Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Aurora-A and BRCA1
Aurora-A and mTOR
Aurora-A and Plk1
CONCLUSION
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2009; 5(7):758-762. doi:10.7150/ijbs.5.758 This issue Cite
Review
Physiological and Oncogenic Aurora-A Pathway
Toshiaki SAEKI1, Mutsuko OUCHI2, Toru OUCHI2 ![]()
1. Department of Breast Oncology, Saitama Medical School, Saitama, JAPAN
2. NUHS, Systems Biology Program, Pritzker School of Medicine, University of Chicago, Evanston, IL 60201, USA
Received 2009-11-2; Accepted 2009-11-24; Published 2009-11-26
Abstract
Aurora family of protein kinases have emerged as crucial factors of, not only mitosis and cytokinesis, but also human carcinogenesis. Among these family members is Aurora-A that is frequently overexpressed in varieties of human cancer. Both in vitro and in vivo studies demonstrated that Aurora-A induces tumorigenesis through genome instability. These studies have further shown that cell signaling cross-talk between Aurora-A and other cellular proteins are essential for fully-transformed phenotypes. This review summarizes recent progress of Aurora-A-associated carcinogenesis.
Keywords: Aurora-A, Plk1, mTOR, Cell Cycle, Checkpoint, Genome Instability, Phosphorylation
Introduction
Aurora-A was discovered in a screen for Drosophila mutations affecting the poles of the mitotic spindle function [1]. Transcription of the Aurora-A gene is cell-cycle regulated. Thus, the promoters of the Aurora-A gene contain specific elements (CDE/CHR sequences), which are responsible for transcription at G2 phase of the cell cycle [2-4]. It has been well documented that activation of Aurora-A is required for mitotic entry, centrosome maturation and separation, and G2 to M transition [5.6]. Interestingly, overexpression of Aurora-A is frequently observed in varieties of human cancer, including breast, colorectal, bladder, pancreatic, gastric, ovarian and esophageal cancer [7-12]. Overexpression of Aurora-A in fibroblasts resulted in cell transformation, supporting a notion that high levels of this protein are correlated to cell malignancy [13].
Potential roles of Aurora-A in cell transformation were also demonstrated from recent studies that this kinase phosphorylates a breast cancer tumor suppressor BRCA1 at Ser308 [14]. Both proteins are localized on centrosome at the beginning of mitosis [15], suggesting that signaling between these two proteins are crucial for regulation of normal cell cycle.
Recent studies added a couple of new insight of how Aurora-A induces cell transformation. Thus, in physiological conditions, Aurora-A and its activator collaborate with Plk1, Polo-like kinase 1, to initiate mitosis. On the other hand, in cells transformed with Aurora-A, mTOR pathway is activated [16,17].
In this review, differential roles of Aurora-A in cell cycle and cell transformation are discussed.
Aurora-A and BRCA1
The Aurora-A gene locus is located in the 20q13 chromosome region, which is frequently amplified in several different types of malignancies such as breast, colorectal, pancreatic, and bladder cancers [7-12]. In particular, 20q11-q13 regions are amplified in 40% of breast cancer cell lines as well as in 12-18% of primary tumors. Aurora-A protein is a member of the Ser/Thr kinase family, and recent studies have shown that the protein is involved in the G2-M checkpoint and commitment to mitosis [18-21]. Furthermore, it has been demonstrated that Aurora-A is inactivated by DNA damage at the end of the G2 phase, and overexpression of Aurora-A abrogates the G2 checkpoint, resulting in the amplified centrosome and cell transformation [18]. Significantly, Aurora-A is recruited to the centrosome early in the G2 phase and becomes phosphorylated and activated in the centrosome late in the G2 phase [6].
Deng's lab demonstrated that ~25% of mouse embryonic fibroblasts (MEFs) derived from the BRCA1 exon 11-deleted mice contains more than two centrosomes, leading to loss of the G2-M checkpoint and aneuploidy [21]. In addition, we and others found that BRCA1 is localized in the centrosome and binds to γ-tubulin [15,22,23].
