Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. Materials and Methods
3. Results
4. Discussion
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2009; 5(7):637-646. doi:10.7150/ijbs.5.637 This issue Cite
Research Paper
An apoA-I mimetic peptide facilitates off-loading cholesterol from HDL to liver cells through scavenger receptor BI
Xuelei Song ![]() , Paul Fischer, Xun Chen, Charlotte Burton, Jun Wang
, Paul Fischer, Xun Chen, Charlotte Burton, Jun Wang ![]()
Merck Research Laboratories, Rahway, New Jersey 07065, USA
Received 2009-8-22; Accepted 2009-10-5; Published 2009-10-9
Abstract
Apolipoprotein A-I (apoA-I) mimetic peptides have been pursued as new therapeutic agents for the treatment of atherosclerosis, yet their precise mechanism responsible for atheroprotection remains unclear. Like apoA-I itself, most of these peptides are capable of stimulating cholesterol efflux from macrophages or foam cells, and some of them stimulate lecithin cholesterol acyltransferase (LCAT) activity in the reverse cholesterol transport (RCT) pathway. However, the ability of mimetic peptides to deliver cholesterol into hepatocytes (off-loading), the last step of the RCT pathway, has not been demonstrated. In this study, we compared a mimetic peptide D-4F to purified apoA-I, to address the role that mimetics play during the off-loading process. Both D-4F and apoA-I formed spherical nano-particles when reconstituted with cholesteryl ester and phospholipids. Compared to apoA-I, D-4F particles were 20 times more efficient in off-loading cholesterol to HepG2 hepatocytes with an apparent Kt (transport) of 0.74 μg/mL. Furthermore, D-4F also facilitated cholesteryl ester offloading from HDL particles into HepG2 cells when it was pre-incubated with these HDL particles. Using an inducible HEK293 cell line, we demonstrated that these nano-particles were able to be taken up through SR-BI, a HDL selective receptor. Cholesterol uptake by HepG2 cells was completely blocked by a neutralizing monoclonal antibody against SR-BI, demonstrating that D-4F particles, similar to HDL, specifically off-loaded cholesterol through SR-BI. Overall our data provides evidence that D-4F is capable of mimicking apoA-I to form HDL-like particles, and off-loads cholesterol for catabolism and excretion, thus completing RCT.
Keywords: HDL, SR-BI, apolipoprotein, RCT, D-4F, mimetics
1. Introduction
Although the precise mechanism by which high-density lipoprotein (HDL) exerts its athero-protective effects remains to be determined, the inverse correlation of HDL cholesterol (HDL-C) levels and cardiovascular risk has been demonstrated in many clinical studies [1]. Recently, torcetrapib, a cholesteryl ester transfer protein (CETP) inhibitor, failed in the Phase III clinical study [2], in which patients receiving torcetrapib showed no significant atherosclerotic lesion change despite substantially higher levels of large size HDL [3]. Since then, the research focus has been dramatically shifted from HDL concentration to HDL compositions and functions that include reverse cholesterol transport (RCT), anti-inflammation, anti-oxidation, anti-thrombosis and vessel relaxation [4-7]. HDL is still believed to play important roles in atherosclerosis protection. However, because of the lesson learned from torcetrapib, a detailed understanding of HDL function and its metabolism becomes essential for the development of new HDL therapies against atherosclerosis.
Numerous studies have demonstrated that apoA-I protein plays an important role in atheroprotection. Overexpression of apoA-I decreased atherosclerosis in animal studies [8,9]. Infusion of complexes of phospholipids and apoA-I or its genetic variant (R173C), apoA-IMilano, reduced the atherosclerotic lesion size in animals and humans [10-13]. The beneficial effects of apoA-I have been demonstrated in all steps of RCT, the process by which excess cholesterol is transported from peripheral tissues, including atherogenic foam cells or macrophages in plaques, to the liver [14]: apoA-I interacts with phospholipids to form pre-beta HDL; it initiates cholesterol efflux through the ATP-binding cassette transporter A1 (ABCA1) to remove excess cholesterol from peripheral cells; it activates lecithin cholesterol acyltransferase (LCAT) to convert cholesterol to cholesteryl ester (CE) to form mature HDL; and it interacts with SR-BI to facilitate the delivery of cholesterol from mature HDL into hepatocytes [15-17].
While there is a clear benefit in apoA-I infusion therapy, the monetary cost of apoA-I therapy would be immense. A search for cheaper alternatives to apoA-I protein yielded a number of apoA-I mimetic peptides [18]. These 18-22 amino acid amphipathic peptides mimic apoA-I's ability to reduce atherosclerotic lesions [19]. They are structurally diverse, allowing the selection of peptides with greater potency and better pharmacokinetic profiles than apoA-I itself. One of these peptides, D-4F, is an 18-amino acid peptide with a helical structure similar to the consensus repeats of the apoA-I protein [20]. The primary structure of D-4F consists of D-amino acids only, thus is resistant to mammalian gastrointestinal enzymes with a good potential for oral administration. D-4F inhibited atherosclerosis in mouse and improved anti-inflammatory function of HDL in mouse and several other species [21]. Like apoA-I itself, exactly how D-4F exerts its atheroprotective effect remains unclear. Both apoA-I and D-4F promoted ABCA1-dependent cholesterol efflux from macrophage, the first step of RCT process. But unlike apoA-I, D-4F did not activate LCAT in vitro [22]. There are no data available to show whether D-4F itself is able to deliver cholesterol to the liver or has any impact on this last step of RCT. In this paper, we demonstrate that D-4F peptide is able to form HDL-size particles. Similar to HDL, these particles are capable of delivering CE into HepG2 hepatocytes specifically through SR-BI. In addition, D-4F facilitates cholesteryl ester offloading from HDL particles into HepG2 cells when pre-incubated with HDL particles.
