Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
High Density Lipoproteins and...
Structural components of HDL
Cholesterol efflux into nascent...
HDL maturation
Lipase activity
Lipoprotein remodeling in...
The duality of the scavenger...
Hepatic bile acid synthesis
Low Density Lipoproteins and...
Exogenous lipid metabolism in...
Movement of exogenous...
VLDL synthesis and maturation...
Intracellular cholesterol...
End product feedback regulation...
Summary
Acknowledgements
Abbreviations
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2009; 5(5):474-488. doi:10.7150/ijbs.5.474 This issue Cite
Review
Lipoproteins, cholesterol homeostasis and cardiac health
Tyler F. Daniels1, Karen M. Killinger2, Jennifer J. Michal 1, Raymond W. Wright Jr. 1, Zhihua Jiang1 ![]()
1. Department of Animal Sciences, Washington State University, Pullman, WA 99164-6351, USA;
2. School of Food Science, Washington State University, Pullman, WA 99164-6376, USA
Received 2009-5-11; Accepted 2009-6-19; Published 2009-6-29
Abstract
Cholesterol is an essential substance involved in many functions, such as maintaining cell membranes, manufacturing vitamin D on surface of the skin, producing hormones, and possibly helping cell connections in the brain. When cholesterol levels rise in the blood, they can, however, have dangerous consequences. In particular, cholesterol has generated considerable notoriety for its causative role in atherosclerosis, the leading cause of death in developed countries around the world. Homeostasis of cholesterol is centered on the metabolism of lipoproteins, which mediate transport of the lipid to and from tissues. As a synopsis of the major events and proteins that manage lipoprotein homeostasis, this review contributes to the substantial attention that has recently been directed to this area. Despite intense scrutiny, the majority of phenotypic variation in total cholesterol and related traits eludes explanation by current genetic knowledge. This is somewhat disappointing considering heritability estimates have established these traits as highly genetic. Thus, the continued search for candidate genes, mutations, and mechanisms is vital to our understanding of heart disease at the molecular level. Furthermore, as marker development continues to predict risk of vascular illness, this knowledge has the potential to revolutionize treatment of this leading human disease.
Keywords: HDL, LDL, Homeostasis, cholesterol, expression, candidate genes, heart disease
Introduction
The clinical manifestation of cholesterol buildup in arteries servicing the heart muscle causes more death and disability than all types of cancer combined (1). This is a remarkable outcome for a common, polycyclic lipid with the humble primary function of maintaining the permeability and fluidity of cell membranes. Nonetheless, cholesterol is required by all eukaryotic cells, which have specialized methods of recruiting and synthesizing the lipid only when it is needed. While effectively maintaining intracellular cholesterol homeostasis, these processes leave excess circulating though the body, leading to atherosclerotic plaque development and subsequent coronary artery disease. Thus, levels of cholesterol and related lipids circulating in plasma are important predictive tools utilized clinically to gauge risk of a cardiac event. For example, a rise in total cholesterol in men from 200 to 240 mg DL-1 is associated with a three-fold increase in death from cardiac disease (2).
While in circulation, cholesterol, being a lipid, requires a transport vesicle to shield it from the aqueous nature of plasma. Complex, micelle-like amalgamations of various proteins and lipids achieve cholesterol transport through the vascular system. These particles, intuitively known as lipoproteins, are heterogeneous in size, shape, composition, function, and perhaps most importantly, their contribution to vascular disease. High density lipoprotein (HDL) particles promote vascular health by extracting cholesterol from tissues (including atherosclerotic plaques) and delivering it back to the liver. Conversely, low-density lipoproteins (LDLs) are the classic antagonists of the circulatory system due to their propensity to bind to connective tissue in the intimal sub-layer of arteries (3). These processes are the motivation for the “good cholesterol,” and “bad cholesterol” stigmas attached to HDL and LDL, respectively. Before any cholesterol reaches a particle of the HDL or LDL sub-fraction, it will usually have undergone a maturation process beginning with the hepatic or intestinal synthesis of very low density lipoprotein (VLDL). VLDLs, LDLs, and HDLs make up three of the six major sub-fractions of lipoproteins, which also include chylomicrons, chylomicron remnants (CRs), and intermediate density lipoproteins (IDLs). Together, lipoprotein particles are heterogenous composites of the stored forms of fatty acids and cholesterol (triglycerides and cholesteryl esters, respectively), amphipathic phospholipids, and apolipoproteins.
High Density Lipoproteins and Reverse Cholesterol Transport
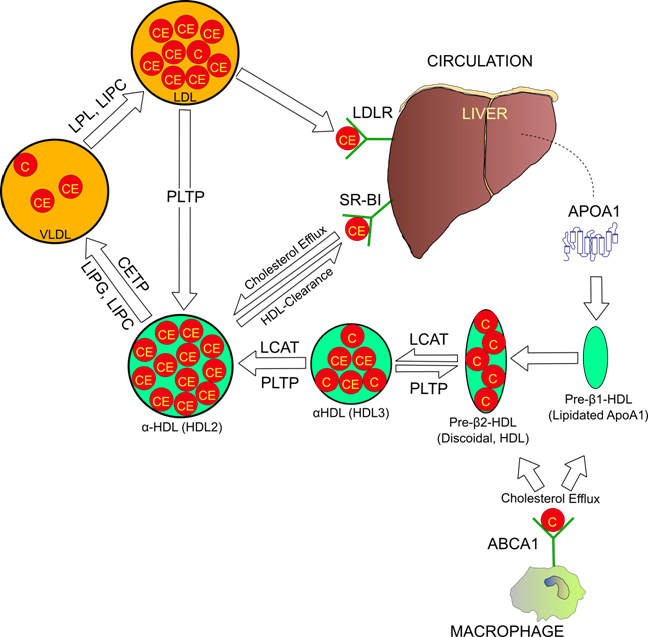
Particles of HDL prevent coronary artery disease by serving as transport particles for excess cholesterol to the liver, where it is converted into bile acids and excreted (Figure 1). In humans, HDL levels are a very well known measurement of cardiac health due to their strong inverse relationship with coronary artery disease (2, 4). The principal HDL pathway, termed reverse cholesterol transport (RCT) is a major component of lipid homeostasis. Genetic variation among the RCT pathway contributes greatly to phenotypic variation in humans. Estimates using twin family studies have determined that nearly 50% of HDL phenotypic variability is genetic (5). These figures are controversial however, as it is possible that common mutations might contribute minor effects to lipid phenotypes (6).
Reverse cholesterol transport pathway. Arrows are indicative of cholesterol movement and particle maturation. Cholesterol molecules are labeled “C”, and cholesterol esters “CE”.
Structural components of HDL
The principal protein components of HDL are dual apolipoprotein (Apo) A1 peptides, which wrap around the particle in an anti-parallel, double-belt structure (7). Secondary structure of ApoA1 consists of ten transmembrane amphipathic α-helices (8). Functionally, ApoA1 forms the initial structure of discoidal HDL (9), and is recognized by some of the most important proteins involved in RCT including: lecithin-cholesterol acyltransferase (LCAT) (10), ATP binding cassette A1 (ABCA1) (11), and the scavenger receptor BI (SR-BI) (12). Therefore, ApoA1 is not only the primary structural component; it serves as the recognition molecule for most of the proteins that interact with HDL. ApoA1-deficient mice do not form normal HDL particles and exhibit 70-80% reductions in both plasma cholesterol and HDL (13). Human studies have concluded that mutations in ApoA1 exhibit the most profound effects on accelerated atherosclerosis compared to other RCT proteins (14). However, some ApoA1 mutations reduce levels of circulating HDL, but are reported to actually decrease risk of cardiac events. Notably, the ApoA1 milano (R173C) mutation has been implicated in improved circulatory health despite leading to reduced HDL and hypertriglyceridemia (15). Franceschini et al. provided a possible explanation for this anomaly when they concluded that mice carrying the ApoA1 milano mutation had more efficient cholesterol efflux capabilities then their wild-type littermates (16). This finding has recently been challenged, as Weibel and colleague's 2007 study compared efflux potentials of ApoA1 wild-type and milano rats from 3 different cell types, and determined that efflux rates were not significantly different (17). Certainly, the elusive details of the potential atheroprotective effects resulting from ApoA1 mutations such as milano are worthy of intense scrutiny.
Cholesterol efflux into nascent HDL
The rate-limiting step in RCT is cholesterol efflux, a process mediated largely by the ABCA1 gene product. ABCA1 promotes cholesterol efflux by catalyzing the transfer of cholesterol and phospholipids from potentially atherogenic cells in peripheral tissues to discoidal HDL (11) (Figure 1). ABCA1 induced cholesterol efflux is especially combatant to vascular disease when cholesterol is taken from macrophages inside artery walls because their cholesterol-induced differentiation into foam cells is the foundation of atherogenesis (18). The efflux process initiates as discoidal HDL binds to ABCA1 on the cell surface, is internalized, and then transported into endocytic vesicles inside the cell (19). While inside endosomes, ABCA1 and the Niemann-Pick C1 protein mediate the transfer of lipid pools into intracellular HDL (20). The complex then returns to the cell surface and dissociates, thus releasing the cholesterol-enriched pre-β2-HDL undegraded (19).
Genetic studies have definitively established the crucial involvement of ABCA1 with HDL genesis. Humans with ABCA1 inactivating mutations have Tangier disease, which is marked by orange, cholesterol ester laden tonsils, peripheral neuropathy, and strong predisposition to coronary artery disease caused by the virtual absence of HDL (18, 21). Several recent studies have identified more common mutations that play important roles in HDL homeostasis. For example, in their study of patients with the low HDL levels, Cohen et al. identified an ABCA1 mutation in 20 of 128 individuals (22). Furthermore, large-population studies have estimated that ABCA1 mutations are responsible for 10% of low HDL cases in the general population (22, 23).