From these observations, we discovered that BRCA1 functionally interacts with Aurora-A [14]. Interestingly, the aa1314-1863 region of BRCA1 was found to bind to Aurora-A directly. Mutagenic analysis and phospho-specific antibodies revealed that S308 of BRCA1 is normally phosphorylated by Aurora-A early in the M phase. Phosphorylation of BRCA1 S308 by Aurora-A was abolished by treating cells with ionizing radiation. Most interestingly, re-expression of the phospho-deficient form of BRCA1, S308N (N=Asn), in BRCA1-mutated MEFs resulted in growth arrest at the G2 phase without any cell stress, indicating that phosphorylation of BRCA1 S308 is necessary for the transition from G2 to M. These results indicate that an unphosphorylated form of BRCA1 at S308 is necessary for G2-M checkpoint. These are the first indications of the roles of the physiological levels of BRCA1 phosphorylation in regulating the cell cycle. Additional evidence of BRCA1/Aurora-A interaction is that Aurora-A regulates inhibition of centrosome microtubule nucleation mediated by BRCA1's E3 ligase activity [24].
Exogenous overexpression of Aurora-A in human cell culture was further studied by transfecting U2OS osteosarcome cell line [17]. Interestingly, in those cells, increased phosphorylation of BRCA1 S308 was not detected [unpublished results]. These results suggest that phosphorylation of BRCA1 S308 may not be necessary for cell transformation. Thus, perhaps there is substrate selectivity by Aurora-A in physiological and malignant conditions.
Aurora-A and mTOR
Most prominent discoveries from MMTV-Aurora-A transgenic mice are constitutive phosphorylation of mTOR Ser2448 and Akt Ser473 in developed mammary tumors [16]. Mammalian target of rapamycin (mTOR) is a protein serine/threonine kinase that controls a broad range of cellular processes. mTOR exists in two distinct complexes; mTOR complex 1 (mTORC1) and complex 2 (mTORC2). mTOR is phosphorylated at multiple sites, including Ser2448, Ser2481, Thr2446 and Ser1261. Phosphorylation at Ser2448 is mediated by p70 ribosomal S6 kinase (S6K) and occurs predominantly to mTOR in mTORC1 [25-27]. mTORC1 is composed of mTOR, mLST8, raptor and PRAS40. Its function is involved in many growth-related processes such as translation, ribosome biogenesis, transcription, autophagy and hypoxic adaptation, and is sensitive to rapamycin. mTORC2 shares both mTOR and mLST8 with mTORC1. Other unique components in mTORC2 are rapamycin-insensitive companion of mTOR (rictor), mammalian stress-activated protein kinase-interacting protein 1(mSIN1) and proline-rich repeat protein-5 (PRR5) or PRR5-like [28-33].
Two major functions have been ascribed to mTORC2, including regulation of Akt and cell cycle-dependent organization of actin cytoskeleton. mTORC2 phosphorylates Akt at Ser473 in its C-terminal hydrophobic motif, which, in conjunction with PDK1-mediated phosphorylation of Thr308, confers full activation of Akt [34]. mTORC2 regulates actin cytoskeleton through a mechanism that involves the small GTPases Rho and Rac, although the molecular details are largely still unclear [8,35]. Interestingly, mTORC2 phosphorylates PKC and SGK1 (serum- and glucocorticoid-induced protein kinase 1), and has been implicated in controlling cell size [36-39].
Elevated phosphorylation of mTOR Ser2448 and Akt Ser473 in Aurora-A transformed cells suggests that Aurora-A can potentially regulate two mTOR pathways, mTORC1 and mTORC2. Since chemical inhibitors of mTOR can abolish transformed phenotypes induced by Aurora-A [17], it is likely that either or both of mTORC1 and 2 is important for Aurora-A transformation.
Of note, mammary tumor development can be observed only after long latency in MMTV-Aurora-A mice [16]. In cell culture system of stable transfectants, cells in early passage numbers do not contain phosphorylated mTOR and Akt, but cells after long passage numbers they show up [17]. As one possible interpretation, overexpression of Aurora-A is not a strong driving force, but some additional events need to happen to accelerate Aurora-A's tumor development. When mTOR pathway is activated under this situation, cells now acquire the full-transforming ability.
Aurora-A and Plk1
Expression of Plk1 is cell cycle-dependent. Levels of the protein increases in late G2 phase, and decreases during mitotic exit [40]. Kinase activity well correlates with levels of the protein, thus it increases at G2/M transition and reaches at the maximal during mitosis. Similar to Plk1, levels of Aurora-A increase during G2 and reach at the maximal in early mitosis [13,41]. 'Activator' proteins for Aurora-A have been identified. Those include TPX2, Ajuba, PAK1, HEF1 and hBora [6,42-47]. Among these Aurora-A interactors, hBora expression peaks during G2 and decreases rapidly during mitosis [48,49]. It has also been shown that hBora forms a complex with Plk1 in G2 phase [48,50,51]. Aurora-A's binding to Bora and its subsequent phosphorylation are required for full activation of Aurora-A. In addition, both proteins are essential for Plk1 activation at the centrosome in G2 phase. In this model, it is thought that Bora binding to Plk1 induces allosteric effects that allow Aurora-A to the Plk1 T-loop of its kinase domain, where Aurora-A phosphorylates Thr210, leading to full activation of Plk1 [51,52].