2. Materials and Methods
2.1. Reagents
All chemical reagents and all cell culture reagents were obtained from Sigma and Invitrogen respectively unless otherwise indicated. Native HDL and DiO-HDL (3, 3'-dioctadecyloxacarbocyanine) were purchased from Biomedical Technologies.
2.2. Generation of HEK293 cells expressing SR-BI regulated by Tet-on inducible system
A DNA fragment corresponding to the coding region of human SR-BI was subcloned into pTRE-tight plasmid (Clontech), at NotI sites, under the control of tetracycline regulated-promoter. Positive E. coli clones with the correct orientation were confirmed by restriction digestion and DNA sequencing. After amplification, pTRE-tight[SR-BI] plasmid was linearized and transfected into the HEK293 Tet-on Advanced Cell Line (Clontech) using Lipofectamine Transfection Kit (Invitrogen). This commercially available cell line stably expresses the optimized Tet-On Advanced reverse transactivator protein (rtTA-Advanced) that can strongly and rapidly activate transcription from a tetracycline-responsive promoter (TRE) in the presence of Doxycycline (Dox, a tetracycline derivative). Stable transfected clones were selected in the presence of G418 (100 μg/mL) and hygromycin (20 μg/mL) to create a stable cell line containing the entire inducible system. Individual clones started to appear after 7-10 days. These individual clones were then isolated and expanded in triplicate plates. One plate was frozen down, while the remaining two plates were grown in the absence and presence of 2 μg/mL of Doxycycline. The positive clones were confirmed by western blot analysis. The resulting cell line is named HEK293[TREtightSR-BI].
2.3. Western blot analysis
After 48 hour incubation with different concentrations of Doxycycline, the HEK293[TREtightSR-BI] cells were lysed in RIPA buffer (Sigma). The protein concentration was determined using BCA kit (Pierce). After adding 2x sample buffer (Invitrogen), 20 μg of cell lysate was boiled, and the lysate was cleared by centrifugation. The supernatant was loaded onto 10% SDS-PAGE gels. Western blot analysis was performed with polyclonal anti-SR-BI antibody (Novus Biologicals, NB400-113). Horseradish peroxidase-conjugated secondary antibody and the enhanced chemiluminescent substrate system (Amersham) were used as a detection system. The signal was visualized by X-ray film (Roche Diagnostic).
2.4. Generation of fluorescent (Bodipy-CE labeled) particles
Reconstituted HDL and synthetic peptide particles were prepared according to Hirz and Scanu [23] with minor modifications. Briefly, 1 mg 2-Oleoyl-1-palmitoyl-sn-glycero-3-phosphocholine (POPC), 0.5 mg triolein, 0.12 mg Bodipy®-CE, and 0.09 mg dabcyl dicetyl amide quencher were combined from solvent stocks in a 2 mL polypropylene screw cap tube. These components were then co-evaporated under a gentle argon stream, resuspended in 1 mL ethanol and dried again under argon to remove any traces of solvent, then thoroughly dried by a brief lyophilization. One mg of apoHDL (delipidated HDL), apoA-I (Biodesign) or D-4F in one mL buffer A (50 mM Tris pH 8.6, 150 mM NaCl, 1 mM EDTA) was added to the dried components and resuspended by vortexing to form a cloudy suspension. The sample was then subjected to two rounds of probe sonication (5 X 1 min) using a Misonix Sonicator 3000 with a microtip set at power level 6.5 in order to induce particle formation. During this procedure, the solution cleared noticeably. The co-sonicate was then fractionated by equilibrium density ultracentrifugation to isolate particles in the density range of 1.063 - 1.21 g/mL. In some of the experiments, samples were then passed through a G-50 column and dialyzed extensively against PBS buffer in the presence of 1 mM EDTA. Particle concentration was expressed in terms of protein concentration as determined by BCA assay (Pierce). Non-fluorescent particles were generated the same way as the fluorescent ones, except the Bodipy-CE is replaced with regular CE. The synthetic HDL particles were stable at 4°C for several months.
2.5. Determination of particle size by native polyacrylamide gel eletrophoresis
Twenty microliters of native HDL, reconstituted HDL and synthetic particles were mixed with 2X native loading dye (Invitrogen). Samples were loaded onto 4-20% gradient Tris-Glycine gel without boiling. The gel was run at 125V for 2.5 hours before stained by the SimplyBlue SafeStain from Invitrogen. The sizes of particles were compared with the High Molecular Weight marker from GE.
2.6. Off-loading Assay
HepG2 and HEK293[TREtightSR-BI] cell lines were maintained in a humidified 37 ºC incubator with 5% CO2. On day 0, 5,000 cells were plated with DMEM/FBS medium and switched to medium A (DMEM + 10% lipoprotein deficient serum+Pen/Strep glutamine) after the cells were attached to the plates. On day 2, cells were washed once with medium B (DMEM+0.5% essential fatty acid-free BSA+ Pen/Strep glutamine) and then pre-incubated in medium B with or without antibody C11 [24] for 30 min. After this pre-incubation, HDL labeled with 3, 3'-dioctadecyloxacarbocyanine (DiO-HDL) (Biomedical Technologies) or Bodipy-CE labeled particles were added to the wells. After an additional incubation at 37 °C for 2 hours, the cells were washed twice with medium B and twice with PBS containing 0.5% essential fatty acid free BSA followed by fixation with Cytofix (Invitrogen) solution at 4ºC for 30 minutes. The cells are then washed and stained with 0.1% Hoechst 33342 nuclear dye (Invitrogen). Imaging studies were performed with an ArrayScan V3.5 (Thermo-Fisher). The nuclei and DiO fluorescence images were acquired sequentially using UV and 488 excitation wavelengths respectively, and an XF100 blue/green dual emission filter. Images were analyzed using the Cellomics Target Activation BioApplication, and a mean relative fluorescence per well was obtained. A minimum of 300 cells per well was evaluated for DiO and Bodipy. The neutralizing monoclonal antibody (C11) used in this study was kindly provided by Dr. Alessandra Vitelli. This antibody is generated by selection of a phage display library using CHO/SR-BI as selector and in turn converted to whole human IgG4 [24].
