International Journal of Biological Sciences 10
Impact Factor
ISSN: 1449-2288
Impact Factor
ISSN: 1449-2288
- Issue 6; 2026
- Issue 5; 2026
- Issue 4; 2026
- Issue 3; 2026
- Issue 2; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Top
Introduction
Expression of SIRT1 in cancer
SIRT1 as a tumor promoter
SIRT1 as a tumor suppressor
Perspective
Acknowledgements
References
Introduction
Expression of SIRT1 in cancer
SIRT1 as a tumor promoter
SIRT1 as a tumor suppressor
Perspective
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2009; 5(2):147-152. doi:10.7150/ijbs.5.147 This issue Cite
Review
SIRT1, Is It a Tumor Promoter or Tumor Suppressor?
Chu-Xia Deng ![]()
Genetics of Development and Disease Branch, 10/9N105, National Institute of Diabetes, Digestive and Kidney Diseases, National Institutes of Health, Bethesda, Maryland, MD 20892, USA
Received 2009-1-8; Accepted 2009-1-20; Published 2009-1-21
Citation:
Deng CX. SIRT1, Is It a Tumor Promoter or Tumor Suppressor?. Int J Biol Sci 2009; 5(2):147-152. doi:10.7150/ijbs.5.147. https://www.ijbs.com/v05p0147.htm
Other stylesAbstract
SIRT1 has been considered as a tumor promoter because of its increased expression in some types of cancers and its role in inactivating proteins that are involved in tumor suppression and DNA damage repair. However, recent studies demonstrated that SIRT1 levels are reduced in some other types of cancers, and that SIRT1 deficiency results in genetic instability and tumorigenesis, while overexpression of SIRT1 attenuates cancer formation in mice heterozygous for tumor suppressor p53 or APC. Here, I review these recent findings and discuss the possibility that activation of SIRT1 both extends lifespan and inhibits cancer formation.
Keywords: SIRT1, tumor promoter, tumor suppressor, DNA damage repair
Introduction
Epigenetic modifications of protein, histone, and chromatin play an important role in regulating gene expression, cancer formation, and lifespan. In budding yeast, Sir2 is a NAD+-dependent histone deacetylase that plays a role in chromatin silencing, longevity and genomic stability [1, 2]. In mammals, seven sirtuin proteins (SIRT1-7) have been found to share homology with Sir2 and are suspected to have many similar functions as Sir2 [1, 3, 4]. SIRT1, a proto member of the sirtuin family, modifies histones through deacetylation of K26 in histone H1 (H1K26), K9 in histone H3 (H3K9) and K16 in histone H4 (H4K16). It also deacetylates many non-histone proteins that are involved in cell growth, apoptosis, neuronal protection, adaptation to calorie restriction, organ metabolism and function, cell senescence, and tumorigenesis [1, 3-5]. However, it remains controversial whether SIRT1 acts as a tumor promoter or tumor suppressor due to recent controversy over SIRT1 regarding: 1) its expression level in human cancers; 2) its activity on tumor suppressors and oncoproteins; 3) its effect on growth arrest, cell death, and DNA damage repair; and, finally, 4) its long-term impact on lifespan and cancer risk.
Expression of SIRT1 in cancer
It has been shown that SIRT1 is significantly elevated in human prostate cancer [6], acute myeloid leukemia [7], and primary colon cancer [8]. Hida et al. examined SIRT1 protein levels in several different types of skin cancer by immunohistochemical analysis [9]. Overexpression of SIRT1 was frequently observed in all kinds of non-melanoma skin cancers including squamous cell carcinoma, basal cell carcinoma, Bowen's disease, and actinic keratosis. Based on the elevated levels of SIRT1 in cancers, it was hypothesized that SIRT1 serves as a tumor promoter [10]. However it does not rule out a possibility that increased expression of SIRT1 is a consequence, rather than a cause, of tumorigenesis. In contrast, Wang et al. analyzed a public database and found that SIRT1 expression was reduced in many other types of cancers, including glioblastoma, bladder carcinoma, prostate carcinoma and ovarian cancers as compared to the corresponding normal tissues [11]. Their further analysis of 44 breast cancer and 263 hepatic carcinoma cases also revealed reduced expression of SIRT1 in these tumors [11]. These data suggest that SIRT1 acts as a tumor suppressor rather than a promoter in these tissues.