It has been speculated that Aurora-A is a target for ubiquitination by CHFR, checkpoint with FHA and RING finger domains. CHFR regulates an early mitotic checkpoint, during prophase, in response to the disruption of microtubule formation or stabilization as assessed after treatment with microtubule inhibitors such as nocodazole, colcemid and taxanes [53]. Interestingly, Aurora-A was overexpressed in CHFR-null mouse embryonic fibroblasts and tissues, strongly supporting that CHFR ubiquitinates Aurora-A [54]. These studies have also demonstrated that the C-terminal cysteine-rich region of CHFR protein interacts with the N-terminus of Aurora-A protein. Similar results were shown from the other studies that siRNA-mediated depletion of CHFR in MCF10A cells resulted in overexpression of Aurora-A [55]. It has been demonstrated that, in HCT116 cells overexpressing CHFR, there was no change in levels of Aurora-A and localization of Aurora-A to the centrosomes, however, nocodazole-induced CHFR-mediated mitotic delay was associated with unphosphorylation of Aurora-A at Thr288 [56].
Studies of CHFR protein further supported functional interaction between Aurora-A and Plk1. It has been shown that overexpression of CHFR mutants which mimic unphosphorylated CHFR can decrease levels and kinase activity of Plk1 [57]. Interestingly, mouse embryonic fibroblasts from CHFR knockout mice express high levels of Plk1, suggesting that CHFR can ubiquitinate Plk1 to target it for degradation [54].
CONCLUSION
Given the high frequency of overexpression of Aurora-A in human cancers, inhibition of Aurora-A with small compounds looks like an attractive cancer-therapeutic strategy. Several compounds have been synthesized and are under clinical trials.
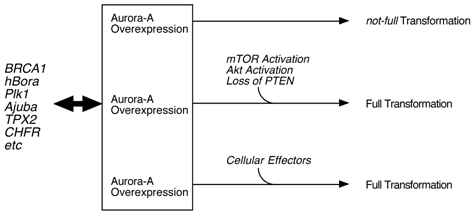
Classical cell biology assay, such as transfection of normal fibroblasts with Aurora-A cDNA, resulted in cell transformation. Transgenic model targeting Aurora-A in mammary glands also support a notion that this kinase is oncogenic. However, quite long latency and low incidence of tumor development in these mice suggest that Aurora-A alone is not a strong driving force of malignancy, but other hits need to occur for full transformation [16]. Thus, it is possible that inhibition of Aurora-A with compounds may not be sufficient for killing Aurora cancer cells. Chromosome instabilities observed in those mammary tumors support this hypothesis that activation or inactivation of 'effector proteins' due to the gross alteration of chromosome structure may result in accelerating tumorigenesis (Fig. 1). In that sense, simultaneous inhibition of this pathway(s) as well as Aurora-A might be necessary for the better treatment of patients. For example, mTOR/Akt pathway might be the one which is crucial for Aurora-A tumorigenesis.
Model of Aurora-A cell transformation. Physiological regulation of Aurora-A kinase activity is by BRCA1, hBora, Ajuba, TPX2 an dPlk1 etc, however, cell transformation by Aurora-A requires additional oncogenic events, such as constitutive activation of mTOR/Akt pathway and loss of PTEN tumor suppressor [17].
Acknowledgements
I would like to thank members of the Ouchi laboratory and Michael Meyer for critical reading and suggestions. Supported by the NIH (CA79892 and CA90631), Susan G. Komen Foundation and AVON foundation.