3. Results
3.1. Mimetic peptides form HDL-like particles
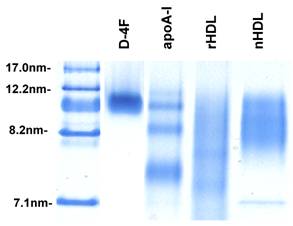
Reconstituted HDL and synthetic particles were prepared as described in Materials and Methods containing protein component of either delipidated HDL (apoHDL), apoA-I or D-4F. The size of particles was determined using native polyacrylamide gel electrophoresis. As shown in Figure 1, the synthetic particles formed by apoA-I and D-4F are quite homogeneous and migrated similarly to spherical native HDL, between 8.2 nm and 12.2 nm markers. The similar density of bands suggested that the incorporation efficiency is similar between apoA-I and D-4F. When separated on FPLC using a Superose 6 column, the reconstituted HDL and synthetic particles eluted in the same fractions as native HDL, and under the electronic microscope (EM) they had a spherical appearance similar to native HDL (Cianetti et al, unpublished data).
Native polyacrylamide gel picture of native HDL and reconstituted particles. To prepare synthetic particles, POPC, TG and CE were mixed proportionally and dried to completion under argon gas. A protein component, apoHDL, apoA-I or D-4F in buffer was then added to the pellet. The sample was sonicated with a microtip sonicator to create the synthetic particles. 20µl of each sample was loaded onto a native acrylamide gel. After running at 90V for 2 hours, the gels were stained with Coomassie blue. Lane 1, marker; Lane 2, synthetic D-4F particles; Lane 3, synthetic apoA-I particles; Lane 4, reconstituted apoHDL (rHDL) particles; Lane 5, native HDL (nHDL).
3.2. Particles formed by mimetic peptide is capable of off-loading CE into liver cells
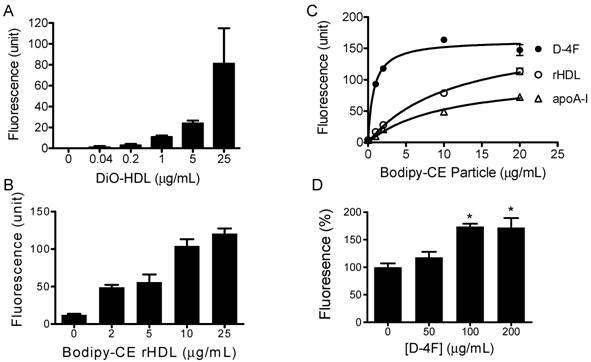
HDL labeled with 3, 3'-dioctadecyloxacarbocyanine (DiO-HDL) was used to test HDL-CE off-loading. When compared with native HDL, DiO-HDL has similar electrophoretic mobility and apolipoprotein content, as well as similar binding properties [25]. Figure 2A shows that DiO-HDL can be taken up by HepG2 cells in a dose-dependent manner. To monitor the cholesteryl ester uptake by HepG2 from reconstituted HDL (rHDL) and synthetic particles, bodipy labeled cholesteryl ester (Bodipy-CE ) was used to make these rHDL or synthetic particles. Figure 2B shows that the reconstituted HDL particles were able to load Bodipy-CE into HepG2 cells. The amount of Bodipy-CE that gets into the cells is expressed as relative fluorescence in cells and correlates well with the titration of particle concentrations. Figure 2C indicates that the synthetic particles formed by apoA-I or D-4F can off-load their CE into HepG2 cells. The off-loading efficiency was analyzed as a function of concentration by nonlinear regression using the Prism™ software. Vmax for Bodipy-CE off-loading, is 176, 107 and 162 µg/mL/h for rHDL, apoA-I and D-4F, respectively. The apparent transport constant Kt for rHDL, apoA-I and D-4F particles is 11.5, 10.4 and 0.74 μg/mL, respectively. Vmax / Kt is 15.3 h-1 for rHDL, 10.28 h-1 for apoA-I and 219 h-1 for D-4F, demonstrating the off-loading for D-4F particles is 20 times more efficient than apoA-I particles and 14.3 times more efficient than rHDL particles. The Kt of rHDL is quite consistent with the reported 16 μg/mL dissociation constant for native HDL [26]. Based on these data, we propose that D-4F can not only bind to HepG2 cells, but can also to form a much more "productive complex" for delivery of CE into HepG2 cells.
Cholesterol uptake by HepG2 cells. HepG2 cells were incubated with indicated amounts of DiO-HDL or Bodipy-CE labeled particles, with or without preincubation of D-4F. The selective uptake was determined as described in Experimental Procedures. Data are presented as mean relative fluorescence units from triplicate or duplicate determinations. Error bars represent ±SD. Panel A), cholesterol uptake of DiO-HDL. Panel B), cholesterol uptake of Bodipy-CE labeled rHDL. Panel C), cholesterol uptake from Bodipy-CE labeled rHDL (○), apoA-I (∆) and D-4F synthetic particles (●). The uptake efficiency for each particle was analyzed as a function of concentration by nonlinear regression using Prism™ software. Panel D), Percentage increase of cholesterol uptake of DiO-HDL (4 µg/mL) with preincubation of D-4F. (*P<0.05 versus no D-4F control).