SIRT1 as a tumor promoter
The first evidence of SIRT1 as a tumor promoter came from experiments showing that SIRT1 physically interacts with p53 and attenuates p53-mediated functions through deacetylation of p53 at its C-terminal Lys382 residue [12, 13]. They further showed that overexpression of SIRT1 represses p53-dependent cell cycle arrest and apoptosis in response to DNA damage and oxidative stress. However, expression of a SIRT1 mutant form that carries a point mutation disrupting its deacetyalase activity, increases the sensitivity of cells to a stress response [12, 13]. More recently, it was shown that inhibition of SIRT1 by a specific inhibitor causes p53 hyperacetylation and increases p53-dependent transcriptional activity [14]. In addition, overexpression of SIRT1 epigenetically represses expression and/or activity of many tumor suppressor genes, and proteins with DNA damage repair functions. This includes forkhead class O transcription factor (FOXO) family members (FOXO1, FOXO3a and FOXO4) [15], p73 [16], RB [17], SFRP1, SFRP2, GATA4, GATA5, CDH1, MLH1 [18], Ku70 [19], NBS1 [20], and WRN [21]. Consistent with these data, a number of studies showed that depletion of SIRT1 by siRNA reduces drug resistance and/or induces growth arrest of cancer cells in vitro [18, 22-24].
On the other front, expression and/or activity of SIRT1 are also subjected to regulation by several tumor suppressors. Hypermethylated in cancer-1 (HIC1) encodes a transcriptional repressor with zinc finger motifs and a N-terminal BTB/POZ domain that cooperates with p53 to suppress age-dependent development of cancer in mice [25]. HIC1 and SIRT1 form a transcriptional repression complex, which directly binds to SIRT1 promoter and represses SIRT1 transcription. In both normal and cancer cells, inactivation of HIC1 results in upregulated SIRT1 expression, which deacetylates and inactivates p53, allowing cells to bypass apoptosis and survive DNA damage [26]. Because p53 trans-activates HIC1, the authors proposed a circular regulatory loop among HIC1, SIRT1, and p53, i.e. HIC1 represses the transcription of SIRT1, SIRT1, in turn, deacetylates p53, and p53, then, activates HIC1, for modulating p53 activity in DNA damage response (DDR), cell cycle progression and apoptosis (Fig. 1A). Specifically, the authors proposed that in normal situations, actively expressed HIC1 represses SIRT1 transcription, thereby maintaining p53 in an active state in controlling growth arrest and apoptosis in response to DNA damage [26]. In cells in which proper repair of DNA damage is accomplished, subsequent decreased p53 can lead to decreased HIC1 and increased SIRT1 expression to keep p53 in an inactive state. However, during the course of aging, the HIC1 promoter undergoes hypermethylation. As a consequence, upregulation of SIRT1 in aging cells due to epigenetic silencing of HIC1 might be a double-edged sword that both promotes survival of aging cells and increases cancer risk in mammals [26].
In addition, two recent studies demonstrated that DBC1 (deleted in breast cancer-1), which was initially cloned from a region (8p21) homozygously deleted in breast cancer, forms a stable complex with SIRT1 and inhibits SIRT1 activity, leading to increased levels of p53 acetylation and upregulation of p53-mediated function. Consistently, knockdown of DBC1 by RNA interference (RNAi) promoted SIRT1-mediated deacetylation of p53 and inhibited p53-mediated apoptosis induced by genotoxic stress. These effects were reversed in cells by concomitant RNAi-mediated knockdown of endogenous SIRT1 [27, 28]. These data suggest that increased SIRT1 expression and/or activity may increase the risk of cancer in mammals by inhibiting p53 and potentially other tumor suppressor genes (Fig. 1A).