HDL maturation
During HDL maturation, discoidal pre-β-HDL is converted into spherical α-HDL via LCAT mediated conversion of cholesterol into cholesteryl esters (Figure 1). The 416 amino acid LCAT protein is synthesized in the liver and secreted into circulation (24), where it exists both bound to lipoproteins and lipid-free (25). On HDL particles, LCAT generates cholesterol esters by catalyzing the transfer of the 2-acyl group of lecithin to the hydroxyl group of cholesterol (26) (Figure 2). LCAT also can esterify cholesterol in LDL, but HDL is the preferred lipoprotein substrate due to the presence of ApoA1, which is a cofactor (27). Cholesterol esters are more hydrophobic than free cholesterol, and accumulate in the center of HDL particles causing a change in the geometry from discoidal to spherical (Figure 1). This process is crucial to cholesterol efflux because it helps maintain a concentration gradient favoring addition of free cholesterol to lipoproteins including HDL (28, 29).
Representation of the molecular activity of the LCAT protein.
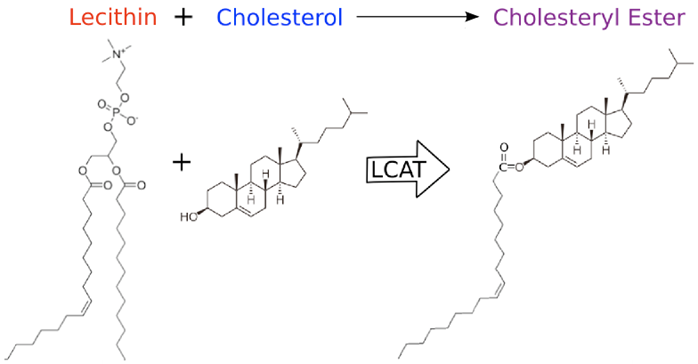
In humans, LCAT inactivating mutations cause familial LCAT deficiency (FLD), which is characterized by severe corneal opacification, low plasma HDL, sharply increased LDL triglycerides, and accumulation of discoidal HDL in plasma (30). Additionally, some LCAT defects disrupt interaction with ApoA1, which leads to fish eye disease, decreased HDL (but not LDL), decreased cholesterol esters, and similar but milder phenotypes compared to FLD (30, 31).
Lipase activity
In order to understand HDL metabolism, it is important to be aware that lipoproteins of each class are heterogeneous particles that heavily interact with, and often change into one another. A common factor influencing lipoprotein interaction is activity of lipase proteins. Lipases are water-soluble enzymes that hydrolyze ester bonds of water-insoluble substrates such as triglycerides, phospholipids, and cholesteryl esters (32). Endothelial lipase (LIPG), hepatic lipase (LIPC), and lipoprotein lipase (LPL) are three vascular lipase proteins that migrate to endothelial cells and anchor to the distal side via interaction with heparin sulfate proteoglycans (33-35). The exposed lipase proteins remodel circulating lipoproteins, generating important effects to lipoprotein metabolism and cholesterol homeostasis. Additionally, some lipase enzymes directly interact with lipoprotein receptors, such as the LDL receptor, enhancing metabolism of circulating lipoproteins.
The majority of the catalytic activity of LIPG is devoted to hydrolysis of phospholipids of VLDL, chylomicrons, and HDL (33). Interestingly, LIPG mediated phospholipid modulation of HDL inhibits cholesterol efflux from SR-BI, but enhances efflux from ABCA1, and HDL uptake in the liver (36). In 2006, Badellino and colleagues (37) established a correlation between LIPG and atherosclerosis, which underscores the importance of SR-BI cholesterol efflux and HDL longevity in ideal HDL function.
The LIPC protein is synthesized primarily by hepatocytes, and then secreted and bound to the extracellular matrix of hepatic endothelial cells (38). LIPC has powerful VLDL and IDL triglyceride hydrolysis capabilities (39), as well as the ability to catalyze conversion of α-HDL subspecies HDL2 to denser HDL3 (40) (Figure 1). The latter functionality has direct implications to RCT because HDL2 is more likely to interact with SR-BI for cholesterol efflux or endocytosis (41). Independently of RCT, LIPC appears to demonstrate pro-atherogenic effects by increasing artery wall retention of VLDL, chylomicrons, and LDL (42). Although mutations in human LIPC are associated with variations in HDL concentration, the connection with coronary artery disease is controversial (43, 44).
LPL is a critical enzyme involved in hydrolysis of triglyceride rich lipoprotein particles in muscle, adipose, and macrophages, a process which generates free fatty acids and glycerol for energy metabolism and storage (45). Expression of LPL has been implicated in atherosclerosis, citing the increased affinity for macrophage phagocytosis (and subsequent foam cell development) on LDL and chylomicron particles after LPL mediated remodeling (46).
Lipoprotein remodeling in circulation
Cholesterol homeostasis is greatly modulated by proteins that catalyze the exchange of cholesterol and other lipids between circulating lipoprotein classes. Mutations in lipid transfer proteins are very important sources of lipoprotein phenotypic variation, as the genes are decidedly polymorphic. In plasma, there are two important lipid transfer proteins: cholesteryl ester transfer protein (CETP), and phospholipid transfer protein (PLTP).
CETP catalyzes the exchange of cholesteryl esters inside HDL for triglycerides of LDL and VLDL (Figure 1) (47). Expression of CETP is fairly ubiquitous, however most is synthesized in the liver, and then excreted and bound to HDL in circulation (48). Theoretically, CETP activity is attributed to coronary artery disease, as the movement of cholesterol from HDL to LDL is certainly pro-atherogenic. Surprisingly, studies have failed to clearly identify the relationship between CETP and atherosclerosis, suggesting additional function of the protein. In mice (which naturally lack CETP), expression of the human ortholog has been found to be both pro-atherogenic (49), and anti-atherogenic (50), depending on the metabolic state of the mouse model. In humans, markedly increased HDL has been a consistent finding in carriers of CETP nullifying mutations (51, 52). Some carriers of inactivated CETP have been linked to increased longevity (51), while other studies have reported no association (53), and still other mutations implicate increased risk of coronary artery disease (54). The CETP gene, located on human chromosome 16q21 is highly polymorphic. One important polymorphism, Taq1B, accounted for 5.8% of of the variation in HDL in a Spanish population (55), and appears associated with coronary artery disease (56).
PLTP regulates the size and composition of HDL both by lipid exchange and particle remodeling. The gene is located on human chromosome 20q12-q13.1, and is expressed primary (but not solely) by adipocytes and hepatocytes (57). In circulation, normal PLTP activity appears protective against atherosclerosis due to its ability to both increase circulating levels of cholesterol efflux prone pre-β-HDL and to maintain levels of mature HDL. PLTP is able to catalyze the fusion of two HDL3 particles, forming one larger HDL2, while releasing lipid poor ApoA1, the precursor to pre-β-HDL (58). Enrichment of HDL with triglycerides enhances this process (59), otherwise known as HDL conversion. In addition to HDL conversion, PLTP facilitates the transfer of phospholipids and to a lesser extent, cholesterol from triglyceride rich lipoproteins such as VLDL and chylomicrons into HDL (60, 61). PLTP inactivated mice have 60-70% lower HDL (62), which suggests that phospholipid migration into HDL impedes catabolism of the particle. Transgenic mice overexpressing PLTP have severely reduced HDL and inhibited RCT, despite increased levels of pre-β-HDL (63, 64). This activity has been linked to increased HDL catabolism (63), and excessive HDL conversion, a conclusion that challenges the theory that pre-β-HDL particles are more important for cholesterol efflux then α-HDL (64). While these studies in animal models generate as many questions as answers, together they indicate that correctly balanced PLTP activity is essential in maintaining normal plasma HDL concentration.
The duality of the scavenger receptor class B type I protein
Ultimately, the protective effects generated by RCT are accomplished through the removal of excess cholesterol (cholesterol efflux), and delivery of cholesterol rich lipoprotein particles to liver hepatocytes for lipid excretion (HDL clearance). The SR-BI protein is very unique because it plays an integral role in both of these processes. The gene is expressed primarily in adrenal tissues and the liver, where it is attached to epithelial surfaces (65). Studies in humans and animal models have established SR-BI as vital to cholesterol homeostasis. Mice with inactivated SR-BI have a twofold increase in plasma cholesterol, which is distributed in unusually large and homogenous HDL particles (66). Furthermore, a separate study linked greatly reduced clearance of HDL from plasma to SR-BI deficiency (67). These observations strongly suggest that SR-BI is required to maintain normal cholesterol.
SR-BI is most notorious for catalyzing uptake of cholesterol from lipid-rich HDL particles into hepatocytes, where it is converted into bile acids. The mechanism of HDL clearance is distinct from ABCA1 mediated cholesterol efflux and low density lipoprotein receptor endocytosis in that internalization of the entire particle does not occur, but rather cholesteryl esters are selectively taken into tissues (68). In this way, mature α-HDL can quickly be recycled back into lipid-poor pre-β-HDL, and RCT can start again. Affinity of lipoprotein/SR-BI interaction is complex and highly related to apolipoprotein composition and particle geometry. SR-BI binds both LDL and HDL; however, preference is strongest for lipoproteins containing ApoA1, which it binds to tightly (69). In fact, three HDL associated apolipoproteins, ApoA1, ApoA2, and ApoC3, each interact with SR-BI, and increase affinity for cholesterol movement (70).