3.3. D-4F facilitates HDL delivery
It has been suggested that D-4F could work independently to exert some of its effects such as increasing endothelium- and eNOS-dependent vasorelaxation, but it needs apoA-I to execute its full function and reduce the vessel wall thickness in vivo [27-29]. Consistent with this hypothesis, when administered into mouse, a majority of D-4F is found to be associated with HDL fractions in mouse plasma [30]. Since integrated D-4F could potentially change the lipoprotein and lipid composition of HDL, and in turn affect its function, D-4F could act either as an extra apoA-I protein to facilitate the uptake of HDL-CE, or as a competitor to prevent the uptake of HDL-CE when administered into animals. To address these two possibilities, we performed the following experiments. The DiO-HDL and rHDL particles were preincubated with different concentrations of D-4F at 37ºC for 1 hour before being added to HepG2 cells. Figure 2D shows preincubation of D-4F leads to increased DiO-HDL uptake in a dose-dependent manner. D-4F did not compete with apoA-I in the particle to block the CE off-loading. The average relative DiO fluorescence in HepG2 cells with D-4F at 200 and 100ug/mL is significantly higher than those with no D-4F (P<0.05).
3.4. Off-loading CE is mediated through SR-BI
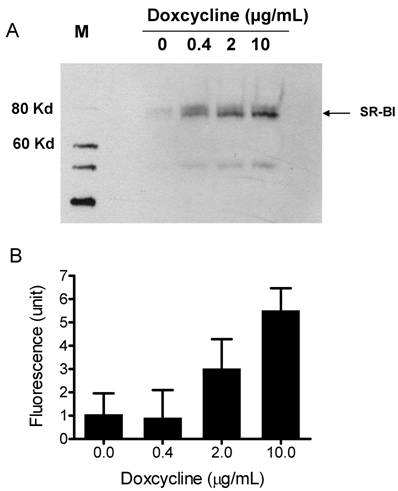
To demonstrate that the synthetic particles deliver their CE core into cells through SR-BI, we used a Doxycycline-regulated inducible SR-BI recombinant cell line. Doxycycline-regulated systems which allow a high degree of control over the expression levels of a protein of interest, provide a tool for studying protein regulation and function in eukaryotic cells [31]. A Doxycycline-inducible HEK293 cell line expressing SR-BI was generated using Clonetech's Tet-on inducible expression system. The expression of SR-BI was induced by adding Doxycycline into culture medium for 48 h. Western blots shown in Figure 3A demonstrate that SR-BI expression is induced in a dose-dependent manner. CE-selective uptake was observed when DiO-HDL particles (or Bodipy-CE labeled particles, Figure 5) were incubated with HEK293 cells expressing SR-BI. Figure 3B shows that the amount of fluorescence getting into the cell was dependent on the amount of SR-BI expressed on the surface of the HEK293 cells. This observation indicates that SR-BI is at least one of the transporters that mediated CE uptake.
A) Induced SR-BI expression in HEK293 cells. Expression of SR-BI was induced by different concentrations of Doxycycline as indicated on the figure. Western blot analysis was performed with anti-SR-BI antibody (Novus Biologicals, NB400-113). Horseradish peroxidase-conjugated secondary antibody and the enhanced chemiluminescent substrate system (Amersham) were used. B) Bodipy-CE uptake is dependent on the expression level of SR-BI. After Doxycycline induction, HEK293[pTRE-tight-SR-BI] cells were incubated with 10 µg/mL DiO-HDL. The selective uptake was determined as described in Experimental Procedures. Data are presented as mean relative fluorescence units from triplicate or duplicate wells. Error bars represent ±SD.
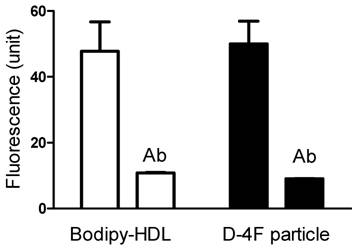
Bodipy-CE uptakes of reconstituted HDL and D-4F synthetic particles are mediated through SR-BI. After Doxycycline induction, HEK293[pTRE-tight-SR-BI] were incubated with 5 µg/ml Bodipy-labeled HDL and D-4F synthetic particles. The selective uptake was determined as described in "Methods". Anti-SR-BI antibody (C11, 10 µg/ml) was used in this study. Data were presented as mean relative fluorescence units from triplicate or duplicate wells. Error bars represent ±SD. The uptake of Bodipy fluorescence was blocked by SR-BI specific antibody.
3.5. Off-loading CE from rHDL or peptide synthetic particles is solely through SR-BI
To determine whether this uptake is solely through SR-BI, we used an SR-BI specific neutralizing antibody, C11. C11 was generated from a phage display library and has very high affinity for SR-BI receptor [24]. C11 effectively blocks SR-BI-mediated cholesterol uptake and cholesterol efflux [24]. Figure 4A and 4B show the actual fluorescent images under microscope. Green color represents Bodipy fluorescence and blue represents Hoechst nuclear dye staining. The Bodipy fluorescence uptake was greatly reduced when antibody C11 was incubated with HepG2 cells at 10 µg/mL. As shown in Figure 4C, C11 antibody at 1 µg/mL and 10 µg/mL significantly reduced Bodipy-CE uptake from rHDL as demonstrated by array scan. At 10 µg/mL, C11 antibody was able to block Bodipy-CE uptake completely. CE uptake from apoA-I and D-4F synthetic particles was also blocked by C11 antibody as shown in Figure 4D, indicating that CE off-loading to the liver cells from these particles are solely mediated by SR-BI. These data were confirmed with C167, another neutralizing monoclonal antibody against SR-BI [24].