Genetically engineered mice have provided a powerful means to assess gene function in vivo [29-31]. However, SIRT1's role in tumorigenesis could not be directly assessed due to embryonic lethality in most Sirt1-/- embryos [11, 32, 33]. Because SIRT1 deacetylates and inactivates p53, it was suspected that SIRT1 deficiency might activate p53, leading to the lethality of mutant mice [33]. However, analysis of SIRT1 mutant embryos failed to detect altered expression p53 downstream genes, while introduction of a p53-null mutation into SIRT1 mutant mice failed to rescue the embryonic lethality of Sirt1-/- embryos [11, 34]. Thus, despite the fact that SIRT1 deacetylates p53, SIRT1 does not affect many p53-mediated biological activities in vivo. Currently, several transgenic mouse stains carrying overexpression of SIRT1 have been established. Overexpresson of SIRT1 reduces energy expenditure, improves metabolism and prevents diabetes in mice, while no tumor formation has been reported in these mice [35-37]. Thus, it remains elusive whether or not increased levels of SIRT1 can enhance cancer risk due to the lack of in vivo data. Continuous monitoring of these mice might eventually provide an answer to this issue.
Figure 1
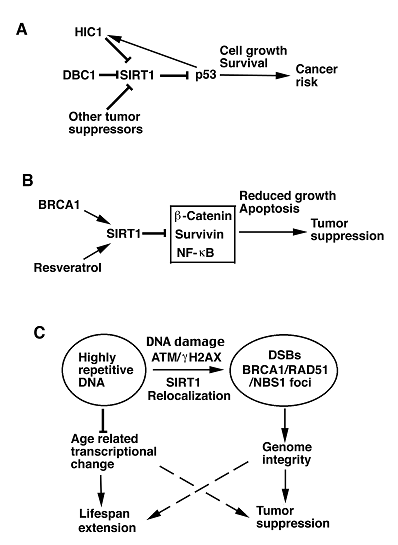
Models illustrating possible functions of SIRT1 in tumor promotion or suppression. A. SIRT1 deacetylates and inactivates p53, leading to down regulation of p53-mediated growth arrest and apoptosis. This may result in increased risk of cancer. Transcription and activity of SIRT1 are also negatively regulated by many tumor suppressor genes, including DBC1, HIC1, and p53, which may regulate SIRT1 through HIC1. B. BRCA1 and resveratrol can positively regulate SIRT1 transcription and activity, respectively. Increased SIRT1, in turn, inhibits expression and/or activity of several oncogenes, leading to reduced cell proliferation, increased apoptosis, and tumor suppression. C. In response to DNA damage, SIRT1 dissociates from highly repetitive DNA and relocalizes to DSBs to promote repair and maintain genome integrity. ATM and γH2AX are required for the efficient recruitment of SIRT1 to DSBs. Moreover, SIRT1 deficiency impaired BRCA1, RAD51 and NBS1 foci formation, suggesting direct or indirect interactions of these proteins in the DNA damage foci. SIRT1 overexpression suppresses the age-related transcriptional changes and tumor formation, suggesting a possibility to extend lifespan and inhibit tumor formation through activation of SIRT1.
SIRT1 as a tumor suppressor
On the other hand, several recent studies have provided convincing evidence that SIRT1 serves as a tumor suppressor. Firestein et al demonstrated that overexpression of SIRT1 in APCmin/+ mice reduces, instead of increases, colon cancer formation [38]. The data demonstrated that the reduction in tumor development is caused by the ability of SIRT1 to deacetylate β-catenin and promote cytoplasmic localization of the nuclear-localized oncogenic form of β-catenin. Their further analysis also uncovered a significant inverse correlation between the presence of nuclear SIRT1 and the oncogenic form of β-catenin in 81 human colon tumor specimens analyzed. Ectopic over-expression of SIRT1 also greatly reduces cell proliferation in a human colon cancer cell line whose growth is driven by active β-catenin [38]. An earlier study also showed that SIRT1 deacetylates the RelA/p65 subunit of NF-κB and sensitizes cells to TNFα-induced apoptosis by inhibiting the transactivation potential of the RelA/p65 protein [39].