The availability of LDLR to commit lipoprotein endocytosis is regulated by Proprotein Convertase Subtilisin Kexin type 9 (PCSK9). In fact, this relationship is important enough to instigate a distinct classification of familial hypercholesterolemia (known as FH3), which is caused by mutations in PCSK9 (71). Secreted PCSK9 competes with lipoprotein-associated apolipoproteins as a ligand for LDLR by binding to the receptor and causing endocytosis and degradation of the complex (72). Additionally, PCSK9 appears to interrupt intracellular receptor recycling by binding to LDLR prior to its placement on the surface of hepatocytes (73). The molecular processes involved in PCSK9 induced degradation are not clear, although Geoghegan et al. have elucidated that PCSK9 exhibits acylenzyme activity on LDLR, in which residue Gln152 of the receptor is esterified by Ser386 of PCSK9 (74). This study, and independent genetic studies have shown that the EGFP domain of LDLR is the active site of PCSK9 interaction (75). The FH3 phenotype is rare, and is derived from missense mutations that increase functionality of PCSK9. However, this effect is not the only clinically important mutation in PCSK9, as loss-of-function mutations appear to generate positive effects on blood lipids. The 2005 Dallas Heart study identified two PCSK9 nonsense mutations among subjects with low plasma LDL levels: Y142X in exon 3, and C679X in exon 12 (76). These mutations led to 28% decreased LDL levels, and significant reductions in risk of coronary artery disease. PCSK9 induced hypocholesterolemia has also been identified in Japanese (77), and Italian (78) patients.
As previously mentioned, SR-BI plays a key role in cholesterol efflux by acting as a cholesterol donor to HDL. Unlike ABCA1 mediated cholesterol efflux, which targets pre-β-HDL, the ideal lipoprotein for SR-BI mediated efflux is large, phospholipid-rich, spherical HDL2 (79, 41). Thus, SR-BI is a bidirectional cholesterol transporter to and from mature HDL, and is one of the most important proteins to RCT.
Hepatic bile acid synthesis
Cholesterol delivered to the liver via HDL enters the bile acid synthesis pathway (also known as the cholesterol catabolic pathway), which begins with the enzymatic modulation of hepatic cholesterol to 7-α-hydroxycholesterol by cholesterol 7-α-hydroxylase (CYP7A1) (80). Transcriptional activity of CYP7A1 dictates the efficacy of the cholesterol catabolic pathway, and is critical to hepatic cholesterol homeostasis. One of many important transcriptional regulators of CYP7A1 is the Farnesoid X receptor (FXR). FXR acts as a bile acid sensor, suppressing CYP7A1 activity when hepatic concentrations of bile salts are high (81). FXR does not work on its target directly, but instead upon activation, it triggers a cascade involving small heterodimer partner 1 (SHP-1), which activates the direct CYPA1 inhibitor, liver receptor homolog 1 (LRH-1) (82). Not surprisingly, homozygous deletion of CYPA1 results in increased hepatic cholesterol content, deficient bile acid secretion, and hypertriglyceridemia (83). The remaining steps in the conversion of cholesterol into conjugated bile salts such as taurocholic and glycocholic acid involve at least 16 enzymes. After synthesis, these bile acids cycle from the liver, to the gall bladder, into the duodenum sub-layer of the intestine, back through enterocytes of the ileum, through the portal vein, and back into hepatocytes several times per day while carrying out their primary function of aiding lipid digestion (84).
Low Density Lipoproteins and Cholesterol Distribution
The mechanisms that manage and utilize LDL are tightly controlled systems evolved to distribute cholesterol through the circulatory system and into cells that require extracellular cholesterol. Unfortunately, LDL-cholesterol does not always reach its most appropriate destination, but rather accumulates in artery walls causing atherosclerosis, the leading cause of death and disability in the developed world (85). For this reason, the quantity of circulating LDL is a well-known risk factor for heart disease, and is the primary focus of most lipid lowering therapies (86). The pathogenicity of LDL and likelihood of atherosclerotic development are heavily influenced by genetic composition of gene products involved with LDL metabolism. Patients with genetic defects that cause severely elevated LDL have familial hypercholesterolemia, which affects approximately 1:500 people (87), and is the consequence of mutations in the low density lipoprotein receptor (LDLR), ApoB, and other genes. In normal individuals, approximately 50% of LDL variation is genetic. This section will address the mechanisms behind exogenous and endogenous cholesterol transport, with emphasis placed on the proteins that participate in this crucial pathway.
Exogenous lipid metabolism in the intestine
The amount of cholesterol absorbed from the diet is a major contributor to levels of cholesterol in circulation. One study has estimated that the complete abolition of dietary cholesterol absorption would reduce plasma cholesterol by up to 62% (88). About 50% of dietary cholesterol is absorbed through intestinal enterocytes, while the rest is excreted through feces (89). This figure however, is extremely variable among individuals and this variation has been established as an inherited trait (90). Thus, the mechanisms behind the entry of dietary cholesterol into the body are critical sources of variation in cholesterol homeostasis.
Early lipid digestion, from the oral cavity to the duodenum sub-layer of the intestine, produce crude emulsions consisting of free cholesterol, triglycerides, free fatty acids, and phospholipids. As these emulsions are delivered into the intestine, they are mixed with bile salt micelles, which are synthesized and secreted into the intestine from the liver (91). Total concentration of bile salt micelles is positively correlated with cholesterol absorption (92), due to catalysis of lipid emulsification into smaller droplets, which interact more readily with lipase enzymes (93). Bile salt emulsified triglycerides and cholesteryl esters are hydrolyzed by pancreatic lipase (PL) (94), and carboxyl ester lipase (CEL) (95), respectively. Not surprisingly, in vivo knockout studies using murine models have confirmed the necessity of both PL (93) and CEL (95) to normal cholesterol absorption, and chylomicron assembly.
Cholesterol absorption is achieved through passage across brush border membranes and into intestinal enterocytes in the jejunum. While details of the mechanism are still unclear, recent studies have identified key proteins. One major breakthrough came with the identification of the substrate for the drug ezetimibe (brand: Zetia), which inhibits cholesterol entry into enterocytes without effecting de novo cholesterol biosynthesis (96). Ezetimibe was approved by the United States Food and Drug Administration for treatment of hyperlipidemia in 2002, however, it took two additional years before Garcia-Calvo and colleagues discovered that the Niemann-Pick C1-Like 1 (NPC1L1) protein is the target of the drug (97). NPC1L1 is a putative transporter of cholesterol, and is expressed on the brush border membranes of enterocytes in a general pattern that parallels that of maximum cholesterol absorption (98). Naturally occurring mutations in human NPC1L1 are associated with reduced cholesterol absorption and circulating levels of LDL (99). In addition to NPC1L1, the ATP binding cassette G5 and G8 proteins (ABCG5/8) appear to negatively regulate cholesterol transport into enterocytes. Mutations in the ABCG5/G8 genes (they are neighbors on human chromosome 2p21) are associated with sitosterolemia, which is characterized by increased absorption of plant sterols. Interestingly, sitosterolemic patients also have increased amounts of dietary cholesterol absorption and premature atherosclerosis (100). This data has been interpreted as evidence that ABCG5/G8 form a complex that promotes secretion of cholesterol back into the intestinal lumen, and that this activity targets plant sterols in normal individuals (101).
Movement of exogenous cholesterol from the intestines to the liver
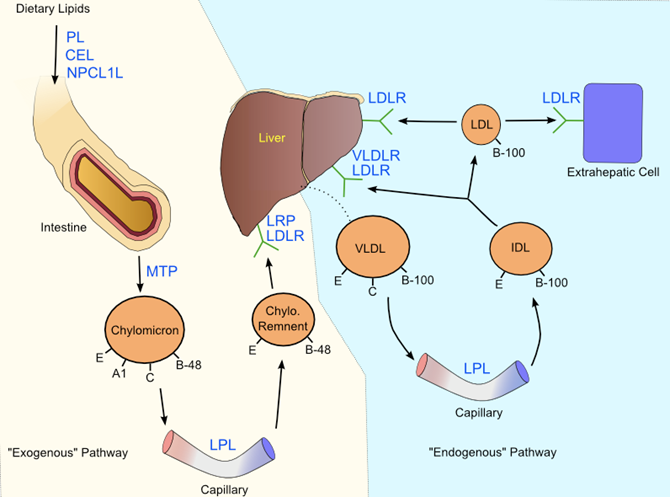
Once inside enterocytes, dietary cholesterol is packaged into chylomicrons, and put into circulation. This process is initiated by the esterification of large amounts of free cholesterol by the cholesteryl transferase protein (102), and the synthesis of triglycerides from free fatty acids by mono- and di-acylglycerol acyltransferases (101). In the endoplasmic reticulum, cholesteryl esters, phospholipids and triglycerides are amalgamated together with ApoB-48 by the microsomal triglyceride transfer protein (MTP) (103). The action of MTP is not localized to enterocytes, or even to chylomicron synthesis, instead it is nearly ubiquitously expressed and a crucial element of VLDL synthesis as well. Inactivating mutations in MTP result in abetalipoproteinemia, in which chylomicrons and VLDL are not synthesized, ApoB containing particles in general are absent, and lipids accumulate in the intestine (104). As nascent chylomicron synthesis nears completion, the particles are transported to the Golgi apparatus where additional triglycerides are recruited, and then the particles are transported via vesicular structures to clatherin-coated pits and exocytosed (105).
