Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2009; 5(7):686-694. doi:10.7150/ijbs.5.686 This issue Cite
Research Paper
Role of Ldb1 in Adult Intestinal Homeostasis
Ipsita Dey-Guha*, Mahua Mukhopadhyay*, Matthew Phillips, Heiner Westphal ![]()
Laboratory of Mammalian Genes and Development, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, HHS, Bethesda, MD 20892, USA.
* These authors have contributed equally to the work.
Received 2009-9-3; Accepted 2009-10-23; Published 2009-10-30
Abstract
Ldb1 is an essential co-regulator of transcription in embryonic development. It acts in conjunction with nuclear LIM-homeodomain and LIM-only proteins to control key events of organogenesis as precursor cells enter lineage specification. Here we ask whether Ldb1 exerts control over stem cell activation and differentiation throughout the life of the organism as required for tissue homeostasis. To help answer this question, we have generated conditional Ldb1 mouse mutants with an Ldb1 floxed/floxed;ROSA26CreER genotype. Tamoxifen treatment of 60 day-old mutant animals results in near-ubiquitous Cre-mediated Ldb1 inactivation. As a consequence, the stem cell microenvironment of intestinal crypts is drastically affected. Cells that normally express Ldb1 together with markers that identify them as lineage progenitors cease to retain bromodeoxyuridine and are gradually lost. Ldb1 inactivation in intestinal crypts and/or in neighboring mesenchymal cells also triggers activation of Wnt signaling in the stem cell niches of the small intestine. Cell proliferation is markedly increased in the epithelia of the small intestine, and Lgr5-expressing stem cells disappear from the base of the crypts. This perturbation of the normal process of tissue homeostasis causes apoptosis, and the animals do not survive. We conclude that Ldb1-mediated transcriptional regulation plays a major role in adult intestinal homeostasis.
Keywords: Ldb1, small intestine, tissue homeostasis, stem cell niche, tamoxifen
1. Introduction
Tissue homeostasis is maintained through periodical replacement of cells. This process originates from special micro environments or niches that contain stem cells able to self-renew or to differentiate into multiple cell lineages. Each tissue has its own replacement cycle. Thus, intestinal epithelia of the mouse completely self-renew approximately every five days1, while the interfollicular epidermis and corneal epithelium are replaced in a span of several weeks2. Balanced canonical Wnt/β-catenin signaling is essential for the maintenance of a stem cell pool able to self renew and to give rise to lineage precursors. Mutations that affect components of this Wnt pathway can result in hyperproliferation of the epithelia3-6, while overexpression of the Wnt inhibitor Dkk1 can cause complete loss of proliferation7-9. Finally, lack of the Wnt inhibitor Dkk2 can alter the fate of corneal stem cells10.
The gut tube of the small intestine is characterized by protrusions towards the lumen, called villi, and by crypts or invaginations into the submucosa. Genetic fate mapping has established that the niche containing multipotent stem cells of the small intestine is located at or near the base of the crypts11, 12. The canonical Wnt signaling pathway is a known regulator of cell proliferation during development and maintenance of the intestinal epithelium. Wnt-activated cells express putative stem cell markers like Musashi-1, but not most cell differentiation markers13. Consistent with this, conditional treatment of adult mice with Dkk1 encoded by adenovirus vectors leads to a precipitous down regulation of the Wnt target genes CD44 and EphB2 in the small intestine and colon. Cell proliferation in these tissues is inhibited, followed by loss of crypts and villi. Several days later, severe defects in glandular structure are observed, leading to substantial mortality from colitis and systemic infection8. The same study showed that decreased reduction of Dkk1 expression at later time points (>10 days) resulted in crypt and villus regeneration. Conversely, ectopic expression of the Wnt inhibitor Dkk1 in the small intestine leads to blockage of the Wnt signaling pathway thus affecting proliferation and differentiation of the epithelial cells in the crypts9. Finally, Wnt signaling in self-renewing tissues such as the intestine is tightly regulated as expression of Wnt ligands, downstream effector genes, Wnt receptors and co-receptors, as well as Wnt-pathway inhibitors has been well documented in both the epithelial and mesenchymal tissues of the small intestine14. Thus it is likely that cross talk between epithelial and mesenchymal cells play a central role in epithelial tissue homeostasis in the small intestine. The need for balanced canonical Wnt signaling has been demonstrated in studies showing that proliferative cells at the bottom of the crypts accumulate nuclear β-catenin15 and activation of Wnt/ β-catenin signaling in mutants leads to adenomatous polyp formation in the mouse intestine16, 17.