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
1. Glover G.M. et al. Mutations in aurora prevent centrosome separation leading to the formation of monopolar spindles. Cell. 1995;81:95-105
2. Kimura M. et al. Cell cycle-dependent expression and spindle pole localization of a novel human protein kinase, Aik, related to Aurora of Drosophila and yeast lpl1. J. Bio. Chem. 1997;272:13766-13771
3. Tanaka M. et al. Cell-cycle-dependent regulation of human Aurora A transcription is mediated by periodic repression of E4TF1. J. Biol. Chem. 2002;277:10719-10726
4. Kimura M. et al. Cell-cycle-dependent regulation of the human Aurora B promoter. Biochem. Biophys. Res. Commun. 2004;316:930-936
5. Fu J. et al. Roles of Aurora kinases in mitosis and tumorigenesis. Mol Cancer Res. 2007;5:1-10
6. Hirota T. et al. Aurora-A and an interacting activator, the LIM protein Ajuba, are required for mitotic commitment in human cells. Cell. 2003;114:585-598
7. Tanaka T. et al. Centrosomal kinase AIK1 is overexpressed in invasive ductal carcinoma of the breast. Cancer Res. 1999;59:2041-2044
8. Nishida N. et al. High copy amplification of the Aurora-a gene is associated with chromosomal instability phenotype in human colorectal cancers. Cancer Biol Ther. 2007;6:525-533
9. Compérat E. et al. Aurora-A/STK-15 is a predictive factor for recurrent behavior in non-invasive bladder carcinoma: a study of 128 cases of non-invasive neoplasms. Virchows Arch. 2007;450:419-424
10. Li D. et al. Overexpression of oncogenic STK15/BTAK/Aurora A kinase in human pancreatic cancer. Clin Cancer Res. 2003;9:991-997
11. Lassmann S. et al. Predictive value of Aurora-A/STK15 expression for late stage epithelial ovarian cancer patients treated by adjuvant chemotherapy. Clin Cancer Res. 2007;13:4083-4091
12. Tong T. et al. Overexpression of Aurora-A contributes to malignant development of human esophageal squamous cell carcinoma. Clin Cancer Res. 2004;10:7304-7310
13. Bischoff J.R. et al. A homologue of Drosophila aurora kinase is oncogenic and amplified in human colorectal cancers. EMBO J. 1998;17:3052-3065
14. Ouchi M. et al. BRCA1 phosphorylation by Aurora-a in the regulation of G2 to M transition. J Biol Chem. 2004;279:19643-19648
15. Okada S, Ouchi T. Cell cycle differences in DNA damage-induced BRCA1 phosphorylation affects its subcellular localization. J Biol Chem. 2003;278:2015-2020
16. Wang X. et al. Overexpression of aurora kinase A in mouse mammary epithelium induces genetic instability preceding mammary tumor formation. Oncogene. 2006;25:7148-7158
17. Taga M. et al. Essential Roles of mTOR/Akt Pathway in Aurora-A Cell Transformation. Int J Biol Sci. 2009;19:444-450
18. Marumoto T. et al. Roles of aurora-a kinase in mitotic entry and G2 checkpoint in mammalian cells. Genes Cell. 2002;7:1173-1182
19. Meraldi P. et al. Aurora-A overexpression reveals tetraploidization as a major route to centrosome amplification in p53-/- cells. EMBO J. 2002;21:483-492
20. Anand S. et al. Aurora-A amplification overrides the mitotic spindle assembly checkpoint, inducing resistance to taxol. Cancer Cell. 2003;3:51-62
21. Xu X. et al. Centrosome amplification and a defective G2-M cell cycle checkpoint induce genetic instability in BRCA1 exon 11 isoform-deficient cells. Mol Cell. 1999;3:389-395
22. Hsu L.C, White R.L. BRCA1 is associated with the centrosome during mitosis. Proc Natl Acad Sci USA. 1998;95:12983-12988
23. Hsu L.C. et al. Identification of a gamma-tubulin-bondong domain of BRCA1. Cancer Res. 2001;61:7713-7718
24. Sankaran S. et al. Aurora-A kinase regulates breast cancer-associated gene 1 inhibition of centrosome-dependent microtubule nucleation. Cancer Res. 2007;67:11186-11194
25. Chiang G.G. et al. Phosphorylation of mammalian target of rapamycin (mTOR) at Ser-2448 is mediated by p70S6 kinase. J Biol Chem. 2005;280:25485-25490
26. Holz M.K, Blenis J. Identification of S6 kinase 1 as a novel mammalian target of rapamycin (mTOR)-phosphorylating kinase. J Biol Chem. 2005;280:26089-26093
27. Copp J. et al. TORC-specific phosphorylation of mammalian target of rapamycin (mTOR): phospho-Ser2481 is a marker for intact mTOR signaling complex 2. Cancer Res. 2009;69:1821-1827