Analyzing published database for SIRT1 expression, Wang et al. found that SIRT1 expression is much lower in the BRCA1-associated breast cancer than BRCA1-wildtype breast cancer in human [40]. They further showed that BRCA1 binds to the SIRT1 promoter and positively regulates SIRT1 gene expression at both the mRNA and protein level, and that BRCA1 deficiency causes reduced SIRT1 levels, which may be responsible for the malignant transformation of BRCA1 mutant cells. Consistently, restoration of SIRT1 levels in BRCA1 mutant cancer cells inhibited proliferation of these cells in vitro and tumor formation in vivo when the cells were implanted into nude mice. In addition, resveratrol, which activates SIRT1 deacetylase activity [41], induced apoptosis in these cells and inhibited tumor formation [40]. These data suggest that SIRT1 not only acts as a tumor suppressor in the context of BRCA1 deficiency, but also that a SIRT1 activator, such as resveratrol, could serve as an excellent strategy for targeted therapy for BRCA1-associated breast cancer. To illustrate the molecular mechanism underlying how activated SIRT1 triggers cell death, the researchers demonstrated that SIRT1 negatively regulates expression of Survivin, which encodes an anti-apoptotic protein, by deacetylating H3K9 within the promoter of Survivin [40]. Altogether, these data suggest that SIRT1 mediates BRCA1 signaling and inhibits tumor growth through repressing transcription of oncogenes or activity of oncoproteins (Fig. 1B).
Two recent investigations reported that SIRT1 plays an important role in DNA damage repair and in maintaining genome integrity. Analyzing SIRT1-deficient mice, Wang et al. found that Sirt1-/- embryos die at middle gestation stages, displaying increased acetylation of H3K9 and H4K16, reduced chromosome condensation, impaired heterochromatin formation, and abnormal mitosis [11]. Sirt1-/- cells displayed chromosome aneuploidy and structural aberrations, conceivably originated from the continuous division of abnormal mitosis. SIRT1 deficiency also causes reduced ability to repair DNA-double strand breaks (DSBs), radiation sensitivity, and impaired DDRs characterized by diminished γH2AX, BRCA1, RAD51 and NBS1 foci formation upon γ-irradiation. Thus, SIRT1 may play a role in recruiting these proteins to DNA damage sites. Of note, Western blot analysis detected reduced level of γH2AX, but not that of BRCA1, RAD51 and NBS1 upon DNA damage in SIRT1 mutant cells compared with wild type cells. To assess whether the reduced γH2AX levels/foci formation is a direct consequence of SIRT1 loss, the researchers transfected SIRT1-deficient cells with a SIRT1 expression vector, and the data indicated that the SIRT1-reconstituted Sirt1-/- cells contained relatively equal numbers of γH2AX foci compared with wild type controls upon γ-irradiation [11]. These observations suggest that γH2AX plays an important role in mediating functions of SIRT1 in DNA damage foci formation and DNA damage repair.
As mentioned earlier that introduction of a p53-null mutation into Sirt1-/- mice does not repress embryonic lethality. However, Sirt1+/-;p53+/- mice started to develop spontaneous tumors in multiple organs from about 5 months of age, and tumor incidence reached about 76% by 20 months of age, while only 2 out of 21 Sirt1+/- mice and 3 out of 22 p53+/- mice developed tumors during the same period of time. Chromosome spreads and spectral karyotyping analysis of primary tumors revealed extensive aneuploidy and chromosomal aberrations, notably translocations, chromosome breaks, deletions, end fusions, and dicentric chromosomes. These results suggest that severe genetic instability could be one of the causes for spontaneous tumorigenesis in SIRT1 and p53 double-heterozygous animals. Interestingly, most tumors developed in the Sirt1+/-;p53+/- mice lost the remaining wild-type allele of p53 yet retained a functional wild-type Sirt1 allele. Accordingly, administration of resveratrol to these mice significantly reduced tumor formation [11]. These data demonstrate that SIRT1 plays an important role in maintaining genome integrity and acts as a haploinsufficient tumor suppressor in mice (Fig. 1C).