Ldb1 binds to the LIM motif of nuclear LIM-homeodomain proteins and acts as their transcriptional co-regulator at multiple stages of embryonic development18, 19. Ldb1 null mutant mice are characterized by early and profound defects in brain and heart formation and severe impairment of mesoderm-derived extraembryonic structures20. Comparable effects of Ldb1 and Dkk2 loss-of-function have been observed in mutant mice showing that both genes are tied in with programs of epithelial fate determination10, 21. Thus, a connection has been established between Ldb1 function and canonical Wnt pathway regulation in epithelia. In the skin the LIM-homeodomain product of the Lhx2 gene, a likely partner of Ldb1-mediated action, is required as the stem cell progeny give rise to the various epidermal cell lineages22. Together, these observations indicate that Ldb1 proteins play an important role in adult tissue homeostasis. Our initial finding that Ldb1 is also readily expressed in the epithelial cells of the villi and the crypts as well as the mesenchyme of the small intestine (E. Makarev, personal communication) strongly suggests that Ldb1 plays a role in tissue renewal of the gut epithelia as well.
Here we report on the function of the Ldb1 gene in epithelial tissue homeostasis of adult mice. In the small intestine Ldb1 is prominently expressed in the epithelial cells of the villi, the crypts and in the mesenchyme. Conditional ablation of Ldb1 function results in a devastating effect on intestinal tissue turnover caused by a loss of slow cycling stem cells from the crypts of the small intestine. These observations reveal a critical role of Ldb1 in the regulation of adult tissue homeostasis.
2. Results and Discussion
2.1 Ldb1 is expressed in the stem cell niche compartments of the small intestines of adult mice
Adult mice, two months of age, were utilized throughout this study. We examined Ldb1 expression in the small intestines of control mice with anti-Ldb1 antibody, which specifically labels nuclei23, 24. The morphology and location of Ldb1-positive cells allow us to conclude that Ldb1 is expressed in the epithelial cells of the villi and the crypts including the Paneth cells and undifferentiated Transiently Amplifying (TA) cells (Fig. 1A). Scattered Ldb1-expression was also detected in the adjacent mesenchymal tissue (Fig. 1B).
Expression pattern of the Ldb1 protein in the small intestine of adult mice. (A) Immunostaining of the small intestine with anti-Ldb1 antibody shows nuclear expression in the crypts and the villi. (B) Magnified image of a crypt stained with anti-Ldb1 antibody. (C) In situ hybridization with Lgr5 shows expression in the crypt base.
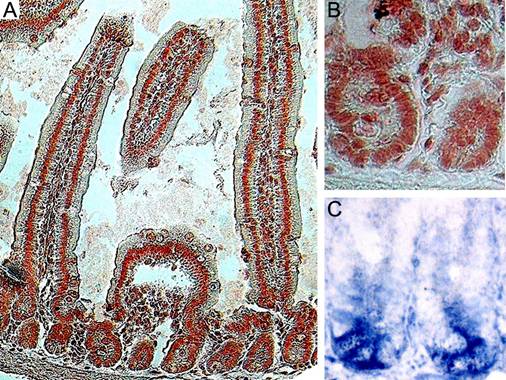
Cells expressing the Wnt target gene Lgr5 (Leucine-rich-repeat-containing G-protein-coupled Receptor 5, also known as Gpr49) are located at the base of the crypts (Fig. 1C). Lineage tracing has identified these cells as stem cells11. A comparison of Figs. 1B and 1C allows us to conclude that the cells expressing Lgr5 transcripts also strongly stain for Ldb1.
2.2 Conditional inactivation of Ldb1 results in loss of stem cells from the niche compartments of the small intestine
We used a loss-of-function approach to study the functional role of Ldb1 in the adult mouse. Our earlier studies established that zygotic ablation of Ldb1 results in early embryonic lethality20. For this reason, we resorted to Cre-mediated inactivation of a floxed Ldb1 allele previously generated in our laboratory25.
The R26CreER mouse can be induced by tamoxifen to express Cre under the control of the ROSA26 promoter in a wide variety of tissues26. The effect of tamoxifen treatment on Ldb1 expression is quite dramatic in the intestines of conditionally mutant (Ldb1fl/fl;R26CreER) compared to control mice (Fig. 2A-C). We refer to both Ldb1fl/+;R26CreER and Ldb1fl/fl genotypes as controls because these mice are indistinguishable from wild type or from R26CreER mice in the experiments described below. For a study of the short-term effects of the tamoxifen-induced Ldb1 deletion, mice were sacrificed 24 hrs after tamoxifen treatment (short-term mutants). A second group of mice were sacrificed 7 days after tamoxifen treatment to study the long-term effect of Ldb1 deletion (long-term mutants). The effect of Ldb1 deletion could not be observed at later time points because mutant mice did not survive more than 7 days after treatment.