ApoB is the major protein component of all lipoproteins except HDL. During chylomicron synthesis in the intestine, ApoB mRNA sequence is altered by the apolipoprotein B mRNA editing enzyme, catalytic polypeptide 1 (APOBEC1) protein, so that the 6666th nucleotide is changed from a cytosine to a uracil (106). This unique process leads to a premature UAA stop codon, and production of a truncated ApoB peptide only 48% the length of full ApoB (106). APOBEC1 inactivated mice demonstrate a complete lack of ApoB-48, 178% more ApoB-100, and decreased HDL cholesterol (107). Mutations in the ApoB gene itself are an important source of phenotypic variation in humans. In a recent review, 132 genetic variants in the ApoB gene are listed including one in the promoter region, one in the 5' untranslated region, 85 in the coding region (22 synonymous), 44 in the various introns, and one in the 3' UTR (108). Notably, genotypes of the very common T2488T and E4154K mutations have crucial implications to LDL homeostasis (109, 110). Considering that ApoB is a direct measurement of potentially atherogenic particles, the levels of ApoB levels in circulation are considered a more appropriate trait to measure risk of cardiovascular disease than LDL (111), however, LDL levels remain more clinically utilized.
While the digestion and packaging of dietary lipids into chylomicrons takes about one hour, the halflife of lipids in chylomicrons is only 4.5 minutes (112). Upon exiting enterocytes, the only protein component of chylomicrons is ApoB-48. After passage through the throacic duct and into the bloodstream, nascent chylomicrons accept ApoC2 and ApoE from HDL, a process that yields mature chylomicrons. As mature chylomicrons circulate, the newly acquired ApoC2 on the particle surface activates LPL, which is bound to epithelial surfaces of capillaries in adipose and muscle tissue where it is differentially expressed according to fed/fasting conditions (45). LPL catalyzes the hydrolysis of triglycerides in chylomicrons, a crucial process that distributes fatty acids to tissues, generates non-esterified fatty acids in plasma, and remodels chylomicrons into CRs (45). The absence of LPL causes familial LPL deficiency, which is characterized by hypertriglyceridemia, decreased HDL and LDL, and massive accumulation of chylomicrons in plasma (113). As chylomicrons shed triglycerides to epithelial cells, ApoC2 is lost (ApoE is retained), and as a result, the CR loses further lipase activation.
As CRs lose triglycerides, they become enriched in ApoE, and consequently are destined for the liver. ApoE peptides are essential for particle uptake into hepatocytes, as demonstrated by ApoE inactivated mice, which demonstrate negligible CR clearance (114). The model for hepatic clearance involves ApoE interaction with hepatocyte cell surface molecules including heparin sulfate proteoglycans and LIPC, followed by an endocytosis step mediated by a complex of LDLR and the low density lipoprotein related 1 protein (LRP1) (115). Another factor influencing CR uptake is cholesterol content of the macromolecule. Chylomicron-like particles, created without cholesterol, undergo triglyceride hydrolysis by LPL but are not taken up by hepatocytes (116). This result has been interpreted as a failure of ApoE to attain the necessary conformation to achieve receptor binding with LRP1 (116). After delivery to the liver, CR lipids are hydrolyzed once again into free fatty acids and free cholesterol for eventual synthesis of VLDL.
VLDL synthesis and maturation into LDL
The assembly of VLDL begins inside the rough endoplasmic reticulum of hepatocytes, at the site of ApoB-100 translation. As the peptide is synthesized by membrane bound ribosomes, it is sent through a protein channel into the cytoplasm. Meanwhile, the aforementioned MTP binds the precursor peptide and recruits a small amount of triglycerides, phospholipids, and cholesteryl esters, allowing ApoB-100 to fold around a small lipid core (117). Next, the bulk of VLDL triglycerides are transferred into the precursor particle, and the now larger precursor (sometimes referred to as VLDL2) is sorted to the Golgi apparatus. Evidence for a two-step maturation system up to this point is given by experiments using Brefeldin A (BFA), which inhibits only the transition into VLDL2, and not the initial lipidation of ApoB-100 (118). BFA selectively inhibits the ADP-ribosylation factor (ARF) protein, suggesting that ARF is a critical factor in VLDL assembly (118). Once in the Golgi, additional lipids are recruited to form the mature VLDL particle, but the mechanism behind this behavior is not yet clear. However, it is certain that the fatty acids used for the synthesis of VLDL are derived from triglycerides stored in cytosolic lipid droplets (119), so it is likely that the concentration of these lipids is the rate limiting property of VLDL biogenesis.
Just like chylomicrons, VLDLs exchange ApoC2 and ApoE with HDL in circulation, and distribute free fatty acids to muscle and adipose tissues expressing LPL. As remodeled VLDL particles lose triglycerides and ApoC2, they become IDLs, which are either removed by the liver or are subject to further lipase activity and develop into LDL. Endocytosis of IDL by the liver includes not only LDLR, but also other receptors in the same class including the very low density lipoprotein receptor (120), and LRP1 (115). Larger, triglyceride-rich IDL particles are generally more susceptible to liver re-uptake by these receptors compared to smaller particles, which are more likely to transition into LDL (121). The triglyceride content (and subsequently, the size and metabolic fate) of IDL particles is heavily influenced by LIPC (39), as well as CETP, (47) and PLTP (60). LDL particles have a relatively long half-life of about 3 days (122), a property attributed to the lack of receptor and lipase activating apolipoproteins, as well as the stability of cholesteryl esters inside the macromolecules.
Variation among VLDL, IDL, LDL, and HDL levels in circulation are highly dependent on the genetic composition of ApoE. Mice lacking functional ApoE have 500% more plasma cholesterol, and rapidly develop severe atherosclerosis compared to wild type mice (114). This effect is presumably from a complete loss of LDLR binding and chylomicron/LDL clearance (123). Due to their tendency to develop atherosclerotic plaques, the ApoE knockout mouse model is a very commonly used model for human atherosclerosis. In humans, ApoE contains two very common SNPs at amino acid positions 112 and 158, which dictate the ApoE2, E3, and E4 genotype nomenclature (124). The ApoE4 appears to be associated with decreased HDL, increased LDL, and increased plasma cholesterol, while the E2 genotype has the opposite effect (125, 126).
Intracellular cholesterol metabolism
LDLR is responsible for uptake of cholesterol carrying lipoproteins. The principal ligand for the receptor is ApoB-100 on LDL, however LDLR can catalyze endocytosis of lipoproteins containing multiple copies of ApoE such as VLDL, IDL, and HDL. The extracellular portion of LDLR consists of three protein modules including a domain with seven contiguous cysteine-rich repeats (referred to as LDLR type A, or LA domains), a 400-amino acid sequence that is strongly homologous to the epidermal growth factor precursor protein (referred to as the EGFP domain), and a 58-residue sequence rich in serine and threonine (Figure 4) (127). Genetic studies have revealed that LA domains 3-7 are required for effective binding of LDL, while only LA5 is required for VLDL uptake (128). Studies utilizing antibody blocking and synthetic peptides have determined that amino acid residues ~3300-3600 of ApoB-100 are the active sites for LDL interaction with LDLR (129, 130).
Transport of exogenous cholesterol, and de novo cholesterol requires a diversity of lipoproteins and proteins, as shown above.
The LDLR structure contains three types of domain: LDLR-type A domains (LA), the epidermal growth factor precursor protein-like domain (EGPF), and a domain rich in serine and threonine residues. The endocytotic process is also shown, which yields free intracellular cholesterol [see (127) with permission].
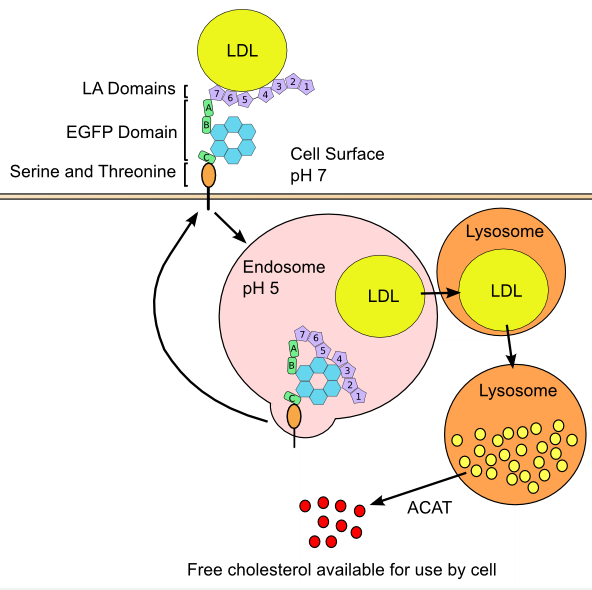
Figure 4 describes the intracellular process in LDL endocytosis. LDLR positioning and subsequent endocytosis of the receptor-ligand complex occurs at clathrin coated pits (131). After endocytosis, acidic conditions of the endosome catalyze disassociation of LDL from the LDLR. Genetic studies have implicated the EGFP domain of LDLR as responsible for release of LDL, as deletion of this region produces a non-separable complex (132). After release of the lipoprotein, the LDLR peptide is recycled back to the membrane in a process also controlled by the EGFP domain (132). Meanwhile, LDL particles that are released from the receptor fuse into lysosomes, and are degraded into lipid components and amino acids by enzymes of the vesicle. Large portions of lipids released are cholesteryl esters, which are hydrolyzed by lysosomal acid lipase (LIPA) into free cholesterol. Patients with complete and heterozygous LIPA deficiency have Wolman disease and cholesteryl ester storage disease, respectively (133). The consequences of inactive LIPA are severe: liver failure, hypercholesterolemia, hypertriglyceridaemia, liver fibrosis, early atherosclerosis, and early death (133). Cholesterol that has been endocytosed and converted to free form is often incorporated into cell membranes, however depending on cell type, it has several other possible fates including efflux to cellular adaptors, conversion back into cholesterol esters, metabolism into bile acids, or synthesis of steroids (134). Regardless of metabolic fate, levels of intracellular cholesterol are the controlling element behind overall cholesterol homeostasis of every cell via their impact on regulating LDLR protein and de novo cholesterol synthesis.