Using mouse embryonic stem cells, Oberdoerffer et al demonstrated that SIRT1 binds to highly repetitive DNA and contributes to the silencing of major satellite repeats [42]. In response to oxidative DNA damage, SIRT1 dissociates from its original loci and relocalizes to DSBs to promote repair and maintain genome integrity. Their data indicated that the efficient recruitment of SIRT1 to DSBs requires DNA damage signalling through ATM and H2AX. Without SIRT1, recruitment of RAD51 and NBS1 to DSBs was delayed and strongly reduced, thus highlighting a key role of SIRT1 in the DNA damage repair process. The researchers further showed that SIRT1 inhibits a functionally diverse set of genes that are dereprssed by oxidative stress. Interestingly, more than two-thirds of these SIRT1-repressed genes are over expressed in 30 months old mice compared with 5 months old mice. Transcripts of major satellite repeat are also increased significantly in the aged mice [42].
To investigate the role of SIRT1 in tumorigenesis, the authors crossed a transgenic strain carrying overexpression of SIRT1 with p53+/- mice and found that SIRT1 overexression in p53+/- mice led to a decreased incidence of thymic lymphoma and increased survival following exposure to γ-irradiation. A similar effect was also observed in p53+/- mice that were treated with resveratrol, which activates SIRT1 [42]. These data provide strong evidence that SIRT1 serves as a tumor suppressor through its role in DDR and DSB repair that are essential for maintaining genome integrity. Because SIRT1 overexpression also suppresses the age-related transcriptional changes, it suggests that activation of SIRT1 might both extend lifespan and reduce cancer risk (Fig. 1C).
Perspective
The controversy over whether SIRT1 serves as a tumor promoter or a tumor suppressor has not been completely solved and the discussion will likely continue. One of the major concerns that SIRT1 activation may increase cancer risk comes from the observation that SIRT1 deacetylates and inactivates p53, and other tumor suppressors [26-28]. However, it is also known that p53 positively regulates SIRT1 transcription and loss of p53 impairs SIRT1 induction [43]. Thus, through a negative feedback loop, p53 inactivation can, in theory, reduce expression level of SIRT1, thereby increasing p53 activity. Similar negative feedback loops are also found in FOXO3a [43], FOXL2 [44] and E2F1 [45], which act as tumor suppressors under certain circumstances. Thus, like p53, the activities of these proteins are not easily inactivated by SIRT1 due to this negative feedback mechanism. Despite the lack of in vivo evidence that activation of SIRT1 may increase cancer risk, however, it remains possible that SIRT1 plays dual functions in different tissue contexts depending on the spatial and temporal distribution and abundance of different SIRT1 downstream targets and factors that regulate SIRT1. Further studies are needed to provide a conclusive answer.
For cancer therapy, irrespective of SIRT1's role as a tumor suppressor or promoter, it is important to identify endogenous genes or small chemicals that inhibit or activate SIRT1. Because Sir2 plays an essential role in lifespan extension in yeast, C. elegans and Drosophila [1], investigating whether SIRT1 has the same function in mammals is important. Aging has been considered as the most potent carcinogen for cancer, as cancer incidence is quickly elevated in the aging population [46]. It is of great interest to define the conditions, in which the activation of SIRT1 can both extend lifespan and inhibits tumor formation. In C. elegans, mutations that increase the lifespan can also inhibit tumor growth [47]. In animal models, caloric restriction, which activates SIRT1, extends lifespan and is also highly protective against cancer [19, 48-50].