Conditional deletion of the Ldb1 gene in the small intestine after 2 days of tamoxifen injections. (A, B) Ldb1 immunostaining in the crypts of a representative control mouse (A) and a mutant mouse one day after tamoxifen treatment (B). (C) Quantification of Ldb1 immunostaining in the crypts of control and mutant mice, n=number of crypts per mice. Data are mean ± standard deviation. (D-F) in situ hybridization for Lgr5 (stem cell marker of the small intestine) in the crypts of a control mouse (D) and mutant mice with short term (E) and long term (F) effects of the deletion of the Ldb1 gene. Abbreviations: control, Ldb1fl/+;R26CreER or Ldb1fl/fl; mutant, Ldb1fl/fl;R26CreER; Ldb+ve, cells positive for Ldb1 antibody staining; short term, 2 days of tamoxifen injections followed by sacrifice after 1 day; long term, 2 days of tamoxifen injections followed by sacrifice after 7 days.
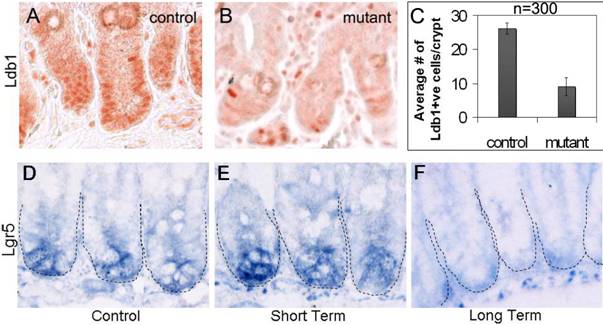
In control intestines over 83% of nuclei in the crypts and the adjacent mesenchyme stain prominently with the anti-Ldb1 antibody (Fig. 2A, C). In contrast, in short-term Ldb1fl/fl;R26CreER mutant intestines, the number of Ldb1-positive cells in the crypts is sharply reduced to 36% (Fig. 2B, C). Therefore, tamoxifen-mediated induction of Cre results in an average 2.3-fold reduction of Ldb1-labeled cells detected in the crypts (Fig. 2C). Quantification of this data is described in experimental procedures and in Figure 2C.
In order to determine the effect of the Ldb1 deletion on stem cells we compared Lgr5 expression in floxed Ldb1 mutant crypts with that of controls. Cells in the niche compartment near the base of the crypts of control mice are clearly marked by Lgr5 in situ hybridization (Fig. 2D). A similar level of Lgr5 signal was seen in the short-term mutant (Fig. 2E). However, after long-term deletion of the Ldb1 gene this signal was markedly reduced and appeared non-specific (Fig. 2F). We conclude that the conditional inactivation of Ldb1 in adult mouse tissues results in a loss of the stem cells from niche compartments in the small intestine.
2.3 Ldb1 controls cell proliferation in the crypts of the small intestine
The prominent expression of Ldb1 in the crypts of the small intestine seen in Fig. 1A, as well as the loss of Lgr5 in the stem cells following Ldb1 deletion, suggested the possibility that Ldb1 plays a role in the regulation of cell proliferation in the stem cell niche of this tissue. We therefore asked whether ablation of Ldb1 would affect the rate of incorporation of the synthetic thymidine analog bromodeoxyuridine (BrdU) into the DNA of replicating intestinal cells. We administered BrdU by intraperitoneal (IP) injection each day for two days along with tamoxifen, and BrdU alone once more on the third day. A group of “short-chase” mice was sacrificed 5 hrs thereafter, while a second “long chase” group was sacrificed after 7 days.
Previous studies have shown that multipotent stem cells of the small intestine are slow cycling but can give rise to rapidly proliferating daughter cells, referred to as TA or Transiently Amplifying cells, which fill the crypts with committed lineage precursor cells (reviewed in 4). In our experiments, a short chase timeframe was sufficient to show that the number of cells staining for anti-Phospho Histone H3 (PH3), a marker for cells in active mitosis, is increased in the mutant crypts, indicating unusually high proliferation (compare Figs. 3A, B). At the same time, the level of BrdU incorporation not only increases in mutant crypt cells (Fig. 3D) but is now also observed in the villi cells, which in control animals do not incorporate BrdU, as they are mainly composed of terminally differentiated, nonproliferating columnar epithelial cells (compare Figs. 3C, D). BrdU-positive cells are also found in the stem cell niches of mutant crypts at a much higher rate than in control crypts (Fig. 3C, D, arrows). These results suggest the slow cycling stem cells of control crypts are proliferating more rapidly in the absence of Ldb1.