Cholesterol uptake by HepG2 cells is blocked by SR-BI specific blocking antibody C11. HepG2 cells were incubated with indicated amount of Bodipy-CE labeled particles, with or without antibody C11. The selective uptake was determined as described in Experimental Procedures. Panel A), picture taken under fluorescent microscope. HepG2 cells were incubated with 25 µg/mL of Bodipy-CE labeled rHDL in the absence of antibody. The green signal represents Bodipy; the Hoechst nuclear staining is in blue. Panel B), picture taken under fluorescent microscopic. HepG2 cells were incubated with 25 µg/mL of Bodipy-CE labeled rHDL in the presence of 10 µg/mL antibody C11. The green signal represents Bodipy; the Hoechst nuclear staining is in blue. Panel C), cholesterol uptake of Bodipy-CE of rHDL particles at different concentrations in the absence of C11 (○), in the presence of 1µg/mL C11(∆) and in the presence of 10µg/mL C11(▲ ). Panel D), cholesterol uptake of Bodipy-CE synthetic particles at 25µg/mL in the presence (empty bar) or absence of 10µg/mL C11 antibody (solid bar). Error bars represent standard deviations of relative fluorescence from triplicate determinations.
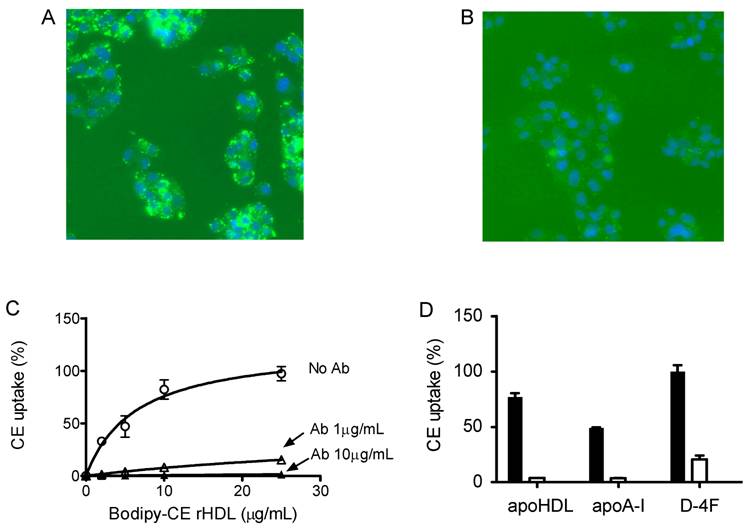
CE uptakes of rHDL and D-4F synthetic particles through SR-BI are confirmed with a SR-BI inducible-expressing cell line. In Figure 5, 5 µg/ml Bodipy-labeled HDL and D-4F synthetic particles were incubated with HEK293[pTRE-tight-SR-BI] after Doxycycline induction. Similar to HepG2 cells, the selective uptake of Bodipy fluorescence was blocked by SR-BI specific antibody.
4. Discussion
HDL mimetic peptides with 18-22 amino acids are designed to mimic apoA-I's ability to form class A amphipathic helices without sharing sequence similarity to apoA-I protein. On one hand, their sequence diversity offers a potential opportunity to surpass apoA-I's atheroprotective effects. On the other hand, mimicking all of the atheroprotective properties of apoA-I protein (243 amino acids) that contains 10 amphipathic helices with one peptide containing a single helix could be a challenge. Both mutagenesis studys and human genetic data suggest that different regions of apoA-I are important for different functions. For example, the mutations at the N-terminal (2-7 helices) are associated with familial amyloidosis while the mutations at the C-terminal (4-10 helices) are associated with LCAT activation, despite the fact that these mutations are all associated with low HDL level in plasma. The binding specificity of apoA-I with SR-BI is not affected substantially by the deletion of the first helix (1-59 amino acid) or of the helices 8-10 (185-243 amino acid) of apoA-I, however the double mutations of apoA-I at helix 4 (D102A/D103A), or at helix 6 (R160V/H162A) alter its interaction properties with SR-BI [32,33]. In this paper, we have assessed whether D-4F, a single amphipathic helix, can represent wild type apoA-I protein to form HDL-like particles and to off-load cholesteryl ester into the liver cells. We prepared the synthetic (apoA-I or peptide) particles containing phospholipids, CE, TG and apoA-I or D-4F. These particles exhibited similar size and shape to native HDL. The off-loading experiments were performed with HepG2 (human) and 1C1C (mouse) hepatoma cell lines (data not shown). Compared to rHDL and the apoA-I particles, D-4F particles are more efficient in delivering its CE core with more than a 14- and 20-fold higher Vmax / Kt, respectively, indicating that D-4F is superior in its ability to deliver CE into HepG2 cells.
Although we demonstrated that D-4F is capable of delivering CE to liver cells, we did not know which receptors are associated with this uptake process. We hypothesized that SR-BI is the major receptor responsible for the uptake since it selectively uptakes HDL cholesterol. To assess the possibility that SR-BI can mediate CE uptake for D-4F particles, we prepared a human embryonic kidney (HEK293) cell line stably expressing SR-BI under control of a tetracycline responsive element. Similar to apoA-I and native HDL particles, D-4F particles were able to deliver CE to the cells after Doxycycline induction and this was completely blocked by anti-SR-BI antibody (Fig. 5), indicating that D-4F is capable of delivering cholesteryl ester though the HDL receptor, SR-BI.
