R.China, and the Gillson Longenbaugh Foundation.
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
1. Dudley ME, Rosenberg SA. Adoptive-cell-transfer therapy for the treatment of patients with cancer. Cancer. 2003;3:666-675
2. Dudley ME, Wunderlich JR, Robbins PF. et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850-854
3. Chang AE, Aruga A, Cameron MJ. et al. Adoptive immunotherapy with vaccine-primed lymph node cells secondarily activated with anti-CD3 and interleukin-2. J Clin Oncol. 1997;15:796-807
4. Chang AE, Li Q, Jiang G. et al. Phase II trial of autologous tumor vaccination, anti-CD3-activated vaccine-primed lymphocytes, and interleukin-2 in stage IV renal cell cancer. J Clin Oncol. 2003;21:884-890
5. Wang L, Chen B, Plautz GE. Adoptive immunotherapy of advanced tumors with CD62 L-selectinlow tumor-sensitized T lymphocytes following ex vivo hyperexpansion. J Immunol. 2002;169:3314-3320
6. Peng L, Kjaergaard JK, Plautz GE. et al. Tumor-induced L-selectinhigh suppressor T cells mediate potent effector T cell blockade and cause failure of otherwise curative adoptive immunotherapy. J Immunol. 2002;169:4811-4821
7. Li Q, Carr A, Donald E. et al. Synergistic effects of IL-12 and IL-18 in skewing tumor-reactive T-cell responses towards a type 1 pattern. Cancer Res. 2005;65:1063-1070
8. Li Q, Grover A, Donald E. et al. Simultaneous targeting of CD3 on T cells and CD40 on B or dendritic cells augments the antitumor reactivity of tumor-primed lymph nodes cells. J Immunol. 2005;175:1424-1432
9. Dissanayake SK, Thompson JA, Bosch JJ. et al. Activation of tumor-specific CD4+ T lymphocytes by major histocompatibility complex class II tumor cell vaccines: a novel cell-based immunotherapy. Cancer Res. 2004;64:1867-1874
10. Gruber TA, Skelton DC, Kohn DB. Requirement for NK cells in CD40 ligand-mediated rejection of Philadelphia chromosome-positive acute lymphoblastic leukemia cells. J Immunol. 2002;168:73-80
11. Kikuchi T, Miyazawa N, Moore M. et al. Tumor regression induced by intratumor administration of adenovirus vector expressing CD40 ligand and naïve dendritic cells. Cancer Res. 2000;60:6391-6395
12. Johnston JV, Malacko AR, Mizuno MT. et al. B7-CD28 costimulation unveils the hierarchy of tumor epitopes recognized by major histocompatibility complex class I-restricted CD8+ cytolytic T lymphocytes. J Exp Med. 1996;183:791-800
13. Foreman KE, Wrone-Smith T, Krueger AE. et al. Expression of costimulatory molecules CD80 and/or CD86 by a Kaposi's Sarcoma tumor cell line induces differential T-cell activation and proliferation. Clin Immunol. 1999;91:345-353
14. Gri G, Gallo E, Di Carlo E. et al. OX40 ligand-transduced tumor cell vaccine synergizes with GM-CSF and requires CD40-Apc signaling to boost the host T cell antitumor response. J Immunol. 2003;170:99-106
15. Giezeman-Smits KM, Okada H, Brissette-Storkus CS. et al. Cytokine gene therapy of gliomas: induction of reactive CD4+ T cells by interleukin-4-transfected 9L gliosarcoma is essential for protective immunity. Cancer Res. 2000;60:2449-2457
16. Zheng G, Chen A, Sterner RE. et al. Induction of antitumor immunity via intratumoral tetra-costimulator protein transfer. Cancer Res. 2001;61:8127-8134
17. Li Q, Normolle DP, Sayre DM, Zeng X, Sun R, Jiang G, Redman BD, Chang AE. Immunological effects of BCG as an adjuvant in autologous tumor vaccines. Clinical Immunology. 2000;94:64-72