Enhanced proliferation in the crypts upon deletion of Ldb1. (A, B) Immunostaining with anti-Phospho HistoneH3 antibody showing enhanced expression in the crypts of a mutant mouse (B) as compared to control (A). Arrows indicate crypts. (C-F) Immunostaining with anti- BrdU antibody after BrdU pulse of 3 days followed by short chase (C, D) shows enhanced proliferation in the crypts (arrows) as well as the villi (arrowheads) of the mutant (D) mice as compared to the control (C). (E, F) After long chase, crypts of control mouse (E) contained a significant number of BrdU-label-retaining cells (LRCs) in their stem cell compartments as opposed to those of mutant mouse (F). Increase in the number of cells populating the mutant crypts is evident in the long chase mutant. (G) Quantification of BrdU immunostaining in the crypts of control and long chase mutant mice, n=number of crypts. Data are mean ± standard deviation. Abbreviations: control, Ldb1fl/+;R26CreER or Ldb1fl/fl; mutant, Ldb1fl/fl;R26CreER; short chase, BrdU pulse of 3 days followed by sacrifice of mice after four hours; long chase, BrdU pulse of 3 days followed by sacrifice of mice after 7 days; PH3, Phospho HistoneH3 antibody and BrdU, bromodeoxyuridine.
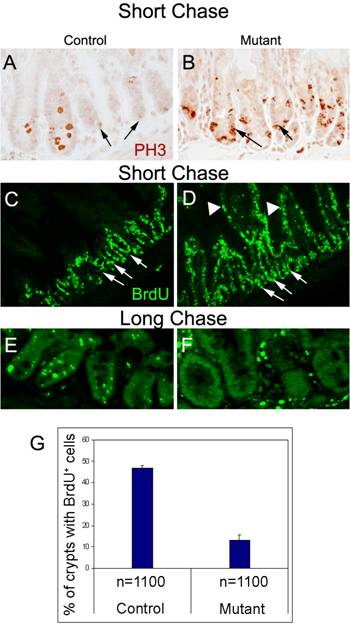
In contrast to the short chase results, after a long chase about 85% of the mutant crypts no longer retain any BrdU signal (Fig. 3F, G) while the label persists in about 50% of the control crypts (Figs. 3E, G). Taken together, these results suggest that the unusual BrdU-labeled cells in the short chase mutant villi are derived from cells of hyperproliferating crypts that have migrated away from the BrdU-label retaining stem cell niche13. We further observe that the initial upregulation of cell division seen in the crypts of the short chase mutants (Fig. 3D) leads to an increase in number of cells populating the mutant crypts after a period of 7days (Fig. 3F). These findings are consistent with the idea that Ldb1 maintains a slow cycling stem cell pool in the crypts that is compromised when the gene is inactivated.
Ldb1 is known to mediate the activity of transcription factors encoded by the Lhx genes, and Lhx2 is known to play a vital role in hair stem cell maintenance in the bulge22. Our findings thus underscore the importance of Ldb1 and associated factors in the transcriptional control of this process.
2.4 The morphology of the small intestine is altered upon prolonged Ldb1 ablation
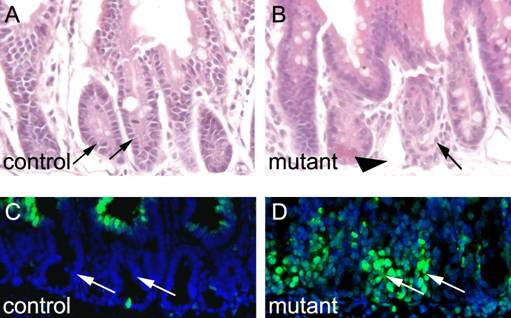
We were interested to know how deletion of Ldb1 affects gut morphology over time. To this end, we treated Ldb1fl/fl;R26CreER mutants and control mice with tamoxifen for two days and sacrificed the animals one week later. Whereas the small intestines of the controls appear morphologically unremarkable (Fig. 4A), the same tissue in the Ldb1 deleted mice appears grossly abnormal (Fig. 4B). Collapse of the mutant small intestine was confirmed through TUNEL staining. Apoptosis was evident in the mutant, but not in control crypts (Figs. 4C and D).