The use of peptide mimetics to improve protein function is a powerful tool, and may lead to novel therapeutic agents for the treatment of various diseases. For example, the GLP-1 mimetic peptide, Albiglutide, has better PK/PD profile than GLP-1 itself and is already in phase II clinical trials. Mimetics could be particularly useful when the target protein is too big or has a short half-life. Amphipathic properties are shared by many different apolipoproteins such as apoA-I, apoE and apoB. An ideal apoA-I mimetic would be able to mimic one, many or all functions of the apoA-I protein selectively, without mimicking the functions of any other apolipoproteins (such as apoB or apoE). ApoB and apoE both deliver cholesterol to the liver through the LDL receptor. To demonstrate that the LDL receptor is not the mechanism by which D-4F delivers CE to liver cells, we performed an off-loading experiment using LDLR-expressing HEK293 cells. D-4F particles were not able to deliver CE to the cells by LDL receptors, while apoB containing LDL particles were capable of delivering CE to cells in a LDLR dependent fashion (data not shown). Many other receptors or transporters on the cell surface besides LDL receptors and SR-BI might also be involved in the peptide delivery. To further rule out such a possibility, we chose an monoclonal antibody which selectively recognizes SR-BI and blocks SR-BI mediated cholesterol delivery [24]. With this antibody, we demonstrated that the off-loading of cholesteryl ester to HepG2 cells was inhibited completely, indicating that D-4F mimics the off-loading function of apoA-I, selectively transporting cholesteryl ester into the liver cells solely through SR-BI.
Data from genetic analysis suggests that the dynamic flow rate of cholesterol flux from peripheral foam cells to the liver, which is influenced by many factors, is considered to be more important than the static HDL mass alone [2]. For example, ApoA-IMilano carriers have low plasma HDL but are nevertheless protected from atherosclerosis, and SR-BI knockout mice have high plasma HDL but more advanced atherosclerotic lesions than their wildtype counterparts. The increase of HDL-C resulting from increased cholesterol efflux from macrophages is considered beneficial, while the increase of HDL-C from reduced off-loading by SR-BI is considered detrimental. D-4F, when dosed in animals, was reported to slow the progression of atherosclerotic lesion development and increase cholesterol excretion without changes in plasma HDL level [34,35]. Our in vitro study demonstrated that compared with apoA-I, D-4F induced cholesterol efflux from macrophage [22] and delivered HDL-C to HepG2 cells in a much more efficient manner. Based on these observations, we propose that D-4F can increase the dynamic flow rate of RCT even though HDL-C level is not changed. Consistent with this hypothesis, human kinetics data have also suggested that the turnover rate of apoA-I is more variable than its production rate [36].
In vivo, the D-4F mechanism is much more complicated. Studies showed that D-4F interacts with endogenous HDL and incorporates into the particles. We wanted to determine whether D-4F, when incorporated into HDL, could have an additive, synergistic or inhibitory effect on the delivery of cholesterol to liver cells. Our data showed that preincubation of D-4F with HDL enhanced the CE off-loading from HDL to HepG2 cells, which is consistent with the observation that additional D-4F could facilitate the HDL-C delivery back to the liver from peripheral tissues in vivo [32]. These data provided additional biochemical evidence to support the working hypothesis that administration of the apoA-I mimetic peptide, D-4F, enhances the reverse cholesterol transport process, and therefore induces lesion regression in patients with acute coronary syndrome.
There are several challenges in drug discovery and development when targeting HDL: 1) the mechanisms of HDL functions, HDL metabolism and HDL compositions/regulators are not completely understood; 2) good animal models have not been identified where both lipoprotein profile and atherosclerotic plaque composition represent the human condition; 3) very long and expensive large scale outcome trials are required because HDL levels are not yet accepted as a surrogate for therapeutic outcome; 4) and no surrogate endpoint has been validated. The total amount of cholesterol uptake by liver may be a better indication since an increased liver cholesterol pool inhibits de novo cholesterol synthesis by down-regulating SREBP-1 and/or enhancing excretion by up-regulating genes involved in bile acid synthesis and secretion (e.g. CYP7α1, BSEP and ABCG5/8).
In summary, we have demonstrated that D-4F is able to deliver cholesteryl ester to HepG2 cells through SR-BI 20 times more efficiently than apoA-I protein. In addition, D-4F facilitates cholesteryl ester off-loading from HDL particles to HepG2 cells when it is pre-incubated with HDL particles. This function of D-4F will help the SR-BI receptor to drive reverse cholesterol transport, transferring more cholesterol from peripheral tissues to the liver. We believe that this property of D-4F, along with its efflux capacity [34], contributes to its athero-protective effect and reduces atherosclerosis in mouse. Understanding this function and details of the mechanism of D-4F will facilitate rational design of other peptides and provide guidance for its proper application in clinical therapy.
Abbreviations
ABCA1: ATP binding cassette transporter A1; apoA-I: apolipoprotein A-I; SR-BI: scavenger receptor BI; LCAT: lecithin cholesterol acyltransferase; RCT: reverse cholesterol transport; CETP: cholesteryl ester transfer protein; HepG2: Human hepatocellular liver carcinoma cell line.
Acknowledgements
We are grateful to Mrs. Suzanne Eveland, Ying Chen and My-Hanh Lam for technical assistance. We thank Dr. Lyndon Mitnaul and Mrs. Jenny Tian for providing delipidated lipoprotein.