End product feedback regulation of LDLR and HMG Co-A reductase
In addition to endocytosis of lipoproteins, mammalian cells increase cholesterol levels through de novo synthesis beginning from acetyl-CoA. The rate limiting reaction of the cholesterol biosynthesis pathway is production of mevalonate by HMG-CoA reductase (135). Expression levels of HMG-CoA reductase as well as LDLR are negatively controlled by intracellular cholesterol, which as a result, commands a powerful system of self-regulation.
Transcription levels of both LDLR and HMG-CoA reductase genes are controlled by promoters with sterol regulatory elements (SREs), which are present on over 30 genes involved with lipid synthesis and uptake (136). Experiments using chimeric LDLR promoter and reporter gene constructs have localized the area of sterol responsiveness to 8bp of palindromic sequence, (5'-CACCCCAC-3') (137). The SRE in the HMG-CoA reductase promoter is homologous to LDLR SRE, with a substitution of a guanine for the central cytosine on the 3' side (138). In both promoters, attachment of transcription factors called SRE binding proteins (SREBPs) is required for efficient production of downstream transcription. There are three types of SREBP, (SREBP1a, SREBP1c, and SREBP2), which are encoded by two genes. Of the three isoforms, SREBP1a and SREBP2 are more important in cholesterol homeostasis than SREBP1c, which mainly alters expression of fatty acid synthesis genes (139). Despite discrepancies in their transcriptional targets, proteolytic activation of each SREBP isoform is regulated by cholesterol through a common mechanism. Complete disruption of SREBP activation in mice hepatocytes results in 75% decrease in sterol and fatty acid synthesis, 50% reduction in LDLR mRNA and LDL clearance, and a significant reduction in total plasma cholesterol (140).
The molecular basis for cholesterol sensitivity of SREBP has been elucidated greatly due to the efforts made by the Brown and Goldstein Lab of the University of Texas Southwestern Medical Center. Each newly translated SREBP is inserted into the ER membrane, where its C-terminal regulatory domain binds to the C-terminal of the SREBP cleavage activating protein (SCAP) (139). While associated with SREBP, SCAP binds to another ER bound protein known as insulin induced gene 1 (INSIG) (141). Interaction between SCAP and INSIG keeps the protein complex firmly in place in the ER. Importantly, this interaction between SCAP and INSIG is cholesterol-sensitive, and thus, acts as the cholesterol sensor of this system (142). In cholesterol abundant conditions, membrane-spanning domains of SCAP bind to cholesterol, causing a conformational change in the cytosolic domain between domains 7 and 8, resulting in strong affinity for INSIG (142). The cholesterol-absent conformation of SCAP is not associated with INSIG, leaving the SREBP/SCAP complex free to leave the ER in COPII vesicles, which then migrate to the Golgi (143). Once there, the N-terminal transcription factor domain of SREBP is cleaved in a two-step process involving proteolytic enzymes site-1 and site-2 proteases (S1P and S2P, respectively) (144). The nuclear form of SREBP migrates into the nucleus and activates genes including HMG CoA reductase and LDLR, ultimately leading to increased cholesterol concentration in the cell.
Mutations in genes involved in the cholesterol biofeedback pathway produce powerful effects on cholesterol homeostasis, especially those in genes that encode cholesterol biosynthetic enzymes. Inactivating mutations in the cholesterol biosynthetic pathway cause accumulation of cholesterol precursors, which manifest other serious health defects besides dyslipidemia. A table of several monogenic diseases in this pathway is included (Table 1).
Monogenic disorders of the cholesterol biosynthesis pathways and the loci that harbor the causative mutations.
| Disorder | Locus | Ref. |
|---|---|---|
| HMG-CoA synthase deficiency | HMG-CoA synthase | (145) |
| Mevalonic aciduria | Mevalonate kinase | (146) |
| Hyperimmunoglobulinemia D syndrome | Mevalonate kinase | (146) |
| Desmosterolosis | 24-dehydrocholesterol reductase | (147) |
| CHILD syndrome | NAD(P) dependent steroid dehydrogenase-like | (148) |
| Conradi-Hunermann syndrome | Emopamil binding protein | (149) |
| Lathosterolosis | Sterol-C5-desaturase | (150) |
| Smith-Lemli-Opitz syndrome | 7-dehydrocholesterol reductase | (151) |
Summary
Cells collect cholesterol molecules through de novo synthesis, or receptor mediated endocytosis of exogenous and endogenous cholesterol packaged in LDL. Extracellular cholesterol is initially packaged into triglyceride rich particles, which are hydrolyzed in circulation to move fatty acids into cells expressing LPL. As lipoproteins undergo triglyceride hydrolysis, they become increasingly dense and cholesterol-laden. Cholesteryl ester-rich lipoproteins interact with the LDLR, which is expressed selectively by the liver, and by cells low in intercellular cholesterol. Meanwhile, the liver is consistently manufacturing HDL particles, which have the critical task of removing excess cholesterol from LDL particles, and the minute spaces that they might accumulate. Together, these systems interact, sharing various proteins and lipids in order to maintain the balance of the famously deadly but undeniably vital cyclic lipid, cholesterol.
The appeal of HDL lies in its ability to undo cholesterol accumulation in tissues. Conversely, LDL demands our attention so that we may comprehend the mechanisms that lead to heart disease. Unfortunately, the extremely polygenic and complex mechanisms that influence cholesterol homeostasis confound experimental design so that only narrow hypotheses may be investigated. However, the research community has established tremendous worth to deciphering these systems. Finally, the advent of new sequencing and analysis technologies assure that these pathways will continue to be investigated until preventative cardiac medicine is a reality.
Acknowledgements
This activity was funded with an Emerging Research Issues Internal Competitive Grant from the Washington State University, College of Agricultural, Human, and Natural Resource Sciences, Agricultural Research Center to Z.J., R.W.W. and K.M.K.
Abbreviations
ABCA1: ATP binding cassette A1; Apo: apolipoprotein; CETP: Cholesteryl ester transfer protein; CR: chylomicron Remnant; HDL: high density lipoproteins; IDL: intermediate density lipoproteins; LCAT: lecithin:cholesterol acyltransferase; LDL: low density lipoproteins; LDLR: low density lipoprotein receptor; LIPC: hepatic lipase; LIPG: endothelial lipase; LPL: lipoprotein lipase; LRP1: low density lipoprotein-related protein 1; MTP: microsomal triglyceride transfer protein; PCSK9: proprotein convertase subtilisin/kexin type 9; PLTP: phospholipid transfer protein; RCT: reverse cholesterol transport; SR-BI: Scavenger receptor class B1; VLDL: very low density lipoproteins.