Apoptosis and morphological alterations in the small intestine after long term deletion of Ldb1. (A, B) H&E staining of the small intestine shows disorganized crypts (arrows) and columnar epithelial cells (arrowhead) in mutant mice (B) as compared to control (A). (C, D) TUNEL assays in the small intestine shows cell death in the crypts (arrows) of mutant mice (D) as compared to control crypts (arrows) (C). Abbreviations: control, Ldb1fl/+;R26CreER or Ldb1fl/fl and mutant, Ldb1fl/fl;R26CreER.
2.5 Ldb1 ablation leads to activation of the Wnt/β-catenin pathway in the intestinal crypts
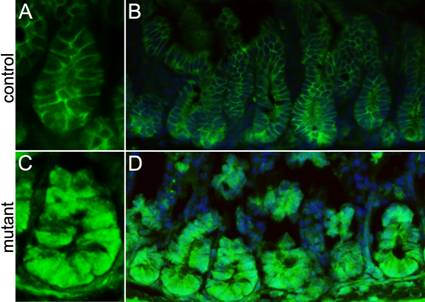
Stem cell division is preceded by activation of the Wnt/β-catenin pathway during epithelial tissue regeneration27. Therefore we examined β-catenin activity in the crypts of the conditionally-deleted Ldb1 mutant mice through immunostaining using anti-β-catenin antibody. Consistent with the published literature13, we observe that in our control mice the Wnt/β-catenin staining is excluded from the nuclei in the crypt cells (Fig. 5A, B). However in the conditionally deleted Ldb1 mutant, abnormal activation of this pathway manifests as nuclear β-catenin immunostaining in almost all the crypt cells (Fig. 5C,D). We conclude that loss of Ldb1 from the small intestine leads to activation of the Wnt/ β-catenin signaling pathway. This is consistent with a previous observation that the Ldb1 gene positively regulates the expression of several Wnt pathway inhibitors20.
Analyzing the level of Wnt/β-catenin signaling in the small intestine of the adult mutant and control mice. (A, B) Anti-β-catenin immunostaining (green) shows membrane staining of β-catenin in cells of control crypts, while in the mutant crypts (C, D) all the cells show nuclear staining of β-catenin. Abbreviations: control, Ldb1fl/+;R26CreER or Ldb1fl/fl and mutant, Ldb1fl/fl;R26CreER.
Our study addresses the function of the transcriptional co-regulator Ldb1 in the small intestine. Ldb1 is prominently expressed in the epithelia of this organ. Tamoxifen-induced activation of Cre acts on floxed Ldb1 alleles and causes their inactivation. The resulting loss of Ldb1 function profoundly affects the stem cell microenvironment of intestinal crypts. Cells in the stem cell niches of these crypts that normally express Ldb1, together with markers that identify them as lineage progenitors, are gradually lost and cease to incorporate BrdU. The small intestine, well known for its rapid turnover of epithelia, is severely affected by this disturbance of the niche environment. We observed a sharp increase of cell proliferation in the gut epithelia and a concomitant loss of slow cycling, Lgr5-expressing stem cells from the base of the crypts.
This perturbation of the normal process of tissue homeostasis is not compatible with cell and tissue viability, hence the eventual onset of apoptosis which the animals did not survive. Additionally, Ldb1 inactivation triggers the canonical Wnt/ß-catenin pathway. We observed a marked increase of anti-ß-catenin immunostaining in the nuclei of the mutant crypt cells including the cells of the niche.
Wnt/ß-catenin signaling plays an indispensable role in the regulation of epithelial stem cells. These signals are involved in the control of stem cell quiescence and activation as well as cell migration and differentiation2. We show that Ldb1 affects the output of these signals in the small intestine. This suggests a mechanism whereby Ldb1 interacts with other transcriptional regulators to control gene products that influence Wnt output levels. Ldb1 is known to positively influence the expression of several Wnt inhibitors10 while Lgr5 is a negative regulator of the Wnt pathway28. It is possible that Wnt inhibitors encoded by Dkk genes are among the target genes for Ldb1-mediated transcriptional control since these genes are known to play crucial roles in epithelial tissue homeostasis. For example, it is known that overexpression of Dkk1 results in a failure to develop hair follicles1, reduces epithelial proliferation and causes a loss of intestinal crypts9. Likewise, the related Dkk2 gene is a key regulator of corneal versus epidermal fate of the ocular surface epithelium10.
Ldb1 knockdown sets off a precipitous wave of proliferation and exit of cells from the niche compartments of the small intestine. Interestingly, previous reports have shown that increased Wnt-signaling is correlated with increased proliferation in the crypts of the small intestine13. In our BrdU pulse-labeled experiments we noted that Ldb1 inactivation causes a substantial increase of BrdU incorporation in epithelial cells extending beyond the crypts and into the villi of the small intestine. The fact that Lgr5-expressing crypt cells are concomitantly lost strongly suggests that at least a portion of the BrdU-positive cells in the villi are descendants of untimely activated niche stem cells.