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
1. Kwiterovich POJr. State-of-the-art update and review: clinical trials of lipid-lowering agents. Am J Cardiol. 1998;82:3U-17U
2. deGoma EM, deGoma RL, Rader DJ. Beyond high-density lipoprotein cholesterol levels evaluating high-density lipoprotein function as influenced by novel therapeutic approaches. J Am Coll Cardiol. 2008;51:2199-211
3. Nissen SE, Tardif JC, Nicholls SJ, Revkin JH, Shear CL, Duggan WT, Ruzyllo W, Bachinsky WB, Lasala GP, Tuzcu EM. Effect of torcetrapib on the progression of coronary atherosclerosis. N Engl J Med. 2007;356:1304-16
4. Shieh SM, Shen MM, Tsai WJ, Shiuan LR, Wang DJ. Serum lipids and lipoprotein abnormalities in patients with thrombotic stroke--with exploring the protective role of HDL subfractions. Proc Natl Sci Counc Repub China B. 1985;9:298-304
5. Matsuda Y, Hirata K, Inoue N, Suematsu M, Kawashima S, Akita H, Yokoyama M. High density lipoprotein reverses inhibitory effect of oxidized low density lipoprotein on endothelium-dependent arterial relaxation. Circ Res. 1993;72:1103-9
6. Hayek T, Oiknine J, Dankner G, Brook JG, Aviram M. HDL apolipoprotein A-I attenuates oxidative modification of low density lipoprotein: studies in transgenic mice. Eur J Clin Chem Clin Biochem. 1995;33:721-5
7. Navab M, Anantharamaiah GM, Fogelman AM. The role of high-density lipoprotein in inflammation. Trends Cardiovasc Med. 2005;15:158-61
8. Rubin EM, Krauss RM, Spangler EA, Verstuyft JG, Clift SM. Inhibition of early atherogenesis in transgenic mice by human apolipoprotein AI. Nature. 1991;353:265-7
9. Paszty C, Maeda N, Verstuyft J, Rubin EM. Apolipoprotein AI transgene corrects apolipoprotein E deficiency-induced atherosclerosis in mice. J Clin Invest. 1994;94:899-903
10. Ameli S, Hultgardh-Nilsson A, Cercek B, Shah PK, Forrester JS, Ageland H, Nilsson J. Recombinant apolipoprotein A-I Milano reduces intimal thickening after balloon injury in hypercholesterolemic rabbits. Circulation. 1994;90:1935-41
11. Miyazaki A, Sakuma S, Morikawa W, Takiue T, Miake F, Terano T, Sakai M, Hakamata H, Sakamoto Y, Natio M. et al. Intravenous injection of rabbit apolipoprotein A-I inhibits the progression of atherosclerosis in cholesterol-fed rabbits. Arterioscler Thromb Vasc Biol. 1995;15:1882-8
12. Chiesa G, Monteggia E, Marchesi M, Lorenzon P, Laucello M, Lorusso V, Di Mario C, Karvouni E, Newton RS, Bisgaier CL, Franceschini G, Sirtori CR. Recombinant apolipoprotein A-I(Milano) infusion into rabbit carotid artery rapidly removes lipid from fatty streaks. Circ Res. 2002;90:974-80
13. Nissen SE, Tsunoda T, Tuzcu EM, Schoenhagen P, Cooper CJ, Yasin M, Eaton GM, Lauer MA, Sheldon WS, Grines CL, Halpern S, Crowe T, Blankenship JC, Kerensky R. Effect of recombinant ApoA-I Milano on coronary atherosclerosis in patients with acute coronary syndromes: a randomized controlled trial. Jama. 2003;290:2292-300
14. Eisenberg S. High density lipoprotein metabolism. J Lipid Res. 1984;25:1017-58
15. Meng QH, Calabresi L, Fruchart JC, Marcel YL. Apolipoprotein A-I domains involved in the activation of lecithin:cholesterol acyltransferase. Importance of the central domain. J Biol Chem. 1993;268:16966-73
16. Acton SL, Scherer PE, Lodish HF, Krieger M. Expression cloning of SR-BI, a CD36-related class B scavenger receptor. J Biol Chem. 1994;269:21003-9
17. Zhang Y, Zanotti I, Reilly MP, Glick JM, Rothblat GH, Rader DJ. Overexpression of apolipoprotein A-I promotes reverse transport of cholesterol from macrophages to feces in vivo. Circulation. 2003;108:661-3
18. Navab M, Anantharamaiah GM, Reddy ST, Hama S, Hough G, Grijalva VR, Yu N, Ansell BJ, Datta G, Garber DW, Fogelman AM. Apolipoprotein A-I mimetic peptides. Arterioscler Thromb Vasc Biol. 2005;25:1325-31
19. Navab M, Anantharamaiah GM, Reddy ST, Fogelman AM. Apolipoprotein A-I mimetic peptides and their role in atherosclerosis prevention. Nat Clin Pract Cardiovasc Med. 2006;3:540-7
20. Datta G, Chaddha M, Hama S, Navab M, Fogelman AM, Garber DW, Mishra VK, Epand RM, Epand RF, Lund-Katz S, Phillips MC, Segrest JP, Anantharamaiah GM. Effects of increasing hydrophobicity on the physical-chemical and biological properties of a class A amphipathic helical peptide. J Lipid Res. 2001;42:1096-104
21. Navab M, Anantharamaiah GM, Hama S, Garber DW, Chaddha M, Hough G, Lallone R, Fogelman AM. Oral administration of an Apo A-I mimetic Peptide synthesized from D-amino acids dramatically reduces atherosclerosis in mice independent of plasma cholesterol. Circulation. 2002;105:290-2