28. Jacinto E. et al. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell. 2006;127:125-137
29. Pearce L.R. et al. Identification of protor as a novel rictor-binding component of mTOR complex-2. Biochem J. 2007;405:513-522
30. Sabassov D.D. et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway tha regulates the cytoskeleton. Curr Bio. 2004;14:1296-1302
31. Thedieck K. et al. PRAS40 and PRR5-like protein are new mTOR interactors that regulate apoptosis. PLoS ONE. 2007;2:1217
32. Woo S.Y. et al. PRR5, a novel component of mTOR complex 2, regulates platelet-derived growth factor receptor beta expression and signaling. J Biol Chem. 2007;282:25604-25612
33. Yang Q. et al. Identification of Sin1 as an essential TORC2 component required for complex formation and kinase activity. Genes Dev. 2006;20:2820-2832
34. Sarbassov D.D. et al. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098-1101
35. Jacinto E. et al. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004;6:1122-1128
36. Facchinetti V. et al. The mammalian target of rapamycin complex 2 controls folding and stability of Akt and protein kinase C. EMBO J. 2008;27:1932-1943
37. Ikenoue T. et al. Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signaling. EMBO J. 2008;27:1919-1931
38. Garcia-Martinez J.M, Alessi D.R. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1). Biochem J. 2008;416:375-385
39. Rosner M. et al. Functional interaction of mTOR complexes in regulating mammalian cell size and cell cycle. Hum Mol Genet. 2009;18:3298-3310
40. Petronczki M. et al. Polo on the Rise-from mitotic entry to cytokinesis with Plk1. Dev Cell. 2008;14:646-659
41. Barr A.R, Gergely F. Aurora-A: the maker and breaker of spindle poles. J Cell Sci. 2007;120:2987-2996
42. Wittmann T. et al. TPX2, a novel xenopus MAP involved in spindle pole organization. J Cell Biol. 2000;149:1405-1418
43. Kufer T.A. et al. Human TPX2 is required for targeting Aurora-A kinase to the spindle. J Cell Biol. 2002;158:617-623
44. Eyers P.A. et al. A novel mechanism for activation of the protein kinase Aurora A. Curr Biol. 2003;13:691-697
45. Bayliss R. et al. Structural basis of Aurora-A activation by TPX2 at the mitotic spindle. Mol Cell. 2003;12:851-862
46. Zhao Z.S. et al. The GIT-associated kinase PAK targets to the centrosome and regulates Aurora-A. Moll Cell. 2005;20:237-249
47. Pugacheva E.N. et al. HEF1-dependent Aurora A activation induces disassembly of the primary cilium. Cell. 2007;129:1351-1363
48. Chan E.H. et al. PLK1 regulates mitotic Aurora A function through βTrCP-dependent degradation of hBora. Chromosoma. 2008;117:457-469
49. Seki A. et al. Plk1- and bTrCP-dependent degradation of Bora controls mitotic progression. J Cell Biol. 2008;181:65-78
50. Macurek L. et al. Polo-like kinase-1 is activated by Aurora A to promote checkpoint recovery. Nature. 2008;455:119-123
51. Seki A. et al. Bora and the kinase Aurora A cooperatively activate the kinase Plk1 and control mitotic entry. Science. 2008;320:1655-1658
52. Macurek L. et al. Aurora-A and hBora jointhe game of polo. Cancer Res. 2009;69:4555-4558
53. Scolnick D.M, Halazonetis T.D. Chfr defines a mitotic stress checkpoint that delays entry into metaphase. Nature. 2000;406:430-435
54. Yu X. et al. Chfr is required for tumor suppression and Aurora A regulation. Nat Genet. 2005;37:401-406
55. Privette L.M. et al. Loss of CHFR expression in mammary epithelial cells causes genomic instability by disrupting the mitotic spindle assembly checkpoint. Neoplasia. 2008;10:643-652
56. Summers M.K. et al. The CHFR mitotic checkpoint protein delays cell cycle progression by excluding cyclin B1 from the nucleus. Oncogene. 2005;24:2589-2598
57. Shtivelman E. Promotion of mitosis by activated protein kinase B after DNA damage involves polo-like kinase 1 and checkpoint protein CHFR. Mol Cancer Res. 2003;1:959-969
Author contact
![]() Correspondence to: Toru Ouchi, University of Chicago, 1001 University Place, Evanston, IL 60201. Tel: 224.364.7687; Fax: 224.364.7402; Email: touchiuchicago.edu
Correspondence to: Toru Ouchi, University of Chicago, 1001 University Place, Evanston, IL 60201. Tel: 224.364.7687; Fax: 224.364.7402; Email: touchiuchicago.edu