We find Ldb1 broadly expressed in the small intestine, in the villi and crypt epithelia as well as in the adjacent mesenchyme. Interaction between these two tissue layers and the involvement of transcription factors in this process has long been recognized as an important determinant of cell fate29. It has been established that such tissue interactions play a central role in tissue homeostasis in the skin and the cornea10, 27. In our experimental setting, all ROSA26CreER-expressing cells are subject to tamoxifen-induced Ldb1 inactivation, including both epithelial and mesenchymal cells. In order to determine if both or one of these tissues are critical for Ldb1-mediated transcriptional control of niche activity, selective conditional Ldb1 inactivation in individual compartments will be required. A recent study showed that incomplete inactivation of the Ascl2 gene activity in epithelial cells of the small intestine leads to a partial loss of stem cells in intestinal crypts, followed by a recovery of the stem cell niche through crypt fission, a repair mechanism believed to be activated after injury or genetic damage30. In contrast, results of our study demonstrate that a 2.3-fold reduction of Ldb1 expression in both epithelial and mesenchymal tissues is sufficient for complete collapse of the small intestinal structure, suggesting that mesenchymal tissue might play a crucial role in the tissue repair process. Current experiments aim at testing the effect of gut mesenchyme-specific Ldb1 ablation by developing a Cre allele that can be conditionally expressed in this tissue.
In conclusion, we show that Ldb1 plays a key regulatory role in transcriptional events that govern the biological process of epithelial tissue homeostasis in mammalian small intestine. The mechanism of this process, as well as its broader implications for homeostasis elsewhere in the adult organism, remain to be elucidated.
3. Experimental procedures
3.1 Mice
The Ldb1 conditional mutant allele was generated in our laboratory by inserting two LoxP sites, one upstream of the protein coding region and the other into the ninth intron of the Ldb1 gene25. This conditional Ldb1fl/fl mutant was mated with Rosa26CreER mice (Jackson Laboratory #004847) to generate Ldb1fl/fl;Rosa26CreER offspring that can be subjected to tamoxifen-mediated Ldb1 inactivation.
3.2 Tamoxifen induction and BrdU injection
Eight week-old mice were given 0.5ml of a 20 mg/ml dilution of tamoxifen (Sigma) in corn oil (Sigma) by oral gavage once a day for two days. Mice were injected intraperitoneally once a day for three days with 0.2ml of BrdU (Sigma) solution in PBS at 10mg/ml. Mice were sacrificed four hours (“short chase”) or seven days (“long chase”) after BrdU injection. Ldb1 deletion was calculated on an average of 1000 cells from 300 crypts per mouse with three mice each for control and mutant. In the controls, 21-32 cells per crypt immunostained for Ldb1 (an average of 25 ± 3.8 out of 30 total cells per crypt) while in the mutants it was 5-15 cells per crypt (averaging 9 ± 4.1 out of 25 total cells per crypt). In a 4µm section, the deletion varied from 40% in 300 crypts, 60% in 200 crypts, 70% in 300 crypts and 90% in 100 crypts.
3.3 Histology
The gut was dissected and fixed in 4% Paraformaldehyde (PFA) in PBS. Samples were then embedded in paraffin blocks, and sectioned at 4μm and 6μm, respectively. For histological analysis, sections were stained with hematoxylin and eosin (H&E) (Sigma).
3.4 Immunohistochemistry
Brightfield: Sections were deparaffinized, rehydrated and washed in 1 X PBS. Endogenous peroxidase activity was quenched with 3% hydrogen peroxide in methanol for 30 min. Heat Induced Epitope Retrieval (HIER) was performed on mouse small intestine sections in Buffer A for 20min utilizing a pressurized de-cloaking chamber (Prestige Medical). The sections were washed and incubated in blocking serum (3%; Vector Laboratories) at room temperature for 60min to block non-specific binding. Thereafter the sections were exposed to primary antibodies, rabbit anti-Ldb1 (kindly provided by Drs. L.-Q. Li and P. Love, NICHD, NIH), and rabbit anti-Phospho-Histone H3 (Ser10, Upstate), overnight at 4oC. After primary antibody incubation, sections were washed in PBS, incubated with biotinylated secondary antibodies (Vector Laboratories) and processed with a Vectastain ABC kit (Vector Laboratories). The sections were developed by using AEC as chromogen (Zymed), counterstained with Hematoxylin, and mounted with Aqua PolyMount (Polysciences).