18. Matsuyama T, Kawai T, Izumi Y. et al. Expression of major histocompatibility complex class II and CD80 by gingival epithelial cells induces activation of CD4+ T cells in response to bacterial challenge. Infection & Immunity. 2005;73:1044-1051
19. Llewelyn M, Sriskandan S, Peakman M. et al. HLA class II polymorhipsms determine responses to bacterial superantigens. J Immunol. 2004;172:1719-1726
20. Yi P, Yu H, Ma W. et al. Preparation of murine B7.1-glycosylphosphatidylinositol and transmembrane-anchored staphylococcal entertoxin. A dual-anchored tumor cell vaccine and its antitumor effect. Cancer. 2005;103:1519-1528
21. Lando PA, Dohlsten M, Hedlund G. et al. Co-stimulation with B7 and targeted superantigen is required for MHC class II-independent T-cell proliferation but not cytotoxicity. Immunol. 1993;80:236-241
22. Teitz-Tennenbaum S, Li Q, Rynkiewicz S, Ito F, Davis MA, Mcginn CJ, Chang AE. Radiotherapy Potentiates the Therapeutic Efficacy of Intratumoral Dendritic Cell Administration. Cancer Research. 2003;63:8466-8475
23. Huang J, Wang Y, Guo J, Lu H, Ma L, Teitz -Tennenbaum S, Chang A E, Li Q. Radiation-induced Apoptosis along with Local and Systemic Cytokine Elaboration is Associated with DC plus Radiotherapy-mediated Renal Cell Tumor Regression. Clinical Immunology. 2007;123:298-310
24. Teitz-Tennenbaum S, Li Q, Okuyama R, Davis MA, Sun R, Knibbs RN, Stoolman LM, Chang AE. Mechanisms involved in radiation enhancement of intratumoral dendritic cell therapy. J. Immunother. 2008;31:345-358
25. Tanigawa K, Takeshita N, Eickhoff GA. Antitumor reactivity of lymph node cells primed in vivo with dendritic cell-based vaccines. J Immunother. 2001;24:493-501
26. Lou Y, Wang G, Lizee G, et all. Dendritic cells strongly boost the antitumor activity of adoptively transferred T cells in vivo. Cancer Res. 2004;64:6783-6790
27. Fields RC, Shimizu K, Mule JJ. Murine dendritic cells pulsed with whole tumor lysates mediate potent antitumor immune responses in vitro and in vivo. Proc Natl Acad Sci USA. 1998;95:9482-9487
28. Li Q, Carr A, Ito F. et al. Polarization effects of 4-1BB during CD28 costimulation in generating tumor-reactive T cells for cancer immunotherapy. Cancer Res. 2003;63:2546-2552
29. Ito F, Carr A, Svensson J. Antitumor reactivity of Anti-CD3/Anti-CD28 bead-activated lymphoid cells; implications for cell therapy in a murine model. J Immunother. 2003;26:222-233
30. Li Q, Yu B, Grover AC. et al. Therapeutic effects of tumor-reactive CD4+ cells generated from tumor-primed lymph nodes using anti-CD3/anti-CD28 monoclonal antibodies. J Immunother. 2002;25:304-313
31. Aruga A, Aruga E, Tanigawa K. et al. Type 1 versus type 2 cytokine release by Vβ T cell subpopulations determines in vivo antitumor reactivity: IL-10 mediates a suppressive role. J Immunol. 1997;159:664-673
32. Hu H, Urba W, Fox B. Gene-modified tumor vaccine with therapeutic potential shifts tumor-specific T cell response from a type 2 to a type 1 cytokine profile. J Immunol. 1998;161:3033-3041
33. Chen JX, Liu JW, Tian YX. et al. Antitumor effects of human ribonuclease inhibitor gene transfected on B16 melanoma cells. Int J Biochem and Cell Bio. 2005;37:1219-1231