22. Chen X, Burton C, Song X, McNamara L, Langella A, Cianetti S, Chang CH, Wang J. An apoA-I mimetic peptide increases LCAT activity in mice through increasing HDL concentration. Int J Biol Sci. 2009;5:489-99
23. Hirz R, Scanu AM. Reassembly in vitro of a serum high-density lipoprotein. Biochim Biophys Acta. 1970;207:364-7
24. Catanese MT, Graziani R, von Hahn T, Moreau M, Huby T, Paonessa G, Santini C, Luzzago A, Rice CM, Cortese R, Vitelli A, Nicosia A. High-avidity monoclonal antibodies against the human scavenger class B type I receptor efficiently block hepatitis C virus infection in the presence of high-density lipoprotein. J Virol. 2007;81:8063-71
25. Pitas RE, Innerarity TL, Weinstein JN, Mahley RW. Acetoacetylated lipoproteins used to distinguish fibroblasts from macrophages in vitro by fluorescence microscopy. Arteriosclerosis. 1981;1:177-85
26. Liadaki KN, Liu T, Xu S, Ishida BY, Duchateaux PN, Krieger JP, Kane J, Krieger M, Zannis VI. Binding of high density lipoprotein (HDL) and discoidal reconstituted HDL to the HDL receptor scavenger receptor class B type I. Effect of lipid association and APOA-I mutations on receptor binding. J Biol Chem. 2000;275:21262-71
27. Ou J, Wang J, Xu H, Ou Z, Sorci-Thomas MG, Jones DW, Signorino P, Densmore JC, Kaul S, Oldham KT, Pritchard KAJr. Effects of D-4F on vasodilation and vessel wall thickness in hypercholesterolemic LDL receptor-null and LDL receptor/apolipoprotein A-I double-knockout mice on Western diet. Circ Res. 2005;97:1190-7
28. Mineo C, Shaul PW. Role of high-density lipoprotein and scavenger receptor B type I in the promotion of endothelial repair. Trends Cardiovasc Med. 2007;17:156-61
29. Feng Y, van Eck M, Van Craeyveld E, Jacobs F, Carlier V, Van Linthout S, Erdel M, Tjwa M, De Geest B. Critical role of scavenger receptor-BI-expressing bone marrow-derived endothelial progenitor cells in the attenuation of allograft vasculopathy after human apo A-I transfer. Blood. 2009;113:755-64
30. Navab M, Anantharamaiah GM, Reddy ST, Van Lenten BJ, Wagner AC, Hama S, Hough G, Bachini E, Garber DW, Mishra VK, Palgunachari MN, Fogelman AM. An oral apoJ peptide renders HDL antiinflammatory in mice and monkeys and dramatically reduces atherosclerosis in apolipoprotein E-null mice. Arterioscler Thromb Vasc Biol. 2005;25:1932-7
31. Berens C, Hillen W. Gene regulation by tetracyclines. Constraints of resistance regulation in bacteria shape TetR for application in eukaryotes. Eur J Biochem. 2003;270:3109-21
32. Liu T, Krieger M, Kan HY, Zannis VI. The effects of mutations in helices 4 and 6 of ApoA-I on scavenger receptor class B type I (SR-BI)-mediated cholesterol efflux suggest that formation of a productive complex between reconstituted high density lipoprotein and SR-BI is required for efficient lipid transport. J Biol Chem. 2002;277:21576-84
33. Zannis VI, Chroni A, Krieger M. Role of apoA-I, ABCA1, LCAT, and SR-BI in the biogenesis of HDL. J Mol Med. 2006;84:276-94
34. Navab M, Anantharamaiah GM, Reddy ST, Hama S, Hough G, Grijalva VR, Wagner AC, Frank JS, Datta G, Garber D, Fogelman AM. Oral D-4F causes formation of pre-beta high-density lipoprotein and improves high-density lipoprotein-mediated cholesterol efflux and reverse cholesterol transport from macrophages in apolipoprotein E-null mice. Circulation. 2004;109:3215-20
35. Van Lenten BJ, Wagner AC, Navab M, Anantharamaiah GM, Hama S, Reddy ST, Fogelman AM. Lipoprotein inflammatory properties and serum amyloid A levels but not cholesterol levels predict lesion area in cholesterol-fed rabbits. J Lipid Res. 2007;48:2344-53
36. Lewis GF, Rader DJ. New insights into the regulation of HDL metabolism and reverse cholesterol transport. Circ Res. 2005;96:1221-32
Author contact
![]() Correspondence to: Xuelei Song, Ph.D., Merck Research Laboratories, RY-80T-A147, P.O. Box 2000, Rahway, NJ 07065. Telephone: 1-732-594-4802; Fax: 1-732-594-4620; E-mail: xuelei_songcom. Jun Wang, Ph.D., Department of Biology, Sundia MediTech Company, Ltd., Building 8, 388 Jialilue Road Zhangjiang Hightech Park, Shanghai 201203, China. Tel: 86-21-51098642 x 128; Fax: 86-21-51903505; E-mail: jwangcom
Correspondence to: Xuelei Song, Ph.D., Merck Research Laboratories, RY-80T-A147, P.O. Box 2000, Rahway, NJ 07065. Telephone: 1-732-594-4802; Fax: 1-732-594-4620; E-mail: xuelei_songcom. Jun Wang, Ph.D., Department of Biology, Sundia MediTech Company, Ltd., Building 8, 388 Jialilue Road Zhangjiang Hightech Park, Shanghai 201203, China. Tel: 86-21-51098642 x 128; Fax: 86-21-51903505; E-mail: jwangcom