Fluorescence: Sections were deparaffinized, rehydrated and washed in 1 X PBS. HIER was performed and sections were incubated in goat or fetal calf blocking serum (3%) (Vector
Laboratories) followed by incubation in primary antibodies, rabbit anti-Ldb1, rabbit anti- Phospho-Histone H3 (Ser10), mitosis marker (Upstate), goat β-catenin (Cell Signaling), overnight at 4oC. Sections were sequentially exposed to appropriate fluorescein-tagged Alexa Fluor secondary antibodies (Molecular Probes). The slides were wet-mounted and counterstained utilizing Vectashield with DAPI (Vector Laboratories) for staining of the nucleus.
3.5 TUNEL Staining
Fluorescein conjugated staining was performed using the “in situ Cell Death Kit” (Roche diagnostics), according to manufacturer's instructions. BrdU Staining Tissue sections of 4 μm thickness were deparaffinized, rehydrated and washed in 1x PBS. Endogenous peroxidase activity was quenched with 3% hydrogen peroxide in methanol for 30 min and HIER was performed. The sections were washed and incubated in 1 N HCl for 10min on ice followed by 2N HCL for 30min at 37oC and final incubation in borate buffer (0.1M) for 10min at room temperature. After washing the sections were incubated in donkey blocking serum (3%, Sigma) at room temperature for 60 min followed by exposure to primary antibody, sheep anti-BrdU (Abcam) at a dilution of 1:100, overnight at 4oC. The sections were incubated in Alexa Fluor 488 donkey anti-sheep secondary antibody (Molecular Probes) for 30 min and wetmounted.
3.6 Microscopic examination
The fluorescent images were captured using an Olympus BX60 microscope fitted with a Zeiss Axiocam MRm camera, using Slidebook v.4.1 software (Intelligent Imaging Innovations, Inc., Denver CO). The brightfield images were captured using the same Olympus microscope fitted with an Olympus Q-Color 6 camera, using QImaging software v.6.0 (Qimaging Corp., Surrey, BC, Canada). All digital images were processed using Photoshop 6 (Adobe Inc., San Jose, CA).
3.7 in situ hybridization
Antisense DIG-labeled Lgr5 probes were synthesized from mouse cDNA (ATCC #17081141) (Barker et al., 2007). The tissue sections were deparaffinized, rehydrated and incubated in 0.2M HCl to reduce background. Proteinase K digestion was carried out using 30μg/ml Proteinase K (Roche) for 13 min at room temperature followed by 7min at 37oC. Postfixation, the sections were treated with acetic anhydride, rinsed and hybridized with the probe for 48 hours at 65oC. The probes were detected using anti-DIG AP antibodies (1:2000) overnight at 4oC, and BM Purple solution (Roche 11093274910 and 11442074001, respectively), overnight at room temperature.
Acknowledgements
We thank Dr. H. Clevers for the in situ hybridization protocol, Drs. Li Qi Li and Paul Love for the Ldb1 antibody, Ms. Rui Lin for genotyping and Dr. Sohyun Ahn for helpful discussions. This research was supported by the Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development.
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
1. Andl T, Reddy ST, Gaddapara T. et al. WNT signals are required for the initiation of hair follicle development. Dev Cell. 2002;2:643-653
2. Blanpain C, Horsley V, Fuchs E. Epithelial stem cells: turning over new leaves. Cell. 2007;128:445-458
3. Andreu P, Colnot S, Godard C. et al. Crypt-restricted proliferation and commitment to the Paneth cell lineage following Apc loss in the mouse intestine. Development. 2005;132:1443-1451
4. Barker N, van de Wetering M, Clevers H. The intestinal stem cell. Genes Dev. 2008;22:1856-1864
5. Harada H, Kettunen P, Jung HS. et al. Localization of putative stem cells in dental epithelium and their association with Notch and FGF signaling. J Cell Biol. 1999;147:105-120
6. Sansom OJ, Reed KR, Hayes AJ. et al. Loss of Apc in vivo immediately perturbs Wnt signaling, differentiation, and migration. Genes Dev. 2004;18:1385-1390
7. Korinek V, Barker N, Willert K. et al. Two members of the Tcf family implicated in Wnt/beta-catenin signaling during embryogenesis in the mouse. Mol Cell Biol. 1998;18:1248-1256