Then, can direct activation of SIRT1 both extend lifespan and reduce cancer risk? Several lines of evidence suggest that it is possible. First, it has been shown that activation of SIRT1 by a low dose of dietary resveratrol partially mimics caloric restriction and retards aging parameters in mice [51]. Similarly, SRT1720, a more potent and specific agonist in activating SIRT1 than resveratrol [52], mimics low energy levels and protects against diet-induced metabolic disorders by enhancing fat oxidation [49]. Secondly, cancer is a metabolic disease. It has been demonstrated that aggressive cancer cells have a high rate of energy-consuming anabolic processes that promote the synthesis of lipids, proteins, and DNA, while decreased energy expenditure prevents tumorigenesis [53, 54]. Finally, as illustrated above that SIRT1 overexpression suppresses the age-related transcriptional changes and reduces formation of colon cancer in APC+/min mice [38], BRCA1-associated mammary cancer [40], spontaneous cancers in multiple tissues of Sirt1+/-;p53+/- mice [11], and γ-irradiation induced lymphoma in p53+/- mice [42]. Thus, through improving metabolic conditions by increasing SIRT1 activity, it is possible to both extend lifespan and reduce cancer risk in humans in the foreseeable future.
Acknowledgements
I thank members of Dr. Deng lab for their critical discussion of this work. This work was supported by the Intramural Research Program of the National Institute of Diabetes, Digestive and Kidney Diseases, National Institutes of Health, USA.
Conflict of Interest
The author has declared that no conflict of interest exists.
References
1. Blander G, Guarente L. The Sir2 family of protein deacetylases. Annu Rev Biochem. 2004;73:417-435
2. Guarente L. Sir2 links chromatin silencing, metabolism, and aging. Genes Dev. 2000;14:1021-1026
3. Saunders LR, Verdin E. Sirtuins: critical regulators at the crossroads between cancer and aging. Oncogene. 2007;26:5489-5504
4. Lavu S, Boss O, Elliott PJ. et al. Sirtuins--novel therapeutic targets to treat age-associated diseases. Nat Rev Drug Discov. 2008;7:841-853
5. Haigis MC, Guarente LP. Mammalian sirtuins--emerging roles in physiology, aging, and calorie restriction. Genes Dev. 2006;20:2913-2921
6. Huffman DM, Grizzle WE, Bamman MM. et al. SIRT1 is significantly elevated in mouse and human prostate cancer. Cancer Res. 2007;67:6612-6618
7. Bradbury CA, Khanim FL, Hayden R. et al. Histone deacetylases in acute myeloid leukaemia show a distinctive pattern of expression that changes selectively in response to deacetylase inhibitors. Leukemia. 2005;19:1751-1759
8. Stunkel W, Peh BK, Tan YC. et al. Function of the SIRT1 protein deacetylase in cancer. Biotechnol J. 2007;2:1360-1368
9. Hida Y, Kubo Y, Murao K. et al. Strong expression of a longevity-related protein, SIRT1, in Bowen's disease. Arch Dermatol Res. 2007;299:103-106
10. Lim CS. SIRT1: Tumor promoter or tumor suppressor? Med Hypotheses. 2006;67:341-344
11. Wang RH, Sengupta K, Li C. et al. Impaired DNA damage response, genome instability, and tumorigenesis in SIRT1 mutant mice. Cancer Cell. 2008;14:312-323
12. Vaziri H, Dessain SK, Ng Eaton E. et al. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149-159
13. Luo J, Nikolaev AY, Imai S. et al. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001;107:137-148
14. Lain S, Hollick JJ, Campbell J. et al. Discovery, in vivo activity, and mechanism of action of a small-molecule p53 activator. Cancer Cell. 2008;13:454-463