34. Arca MJ, Krauss JC, Aruga A. et al. Therapeutic efficacy of T cells derived from lymph nodes draining a poorly immunogenic tumor transduced to secrete granulocyte-macrophage colony-stimulating factor. Cancer Gene Ther. 1996;3:39-47
35. Wahl WL, Stome SE, Nabel GJ. et al. Generation of therapeutic T-lymphocytes after in vivo tumor transfection with an allogeneic class I major histocompatibility complex gene. J Immunother. 1995;17:1-11
36. Arca MJ, Krauss JC, Strome SE. et al. Diverse manifestations of tumorigenicity and immunogenicity displayed by the poorly immunogenic B16-BL6 melanoma transduced with cytokine genes. Cancer Immunol Immunother. 1996;42:237-245
37. Chang AE, Li Q, Bishop DK. et al. Immunogenetic therapy of human melanoma utilizing autologous tumor cells transduced to secrete granulocyte-macrophage colony-stimulating factor. Human Gene Ther. 2000;11:839-850
38. Nabel GJ, Gordon D, Bishop DK. et al. Immune response in human melanoma after transfer of an allogeneic class I major histocompatibility complex gene with DNA-liposome complexes. Proceedings of the Nat Acad Sci of USA. 1996;93:15388-15393
39. Litton MJ, Dohlsten M, Rosendahl A. et al. The distinct role of CD4+ and CD8+ T-cells during the anti-tumour effects of targeted superantigens. British J of Cancer. 1999;81:359-366
40. Lu W, Achour A, Arlie M. et al. Enhanced dendritic cell-driven proliferation and anti-HIV activity of CD8+ T cells by a new phenothiazine derivative, aminoperazine. J Immunol. 2001;167:2929-2935
41. Chikamatsu K, Albers A, Stanson J. et al. p53110-124-specific human CD4+ T-helper cells enhance in vitro generation and antitumor function of tumor-reactive CD8+ T cells. Cancer Res. 2003;63:3675-3681
42. Feuerer M, Beckhove P, Bai L. et al. Therapy of human tumors in NOD/ SCID mice with patient-derived reactivated memory T cells from bone marrow. Nat Med. 2001;7:452-458
43. Wang QJ, Hanada K, Perry-Lalley D. et al. Generating renal cancer-reactive T cells using dendritic cells (DCs) to present autologous tumors. J Immunother. 2005;28:551-559
44. Li Q, Furman SA, Bradford CR, Chang AE. Expanded tumor-reactive CD4+ T cell responses to human cancers induced by secondary anti-CD3/anti- CD28 activation. Clinical Cancer Research. 1999;5:461-469
45. Iuchi T, Teitz-Tennenbuam S, Huang J, Redman BG, Hughes SD, Li M, Jiang G, Chang AE, Li Q. IL-21 augments the efficacy of T cell therapy by eliciting concurrent cellular and humoral responses. Can. Res. 2008;68:4431-41
46. Zhao X, Wei Y, Peng Z. Induction of T cell responses against autologous ovarian tumors with whole tumor cell lysate-pulsed dendritic cells. Immunol Invest. 2001;30:33-45
Author contact
![]() Correspondence to: Dr. Qiao Li, 1520 MSRB-1, 1150 W. Medical Center Dr., Ann Arbor, MI 48109-0666. Phone: 734-615-1977, Fax: 734-763-4135. Email: qiaoliedu; or Hongbin Song, Institute of Disease Control and Prevention, Academy of Military Medical Sciences, 100071, Beijing, China. Phone/Fax: +86-010-66933358. Email: hongbinsongnet.
Correspondence to: Dr. Qiao Li, 1520 MSRB-1, 1150 W. Medical Center Dr., Ann Arbor, MI 48109-0666. Phone: 734-615-1977, Fax: 734-763-4135. Email: qiaoliedu; or Hongbin Song, Institute of Disease Control and Prevention, Academy of Military Medical Sciences, 100071, Beijing, China. Phone/Fax: +86-010-66933358. Email: hongbinsongnet.