8. Kuhnert F, Davis CR, Wang HT. et al. Essential requirement for Wnt signaling in proliferation of adult small intestine and colon revealed by adenoviral expression of Dickkopf-1. Proc Natl Acad Sci U S A. 2004;101:266-271
9. Pinto D, Gregorieff A, Begthel H. et al. Canonical Wnt signals are essential for homeostasis of the intestinal epithelium. Genes Dev. 2003;17:1709-1713
10. Mukhopadhyay M, Gorivodsky M, Shtrom S. et al. Dkk2 plays an essential role in the corneal fate of the ocular surface epithelium. Development. 2006;133:2149-2154
11. Barker N, van Es JH, Kuipers J. et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449:1003-1007
12. Sangiorgi E, Capecchi MR. Bmi1 is expressed in vivo in intestinal stem cells. Nat Genet. 2008;40:915-920
13. Davies PS, Dismuke AD, Powell AE. et al. Wnt-reporter expression pattern in the mouse intestine during homeostasis. BMC Gastroenterol. 2008;8:57
14. Gregorieff A, Pinto D, Begthel H. et al. Expression pattern of Wnt signaling components in the adult intestine. Gastroenterology. 2005;129:626-638
15. Batlle E, Henderson JT, Beghtel H. et al. Beta-catenin and TCF mediate cell positioning in the intestinal epithelium by controlling the expression of EphB/ephrinB. Cell. 2002;111:251-263
16. Moser AR, Pitot HC, Dove WF. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science. 1990;247:322-324
17. Shibata H, Toyama K, Shioya H. et al. Rapid colorectal adenoma formation initiated by conditional targeting of the Apc gene. Science. 1997;278:120-123
18. Hobert O, Westphal H. Functions of LIM-homeobox genes. Trends Genet. 2000;16:75-83
19. Matthews JM, Visvader JE. LIM-domain-binding protein 1: a multifunctional cofactor that interacts with diverse proteins. EMBO Rep. 2003;4:1132-1137
20. Mukhopadhyay M, Teufel A, Yamashita T. et al. Functional ablation of the mouse Ldb1 gene results in severe patterning defects during gastrulation. Development. 2003;130:495-505
21. Xu X, Mannik J, Kudryavtseva E. et al. Co-factors of LIM domains (Clims/Ldb/Nli) regulate corneal homeostasis and maintenance of hair follicle stem cells. Dev Biol. 2007;312:484-500
22. Rhee H, Polak L, Fuchs E. Lhx2 maintains stem cell character in hair follicles. Science. 2006;312:1946-1949
23. Dey-Guha I, Malik N, Lesourne R. et al. Tyrosine phosphorylation controls nuclear localization and transcriptional activity of Ssdp1 in mammalian cells. J Cell Biochem. 2008;103:1856-1865
24. Tzchori I, Day TF, Carolan PJ. et al. LIM homeobox transcription factors integrate signaling events that control three-dimensional limb patterning and growth. Development. 2009;136:1375-1385
25. Zhao Y, Kwan KM, Mailloux CM. et al. LIM-homeodomain proteins Lhx1 and Lhx5, and their cofactor Ldb1, control Purkinje cell differentiation in the developing cerebellum. Proc Natl Acad Sci U S A. 2007;104:13182-13186
26. Hirrlinger PG, Scheller A, Braun C. et al. Temporal control of gene recombination in astrocytes by transgenic expression of the tamoxifen-inducible DNA recombinase variant CreERT2. Glia. 2006;54:11-20
27. Blanpain C, Fuchs E. Epidermal stem cells of the skin. Annu Rev Cell Dev Biol. 2006;22:339-373
28. Garcia MI, Ghiani M, Lefort A. et al. LGR5 deficiency deregulates Wnt signaling and leads to precocious Paneth cell differentiation in the fetal intestine. Dev Biol. 2009;331:58-67
29. Birchmeier C, Birchmeier W. Molecular aspects of mesenchymal-epithelial interactions. Annu Rev Cell Biol. 1993;9:511-540
30. Van der Flier LG, Sabates-Bellver J, Oving I. et al. The Intestinal Wnt/TCF Signature. Gastroenterology. 2007;132:628-632
Author contact
![]() Correspondence to: Heiner Westphal, M.D., Mailing address: Laboratory of Mammalian Genes and Development, NICHD, NIH, Bethesda, MD 20892-2790, USA. Phone: (301) 496-1855; Fax: (301) 402-0543; E-mail: hwnih.gov
Correspondence to: Heiner Westphal, M.D., Mailing address: Laboratory of Mammalian Genes and Development, NICHD, NIH, Bethesda, MD 20892-2790, USA. Phone: (301) 496-1855; Fax: (301) 402-0543; E-mail: hwnih.gov