15. Motta MC, Divecha N, Lemieux M. et al. Mammalian SIRT1 represses forkhead transcription factors. Cell. 2004;116:551-563
16. Dai JM, Wang ZY, Sun DC. et al. SIRT1 interacts with p73 and suppresses p73-dependent transcriptional activity. J Cell Physiol. 2007;210:161-166
17. Wong S, Weber JD. Deacetylation of the retinoblastoma tumour suppressor protein by SIRT1. Biochem J. 2007;407:451-460
18. Pruitt K, Zinn RL, Ohm JE. et al. Inhibition of SIRT1 reactivates silenced cancer genes without loss of promoter DNA hypermethylation. PLoS Genet. 2006;2:e40
19. Cohen HY, Miller C, Bitterman KJ. et al. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science. 2004;305:390-392
20. Yuan Z, Zhang X, Sengupta N. et al. SIRT1 regulates the function of the Nijmegen breakage syndrome protein. Mol Cell. 2007;27:149-162
21. Li K, Casta A, Wang R. et al. Regulation of WRN protein cellular localization and enzymatic activities by SIRT1-mediated deacetylation. J Biol Chem. 2008;283:7590-7598
22. Liang XJ, Finkel T, Shen DW. et al. SIRT1 contributes in part to cisplatin resistance in cancer cells by altering mitochondrial metabolism. Mol Cancer Res. 2008;6:1499-1506
23. Ford J, Jiang M, Milner J. Cancer-specific functions of SIRT1 enable human epithelial cancer cell growth and survival. Cancer Res. 2005;65:10457-10463
24. Kojima K, Ohhashi R, Fujita Y. et al. A role for SIRT1 in cell growth and chemoresistance in prostate cancer PC3 and DU145 cells. Biochem Biophys Res Commun. 2008;373:423-428
25. Chen WY, Zeng X, Carter MG. et al. Heterozygous disruption of Hic1 predisposes mice to a gender-dependent spectrum of malignant tumors. Nat Genet. 2003;33:197-202
26. Chen WY, Wang DH, Yen RC. et al. Tumor suppressor HIC1 directly regulates SIRT1 to modulate p53-dependent DNA-damage responses. Cell. 2005;123:437-448
27. Zhao W, Kruse JP, Tang Y. et al. Negative regulation of the deacetylase SIRT1 by DBC1. Nature. 2008;451:587-590
28. Kim JE, Chen J, Lou Z. DBC1 is a negative regulator of SIRT1. Nature. 2008;451:583-586
29. Deng CX, Brodie SG. Knockout mouse models and mammary tumorigenesis. Semin Cancer Biol. 2001;11:387-394
30. Cardiff RD, Anver MR, Gusterson BA. et al. The mammary pathology of genetically engineered mice: the consensus report and recommendations from the Annapolis meeting. Oncogene. 2000;19:968-988
31. Deng CX. BRCA1: cell cycle checkpoint, genetic instability, DNA damage response, and cancer evolution. Nucleic Acids Res. 2006;34:1416-1426
32. McBurney MW, Yang X, Jardine K. et al. The absence of SIR2alpha protein has no effect on global gene silencing in mouse embryonic stem cells. Mol Cancer Res. 2003;1:402-409
33. Cheng HL, Mostoslavsky R, Saito S. et al. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc Natl Acad Sci U S A. 2003;100:10794-10799
34. Kamel C, Abrol M, Jardine K. et al. SirT1 fails to affect p53-mediated biological functions. Aging Cell. 2006;5:81-88
35. Banks AS, Kon N, Knight C. et al. SirT1 gain of function increases energy efficiency and prevents diabetes in mice. Cell Metab. 2008;8:333-341
36. Zhang QJ, Wang Z, Chen HZ. et al. Endothelium-specific overexpression of class III deacetylase SIRT1 decreases atherosclerosis in apolipoprotein E-deficient mice. Cardiovasc Res. 2008;80:191-199
37. Pfluger PT, Herranz D, Velasco-Miguel S. et al. Sirt1 protects against high-fat diet-induced metabolic damage. Proc Natl Acad Sci U S A. 2008;105:9793-9798