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
1. Lloyd-Jones D, Adams R, Carnethon M. et al. Heart disease and stroke statistics--2009 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2009;119:480-486
2. Stamler J, Daviglus ML, Garside DB. et al. Relationship of baseline serum cholesterol levels in 3 large cohorts of younger men to long-term coronary, cardiovascular, and all-cause mortality and to longevity. JAMA. 2000;284:311-318
3. Mourão PA, Bracamonte CA. The binding of human aortic glycosaminoglycans and proteoglycans to plasma low density lipoproteins. Atherosclerosis. 1984;50:133-146
4. Wilson PW, Abbott RD, Castelli WP. High density lipoprotein cholesterol and mortality. The Framingham Heart Study. Arteriosclerosis. 1988;8:737-741
5. O'Connell DL, Heller RF, Roberts DC. et al. Twin study of genetic and environmental effects on lipid levels. Genet Epidemiol. 1988;5:323-341
6. Boekholdt SM, Souverein OW, Tanck MT. et al. Common variants of multiple genes that control reverse cholesterol transport together explain only a minor part of the variation of HDL cholesterol levels. Clinical Genetics. 2006;69:270-273
7. Wu Z, Wagner MA, Lemin Z. et al. The refined structure of nascent HDL reveals a key functional domain for particle maturation and dysfunction. Nat Struct Mol Biol. 2007;14:861-868
8. Zannis VI, Chroni A, Krieger M. Role of ApoA1, ABCA1, LCAT, and SR-BI in the Biogenesis of HDL. J Mol Med. 2006;84:276-294
9. Castro GR, Fielding CJ. Early incorporation of cell-derived cholesterol into pre-beta-migrating high density lipoprotein. Biochemistry. 1988;27:25-29
10. Fielding CJ, Shore VG, Fielding PE. A protein cofactor of lecithin:cholesterol acytransferase. Biochem Biophys Res. 1972;46:1493-1498
11. Wang N, Silver DL, Costet P. Specific binding of ApoA1, enhanced cholesterol efflux and altered plasma membrane morphology in cells expressing ABCA1. J Biol Chem. 2000;275:33053-33058
12. Rigotti A, Trigatti B, Babitt J. et al. Scavenger receptor BI - a cell surface receptor for high density lipoprotein. Curr Opin Lipidol. 1997;8:181-188
13. Williamson R, Lee D, Hagaman J, Maeda N. Marked reduction of high density lipoprotein cholesterol in mice genetically modified to lack apolipoprotein A-I. Natl Acad Sci U S A. 1992;89:7134-7138
14. Hovingh GK, de Groot E, van der Steeg W. et al. Inherited disorders of HDL metabolism and atherosclerosis. Curr Opin Lipidol. 2005;16:139-145
15. Franceschini G, Sirtori CR, Capurso A. et al. A-I Milano apoprotein. Decreased high density lipoprotein cholesterol levels with significant lipoprotein modifications and without clinical atherosclerosis in an Italian family. J Clin Invest. 1980;66:892-900
16. Franceschini G, Calabresi L, Chiesa G. et al. Increased cholesterol efflux potential of sera from ApoAI milano carriers and transgenic mice. Arterioscler Thromb Vasc Biol. 1999;19:1257-1262
17. Weibel GL, Alexander ET, Joshi MR. et al. Wild-type ApoA-I and the Milano variant have similar abilities to stimulate cellular lipid mobilization and efflux. Arterioscler Thromb Vasc Biol. 2007;9:2022-2029
18. Schmitz G, Assmann G, Robenek H. et al. Tangiers disease: A disorder of intracellular membrane traffic. Proc Natl Acad Sci U S A. 1985;82:6305-6309
19. Schmitz G, Robenek H, Lohmann U, Assmann G. Interaction of high density lipoproteins with cholesteryl ester-laden macrophages: biochemical and morphological characterization of cell surface receptor binding, endocytosis and resecretion of high density lipoproteins by macrophages. EMBO. 1985;4:613-622
20. Choi HY, Karten B, Chan T. et al. Impaired ABCA1-dependent Lipid Efflux and Hypoalphalipoproteinemia in Human Niemann-Pick type C Disease. J Biol Chem. 2003;278:32569-32577
21. Bodzioch M, Orso E, Klucken J. et al. The gene encoding ATP-binding cassette transporter 1 is mutated in Tangier disease. Nat genet. 1999;22:347-351
22. Cohen JC, Kiss RC, Pertsemlidis A. et al. Multiple rare alleles contribute to low levels of plasma HDL cholesterol. Science. 2004;305:869-872
23. Frikke-Schmidt R, Nordestgaard BG, Jensen GB, Tybjaerg-Hansen A. Genetic variation in ABC transporter A1 contributes to HDL cholesterol in the general population. J Clin Invest. 2004;114:1343-1353
24. Francone OL, Gurakar A, Fielding C. Distribution and functions of lecithin:cholesterol acyltransferase and cholesteryl ester transfer protein in plasma lipoproteins. J Biol Chem. 1989;264:7066-7072
25. Mclean J, Fielding CJ, Drayna D. et al. Cloning and expression of human lecithin-cholesterol acyltransferase cDNA. Proc Natl Acad Sci U S A. 1986;83:2335-2339
26. Jauhiainen M, Dolphin PJ. Human plasma lecithin-cholesterol acyltransferase. An elucidation of the catalytic mechanism. J Biol Chem. 1986;261:7032-7043
27. Norum KR, Glomset JA, Nichols AV, Forte T. Plasma lipoproteins in familial lecithin: cholesterol acyltransferase deficiency: physical and chemical studies of low and high density lipoproteins. J Clin Invest. 1971;50:1131-1140
28. Francone OL, Haghpassand M, Bennett JA. et al. Expression of human lecithin:cholesterol acyltransferase in transgenic mice: effects on cholesterol efflux, esterification, and transport. J Lipid Res. 1997;38:813-822
29. Fielding CJ. The origin and properties of free cholesterol potential gradients in plasma, and their relation to atherogenesis. J Lipid Res. 1984;25:1624-1628
30. Kuivenhoven JA, Pritchard H, Hill J. et al. The molecular pathology of lecithin:cholesterol acyltransferase (LCAT) deficiency syndromes. J Lipid Res. 1997;38:191-205
31. Funke H, von Eckardstein A, Pritchard PH. A molecular defect causing fish eye disease: an amino acid exchange in lecithin-cholesterol acyltransferase (LCAT) leads to the selective loss of alpha-LCAT activity. Proc Natl Acad Sci U S A. 1991;88:4855-4859
32. Wong H, Schotz MC. The Lipase Gene Family. J Lipid Res. 2002;43:993-999
33. McCoy MG, Sun GS, Marchadier D. et al. Characterization of the lipolytic activity of endothelial lipase. J Lipid Res. 2002;43:921-929
34. Sendak RA, Bensadoun A. Identification of a heparin-binding domain in the distal carboxyl- terminal region of lipoprotein lipase by site-directed mutagenesis. J Lipid Res. 1998;39:1310-1315
35. Sendak RA, Berryman DE, Gellman G. et al. Binding of hepatic lipase to heparin: Identification of specific heparin-binding residues in two distinct positive charge clusters. J. Lipid Res. 2000;41:260-268
36. Yancey PG, Kawashiri MA, Moore R. et al. In vivo modulation of HDL phospholipid has opposing effects on SR-BI- and ABCA1-mediated cholesterol efflux. J Lipid Res. 2004;45:337-346
37. Badellino KO, Wolfe ML, Reilly MP. et al. Endothelial lipase concentrations are increased in metabolic syndrome and associated with coronary atherosclerosis. PLoS Med. 2006:e22
38. Rea TJ, DeMattos RB, Pape ME. Hepatic expression of genes regulating lipid metabolism in rabbits. J Lipid Res. 1993;34:1901-1910
39. Grosser J, Schrecker O, Greten H. Function of hepatic triglyceride lipase in lipoprotein metabolism. J Lipid Res. 1981;22:437-442
40. Kuusi T, Saarinen P, Nikkilä EA. Evidence for the role of hepatic endothelial lipase in the metabolism of plasma high density lipoprotein2 in man. Atherosclerosis. 1980;36:589-593
41. Catalano G, Duchene E, Zélie J. et al. Cellular SR-BI and ABCA1-mediated cholesterol efflux are gender-specific in healthy subjects. J Lipid Res. 2008;49:635-643
42. Gonzalez-Navarro H, Nong Z, Freeman L. et al. Identification of mouse and human macrophages as a site of synthesis of hepatic lipase. J Lipid Res. 2002;43:671-675
43. Whiting BM, Anderson JL, Muhlestein JB. Candidate gene susceptibility variants predict intermediate end points but not angiographic coronary artery disease. Am Heart J. 2005;150:243-250
44. Eller P, Schgoer W, Mueller T. et al. Hepatic lipase polymorphism and increased risk of peripheral arterial disease. J Intern Med. 2005;258:34434-8
45. Goldberg IJ. Lipoprotein lipase and lipolysis: central roles in lipoprotein metabolism and atherogenesis. J Lipid Res. 1996;37:693-707
46. Zilversmitz DB. Lipase with Triglyceride-Rich Lipoproteins: A Proposal Linking Atherogenesis to the Interaction of Endothelial Lipoprotein. Circ Res. 1973;33:633-638
47. Pattnaik NM, Montes A, Hughes LB, Zilversmit DB. Cholesteryl ester exchange protein in human plasma isolation and characterization. Biochim Biophys Acta. 1978;530:428-438
48. Quinet E, Tall A, Ramakrishnan R, Rudel L. Plasma lipid transfer protein as a determinant of the atherogenicity of monkey plasma lipoproteins. J Clin Invest. 1991;87:1559-1566
49. Marotti KR, Castle CK, Boyle TP. et al. Severe atherosclerosis in transgenic mice expressing simian cholesteryl ester transfer protein. Nature. 1993;364:73-77
50. Hayek T, Masucci-Magoulas L, Jiang X. et al. Decreased early atherosclerotic lesions in hypertriglyceridemic mice expressing cholesteryl ester transfer protein transgene. J Clin Invest. 1995;96:2071-2074
51. Barzilai N, Atzmon G, Schechter C. et al. Unique lipoprotein phenotype and genotype associated with exceptional longevity. JAMA. 2003;290:2030-2040
52. Inazu A, Jiang XC, Haraki T. Genetic cholesteryl ester transfer protein deficiency caused by two prevalent mutations as a major determinant of increased levels of high density lipoprotein cholesterol. J Clin Invest. 1994;94:1872-1882
53. Ken-ichi H, Shizuya Y, Norimichi N. et al. Genetic Cholesteryl Ester Transfer Protein Deficiency Is Extremely Frequent in the Omagari Area of Japan. Arterioscler Thromb Vasc Biol. 1997;17:1053-1059
54. Zhong S, Sharp DS, Grove JS. et al. Increased coronary heart disease in Japanese-American men with mutation in the cholesterol ester trandfer protein despite increased HDL. J Clin Invest. 1996;97:2917-2923
55. Corella D, Saiz C, Guillen M. et al. Association of TaqIB polymorphism in the cholesteryl ester transfer protein gene with plasma lipid levels in a healthy Spanish population. Atherosclerosis. 2000;152:367-376
56. Ordovas JM, Cupples A, Corella D. et al. Association of cholesteryl ester transfer protein-TaqIB polymorphism with variations in lipoproteins subclasses and coronary heart disease risk. Thromb Vasc Biol. 2000;20:1323-1329
57. Dusserre E, Moulin P, Vidal H. Differences in mRNA expression of the proteins secreted by the adipocytes in human subcutaneous and visceral adipose tissues. Biochim Biophys Acta. 2000;1500:88-96
58. Jauhiainen M, Metso J, Pahlman R. et al. Human plasma phospholipid transfer protein causes high density lipoprotein conversion. J Biol Chem. 1993;268:4032-4036
59. Settasatian N, Duong M, Curtiss LK. et al. The mechanism of the remodeling of high density lipoproteins by phospholipid transfer protein. J Biol Chem. 2001;276:26898-26905
60. Rao R, Albers JJ, Wolfbauer G, Pownall HJ. Molecular and macromolecular specificity of human plasma phospholipid transfer protein. Biochemistry. 1997;36:3645-3653
61. Nishida HI, Nishida T. Phospholipid transfer protein mediates transfer of not only phosphatidylcholine but also cholesterol from phosphatidylcholine-cholesterol vesicles to high density lipoproteins. J Biol Chem. 1997;272:6959-6964
62. Qin S, Kawano K, Bruce C. et al. Phospholipid transfer protein gene knock-out mice have low high density lipoprotein levels, due to hypercatabolism, and accumulate apoA-IV -rich lamellar lipoproteins. J Lipid Res. 2000;41:269-276
63. Föger B, Santamarina-Fojo S, Shamburek RD. et al. Plasma phospholipid transfer protein. Adenovirus-mediated overexpression in mice leads to decreased plasma high density lipoprotein (HDL) and enhanced hepatic uptake of phospholipids and cholesteryl esters from HDL. J Biol Chem. 1997;272:27393-27400
64. Samyn H, Moerland M, van Gent T. et al. Elevation of systemic PLTP, but not macrophage-PLTP, impairs macrophage reverse cholesterol transport in transgenic mice. Atherosclerosis. 2009;204:429-434
65. Acton SL, Scherer PE, Lodish HF. et al. Expression cloning of SR-BI, a CD36-related class B scavenger receptor. J Biol Chem. 1994;269:21003-21009
66. Rigotti A, Trigatti BL, Penman M. et al. A targeted mutation in the murine gene encoding the high density lipoprotein (HDL) receptor scavenger receptor class B type I reveals its key role in HDL metabolism. Proc Natl Acad Sci U S A. 1997;94:12610-12615
67. Out R, Hoekstra M, Spijkers JA. Scavenger receptor class B type I is solely responsible for the selective uptake of cholesteryl esters from HDL by the liver and the adrenals in mice. J Lipid Res. 2004;45:2088-2095
68. Acton S, Rigotti A, Landschulz KT. et al. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science. 1996;271:518-520
69. Liadaki KN, Liu T, Xu S. et al. Binding of High Density Lipoprotein (HDL) and Discoidal Reconstituted HDL to the HDL Receptor Scavenger Receptor Class B Type I. J Biol Chem. 2000;275:21262-21271
70. Xu S, Laccotripe M, Huan X. et al. Apolipoproteins of HDL can directly mediate binding to the scavenger receptor SR-BI, an HDL receptor that mediates selective lipid uptake. J Lipid Res. 1997;38:1289-1298
71. Homer VM, Marais AD, Charlton F. et al. Identification and characterization of two non-secreted PCSK9 mutants associated with familial hypercholesterolemia in cohorts from New Zealand and South Africa. Atherosclerosis. 2008;196:659-666
72. Lagace TA, Curtis DE, Garuti R. et al. Secreted PCSK9 decreases the number of LDL receptors in hepatocytes and in livers of parabiotic mice. J Clin Invest. 2006;116:2995-3005
73. Homer VM, Marais AD, Charlton F. et al. Identification and characterization of two non-secreted PCSK9 mutants associated with familial hypercholesterolemia in cohorts from New Zealand and South Africa. Atherosclerosis. 2008;196:659-666
74. Geoghegan K, Boyd J, Hoth L. et al. Binding to Low-Density Lipoprotein Receptor Accelerates Futile Catalytic Cycling in PCSK9 and Raises the Equilibrium Level of Intramolecular Acylenzyme. Biochemistry. 2009 [Epub ahead of print]
75. Zhang DW, Lagace TA, Garuti R. et al. Binding of proprotein convertase subtilisin/kexin type 9 to epidermal growth factor-like repeat A of low density lipoprotein receptor decreases receptor recycling and increases degradation. J Biol Chem. 2007;282:18602-18612
76. Cohen J, Pertsemlidis A, Kotowski IK. et al. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat Genet. 2005;37:161-165
77. Miyake Y, Kimura R, Kokubo Y. et al. Genetic variants in PCSK9 in the Japanese population: rare genetic variants in PCSK9 might collectively contribute to plasma LDL cholesterol levels in the general population. Atherosclerosis. 2008;196:29-36
78. Fasano T, Cefalù AB, Di Leo E. et al. A novel loss of function mutation of PCSK9 gene in white subjects with low-plasma low-density lipoprotein cholesterol. Arterioscler Thromb Vasc Biol. 2007;27:677-681
79. de la Llera-Moya M, Rothblat GH, Connelly MA. et al. Scavenger receptor BI (SR-BI) mediates free cholesterol flux independently of HDL tethering to the cell surface. J Lipid Res. 1999;40:575-580
80. Russell DW, Setchell KD. Bile acid biosynthesis. Biochemistry. 1992;31:4737-4749
81. Wang H, Chen J, Hollister K. et al. Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol Cell. 1999;3:543-553
82. Goodwin B, Jones SA, Price RR. et al. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol Cell. 2000;6:517-526
83. Pullinger CR, Eng C, Salen G. et al. Human cholesterol 7alpha-hydroxylase (CYP7A1) deficiency has a hypercholesterolemic phenotype. J Clin Invest. 2002;110:109-117
84. Russell DW. Fifty years of advances in bile acid synthesis and metabolism. J Lipid Res. 2009;50(Suppl):S120-125
85. Yusuf S, Reddy S, Ounpuu S. et al. Global burden of cardiovascular diseases part I: general considerations, te epidemiologic transition, risk factors, and impact of urbanization. Circulation. 2001;104:2746-2753
86. National Cholesterol Education Program (NCEP). Executive Summary of The Third Report of The NCEP Expert Panel on Detection, Evaluation, And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III). JAMA. 2001;285:2486-2497
87. Koivisto UM, Hamalainen L, Taskinen MR. et al. Prevalence of familial hypercholesterolemia among young North Karelian patients with coronary heart disease: a study based on diagnosis by polymerase chain reaction. J Lipid Res. 1993;34:269-277
88. Gylling HM, Mietinnen TA. The effect of cholesterol absorption inhibition on low density lipoprotein cholesterol level. Atherosclerosis. 1995;117:305-308
89. Ostland RE, Bosner MS, Stenson WF. Cholesterol absorption efficiency declines at moderate dietary doses in normal human subjects. J Lipid Res. 1999;40:1453-1458
90. Gylling HM, Miettinen TA. Inheritance of cholesterol metabolism of probands with high or low cholesterol absorption. J Lipid Res. 2002;43:1472-1476
91. Yao L, Heubi JE, Buckley DD. et al. Separation of micelles and vesicles within luminal aspirates from healthy humans: solubilization of cholesterol after a meal. J Lipid Res. 2002;43:654-660
92. Ponz de Leon M, Loria P, Iori R. et al. Cholesterol absorption in cirrhosis: the role of total and individual bile acid pool size. Gastroenterology. 1981;80:1428-1437
93. Young SC, Hui DY. Pancreatic lipase-colipase mediated triglyceride hydrolysis is required for cholesterol transport from lipid emulsions to intestinal cells. Biochem J. 1999;339:615-620
94. Lowe ME. Pancreatic triglyceride lipase and colipase: insights into dietary fat digestion. Gasteroenterology. 1994;107:1524-1536
95. Howles PN, Carter CP, Hui DY. et al. Dietary free and esterified cholesterol absorption in cholesterol esterase (bile salt-stimulated lipase) gene-targeted mice. J Biol Chem. 1996;271:7196-7202
96. van Heck M, Farley C, Compton DS. et al. Ezetimibe inhibits cholesterol absorption but does not affect acute hepatic or intestinal cholesterol synthesis in rats. Br J Pharmacol. 2003;138:1459-1464
97. Garcia-Calvo M, Lisnock J, Bull HG. et al. The target of ezetimibe is Niemann-Pick C1-Like 1 (NPC1L1). Proc Natl Acad Sci U S A. 2005;102:8132-8137
98. Altmann SW, Davis HR, Zhu LJ. et al. Niemann-Pick C1 like 1 protein is critical for intestinal cholesterol absorption. Science. 2004;303:1201-1214
99. Cohen JC, Pertsemlidis A, Fahmi S. et al. Multiple rare variants in NPC1L1 associated with reduced sterol absorption and plasma low-density lipoprotein levels. Proc Natl Acad Sci U S A. 2006;103:1810-1815