38. Firestein R, Blander G, Michan S. et al. The SIRT1 deacetylase suppresses intestinal tumorigenesis and colon cancer growth. PLoS ONE. 2008;3:e2020
39. Yeung F, Hoberg JE, Ramsey CS. et al. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004;23:2369-2380
40. Wang RH, Zheng Y, Kim HS. et al. Interplay among BRCA1, SIRT1, and Survivin during BRCA1-associated tumorigenesis. Mol Cell. 2008;32:11-20
41. Howitz KT, Bitterman KJ, Cohen HY. et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003;425:191-196
42. Oberdoerffer P, Michan S, McVay M. et al. SIRT1 redistribution on chromatin promotes genomic stability but alters gene expression during aging. Cell. 2008;135:907-918
43. Nemoto S, Fergusson MM, Finkel T. Nutrient availability regulates SIRT1 through a forkhead-dependent pathway. Science. 2004;306:2105-2108
44. Benayoun BA, Batista F, Auer J. et al. Positive and negative feedback regulates the transcription factor FOXL2 in response to cell stress: evidence for a regulatory imbalance induced by disease-causing mutations. Hum Mol Genet. 2008 [Epub ahead of print]
45. Wang C, Chen L, Hou X. et al. Interactions between E2F1 and SirT1 regulate apoptotic response to DNA damage. Nat Cell Biol. 2006;8:1025-1031
46. DePinho RA. The age of cancer. Nature. 2000;408:248-254
47. Pinkston JM, Garigan D, Hansen M. et al. Mutations that increase the life span of C. elegans inhibit tumor growth. Science. 2006;313:971-975
48. Boily G, Seifert EL, Bevilacqua L. et al. SirT1 regulates energy metabolism and response to caloric restriction in mice. PLoS ONE. 2008;3:e1759
49. Feige JN, Lagouge M, Canto C. et al. Specific SIRT1 Activation Mimics Low Energy Levels and Protects against Diet-Induced Metabolic Disorders by Enhancing Fat Oxidation. Cell Metab. 2008;8:347-358
50. Spindler SR. Rapid and reversible induction of the longevity, anticancer and genomic effects of caloric restriction. Mech Ageing Dev. 2005;126:960-966
51. Barger JL, Kayo T, Vann JM. et al. A low dose of dietary resveratrol partially mimics caloric restriction and retards aging parameters in mice. PLoS ONE. 2008;3:e2264
52. Milne JC, Lambert PD, Schenk S. et al. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature. 2007;450:712-716
53. Brunet J, Vazquez-Martin A, Colomer R. et al. BRCA1 and acetyl-CoA carboxylase: the metabolic syndrome of breast cancer. Mol Carcinog. 2008;47:157-163
54. Surmacz E. Obesity hormone leptin: a new target in breast cancer? Breast Cancer Res. 2007;9:301
Author contact
![]() Correspondence to: Chu-Xia Deng, PhD, Tel: (301) 402-7225; Fax: (301) 480-1135; Email: chuxiadniddk.nih.gov
Correspondence to: Chu-Xia Deng, PhD, Tel: (301) 402-7225; Fax: (301) 480-1135; Email: chuxiadniddk.nih.gov
Citation styles
APA
Deng, C.X. (2009). SIRT1, Is It a Tumor Promoter or Tumor Suppressor?. International Journal of Biological Sciences, 5(2), 147-152. https://doi.org/10.7150/ijbs.5.147.
ACS
Deng, C.X. SIRT1, Is It a Tumor Promoter or Tumor Suppressor?. Int. J. Biol. Sci. 2009, 5 (2), 147-152. DOI: 10.7150/ijbs.5.147.
NLM
Deng CX. SIRT1, Is It a Tumor Promoter or Tumor Suppressor?. Int J Biol Sci 2009; 5(2):147-152. doi:10.7150/ijbs.5.147. https://www.ijbs.com/v05p0147.htm
CSE
Deng CX. 2009. SIRT1, Is It a Tumor Promoter or Tumor Suppressor?. Int J Biol Sci. 5(2):147-152.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.