100. Berge KE, Tian H, Grad GA. et al. Accumulation of dietary cholesterol in sitosterolemia caused by mutations in adjacent ABC transporters. Science. 2000;290:1771-1775
101. Hui DY, Howles PN. Molecular mechanisms of cholesterol absorption and transport in the intestine. Semin Cell Dev Biol. 2005;16:183-1892
102. Purdy BH, Field FJ. Regulation of acylcoenzyme A. Cholesterol acyltransferase and 3-hydroxy-3-methylglutaryl coenzyme A reductase activity by lipoproteins in the intestine of parabiont rats. J Clin Invest. 1984;74:351-357
103. Gordon DA, Wetterau JR, Gregg RE. Microsomal triglyceride transfer protein: a protein complex required for the assembly of lipoprotein particles. Trends Cell Biology. 1995;5:317-321
104. Lackner KJ, Monge JC, Gregg RE. et al. Analysis of the apolipoprotein B gene and messenger ribonucleic acid in abetalipoproteinemia. J Clin Invest. 1986;78:1707-1712
105. Sabesin SM, Frase S. Electron microscopic studies of the assembly, intracellular transport, and secretion of chylomicrons by rat intestine. J lipid res. 1977;18:456-511
106. Tennyson GE, Sabatos CA, Higuchi K. et al. Expression of apolipoprotein B mRNAs encoding higher- and lower-molecular weight isoproteins in rat liver and intestine. Proc Natl Acad Sci U S A. 1989;86:500-504
107. Nakamuta M, Chang BH, Zsigmond E. et al. Complete phenotypic characterization of apobec-1 knockout mice with a wild-type genetic background and a human apolipoprotein B transgenic background, and restoration of apolipoprotein B mRNA editing by somatic gene transfer of Apobec-1. J Biol Chem. 1996;271:25981-25988
108. Benn M. Apolipoprotein B levels, APOB alleles, and risk of ischemic cardiovascular disease in the general population, a review. Atherosclerosis. 2009 [Epub ahead of print]
109. Benn M, Nordestgaard BG, Jensen JS. et al. Polymorphism in APOB associated with increased low-density lipoprotein levels in both genders in the general population. J Clin Endocrinol Metab. 2005;90:5797-5803
110. Benn M, Nordestgaard BG, Jensen JS, Tybjærg-Hansen A. Polymorphisms in apolipoprotein B and risk of ischemic stroke. J Clin Endocrinol Metab. 2007;92:3611-3617
111. Talmud PJ, Hawe E, Miller GJ, Humphries SE. Nonfasting apolipoprotein B and triglyceride levels as a useful predictor of coronary heart disease risk in middle-aged UK men. Arterioscler Thromb Vasc Biol. 2002;22:1918-1923
112. Grundy SM, Mok HY. Chylomicron clearance in normal and hyperlipidemic man. Metabolism. 1976;25:1225-1239
113. Takagi A, Ikeda Y, Takeda E, Yamamoto A. A newly identified lipoprotein lipase (LPL) gene mutation (F270L) in a Japanese patient with familial LPL deficiency. Biochim Biophys Acta. 2000;1502:433-434
114. Zhang SH, Reddick RL, Piedrahita JA, Maeda N. Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science. 1992;258:468-471
115. Herz J, Qiu SQ, Oesterle A. et al. Initial hepatic removal of chylomicron remnants is unaffected but endocytosis is delayed in mice lacking the low density lipoprotein receptor. Proc Natl Acad Sci U S A. 1995;92:4611-4615
116. Redgrave TG, Vassiliou GG, Callow MJ. Cholesterol is necessary for triacylglycerol-phospholipid emulsions to mimic the metabolism of lipoproteins. Biochim Biophys Acta. 1987;921:154-159
117. Hebbachi AM, Gibbons GF. Microsomal membrane-associated apoB is the direct precursor of secreted VLDL in primary cultures of rat hepatocytes. J Lipid Res. 2001;42:1609-1617
118. Rustaeus S, Lindburg K, Borén J, Olofsson SO. Brefeldin A Reversibly Inhibits the Assembly of ApoB Containing Lipoproteins in McA-RH7777 Cells. J Biol Chem. 1995;48:28879-28886
119. Gibbons GF, Islam K, Pease RJ. Mobilization of triacylglycerol stores. Biochim Biophys Acta. 2000;1483:37-57
120. Kobayashi K, Oka K, Forte T. et al. Reversal of Hypercholesterolemia in Low Density Lipoprotein Receptor Knockout Mice by Adenovirus-mediated Gene Transfer of the Very Low Density Lipoprotein Receptor. J Biol Chem. 1996;271:6852-6860
121. Ginsberg HN. Lipoprotein metabolism and its relationship to atherosclerosis. Med Clin North Am. 1994;78:1-20
122. Langer T, Strober W, Levy RI. The Metabolism of Low Density Lipoprotein in Familial Type II Hyperlipoproteinemia. J Clin Invest. 1972;51:1528-1536
123. Mahley RW. Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science. 1988;240:622-630
124. Kim YS, Paeng JR, Woo JT. et al. Apolipoprotein E genotypes of normal and hyperlipidemic patients. Kor Med Sci. 1993;8:262-266
125. Mahley RW, Rall SC. Apolipoprotein E: far more than a lipid transport protein. Annu Rev Genomics Hum Genet. 2000;1:507-537
126. Wu K, Bowman R, Welch AA. et al. Apolipoprotein E polymorphisms, dietary fat and fibre, and serum lipids: the EPIC Norfolk study. Eur Heart J. 2007;28:2930-2936
127. Jeon H, Blacklow SC. Structure and physiologic function of the low-density lipoprotein receptor. Annu Rev Biochem. 2005;74:535-562
128. Russell DW, Brown MS, Goldstein JL. Different combinations of cysteine-rich repeats mediate binding of low density lipoprotein receptor to two different proteins. J Biol Chem. 1989;264:21682-21688
129. Yang CY, Chen SH, Gianturco SH. et al. Sequence, structure, receptor-binding domains and internal repeats of human apolipoprotein B-100. Nature. 1986;323:738-742
130. Milne R, Threolis R, Maurice R. et al. The use of monoclonal antibodies to localize the low density lipoprotein receptor-binding domain of apolipoprotein B. J Biol Chem. 1989;264:19754-19760
131. Anderson RG, Vasile E, Mello RJ. et al. Immunocytochemical visualization of coated pits and vesicles in human fibroblasts: relation to low density lipoprotein receptor distribution. Cell. 1978;15:919-933
132. Davis CG, Goldstein JL, Südhof TC. et al. Acid-dependent ligand dissociation and recycling of LDL receptor mediated by growth factor homology region. Nature. 1987;326:760-765
133. Hooper AJ, Tran HA, Formby MR, Burnett JR. A novel missense LIPA gene mutation, N98S, in a patient with cholesteryl ester storage disease. Clin Chim Acta. 2008;398:152-154
134. Liscum L, Underwood KW. Intracellular cholesterol transport and compartmentation. J Biol Chem. 1995;270:15443-15446
135. Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990;343:425-430
136. Horton JD, Shah NA, Warrington JA. et al. Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proc Natl Acad Sci U S A. 2003;100:12027-12032
137. Smith JR, Osborne TF, Goldstein JL, Brown MS. Identification of nucleotides responsible for enhancer activity of sterol regulatory element in low density lipoprotein receptor gene. J Biol Chem. 1990;265:2306-2310
138. Osborne TF, Gil G, Goldstein JL, Brown MS. Operator constitutive mutation of 3-hydroxy-3-methylglutaryl coenzyme A reductase promoter abolishes protein binding to sterol regulatory element. J Biol Chem. 1988;263:3380-3387
139. Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109:1125-1131
140. Yang J, Goldstein JL, Hammer RE. et al. Decreased lipid synthesis in livers of mice with disrupted Site-1 protease gene. Proc Natl Acad Sci U S A. 2001;98:13607-13612
141. Yang T, Espenshade PJ, Wright ME. et al. Crucial steps in cholesterol homeostasis: Sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell. 2002;110:489-500
142. Brown AJ, Sun L, Feramisco JD. et al. Cholesterol Addition to ER Membranes Alters Conformation of SCAP, the SREBP Escort Protein that Regulates Cholesterol Metabolism. Mol Cell. 2002;10:237-245
143. Goldstein JL, DeBose-Boyd RA, Brown MS. Protein sensors for membrane sterols. Cell. 2006;124:35-46
144. Nohturfft A, Tabe D, Goldstein JL. et al. Regulated step in cholesterol feedback localized to budding of SCAP from ER membranes. Cell. 2000;102:315-323
145. Zschocke J, Penzien JM, Bielen R. et al. The diagnosis of mitochondrial HMG-CoA synthase deficiency. J Pediatr. 2002;140:78-80
146. Haas D, Hoffmann GF. Mevalonate kinase deficiencies: from mevalonic aciduria to hyperimmunoglobulinemia D syndrome. Orphanet J Rare Dis. 2006;1:13
147. Waterham HR, Koster J, Romeijn GJ. et al. Mutations in the 3beta-hydroxysterol Delta24-reductase gene cause desmosterolosis, an autosomal recessive disorder of cholesterol biosynthesis. Am J Hum Genet. 2001;69:685-694
148. Hummel M, Cunningham D, Mullett CJ. Left-sided CHILD syndrome caused by a nonsense mutation in the NSDHL gene. Am J Med Genet A. 2003;122A:246-251
149. Braverman N, Lin P, Moebius FF. et al. Mutations in the gene encoding 3 beta-hydroxysteroid-delta 8, delta 7-isomerase cause X-linked dominant Conradi-Hünermann syndrome. Nat Genet. 1999;22:291-294
150. Krakowiak PA, Wassif CA, Kratz L. Lathosterolosis: an inborn error of human and murine cholesterol synthesis due to lathosterol 5-desaturase deficiency. Hum Mol Genet. 2003;12:1631-141
151. Jezela-Stanek A, Ciara E, Malunowicz EM. Mild Smith-Lemli-Opitz syndrome: further delineation of 5 Polish cases and review of the literature. Eur J Med Genet. 2008;51:124-140
Author contact
![]() Correspondence to: Zhihua Jiang, Tel: +509 335 8761; Fax: +509 335 4246; E-mail: jiangzedu
Correspondence to: Zhihua Jiang, Tel: +509 335 8761; Fax: +509 335 4246; E-mail: jiangzedu