Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2010; 6(7):682-690. doi:10.7150/ijbs.6.682 This issue Cite
Short Research Communication
Liver Steatosis and Increased ChREBP Expression in Mice Carrying a Liver Specific SIRT1 Null Mutation under a Normal Feeding Condition
Rui-Hong Wang, Cuiling Li, Chu-Xia Deng ![]()
Genetics of Development and Disease Branch, 10/9N105, National Institute of Diabetes, Digestive and Kidney Diseases, National Institutes of Health, Bethesda, Maryland, MD 20892, USA
Received 2010-11-9; Accepted 2010-11-16; Published 2010-11-16
Abstract
SIRT1, a homolog of yeast Sir2, is a type III NAD+ dependent histone and protein deacetylase. Previous studies of mice carrying liver specific deletion of exon 4 of the Sirt1 gene revealed opposite responses of mutant mice to a high-fat diet in terms of fatty liver formation, which obscures the function of SRIT1 in liver development and lipid metabolism. To investigate this, we deleted exons 5 and 6 of Sirt1 in the liver by using a Cre-loxP approach. Western blot using an antibody to N-terminal SIRT1 does not detect a truncated protein in the liver of the mutant mice (Sirt1flox5-6/flox5-6;Alb-Cre), suggesting a null mutation for SIRT1 is generated in the liver. Unlike the previously reported phenotypes, the Sirt1flox5-6/flox5-6;Alb-Cre mice develop fatty liver under a normal feeding condition. The disease starts at two months of age and incidence increases as the animals become older, affecting 78% of them when they are over one year of age. We showed that the steatosis is accompanied by altered expression of a number of genes, including increased expression of ChREBP, which acts as one of the central determinants of lipid synthesis in the liver. This data uncovers an important role of SIRT1 in regulating lipid metabolism in the liver, and the SIRT1 mutant mice may serve as an animal model for studying human fatty liver disease and facilitate the development of effective therapeutic approach for the disease.
Keywords: SIRT1, SREBP-1c, ChREBP, fatty liver, mice
Introduction
SIRT1 is a founding member of a family of 7 Sirtuins (SIRT1-7), all of which possess NAD+ dependent deacetylase activity and/or mono-ADP ribosylase activity [1-4]. Previous studies revealed important functions of these genes, with a focus largely on SIRT1, in many biological processes including cell growth, apoptosis, senescence, neuronal protection, adaptation to calorie restriction, organ metabolism and disease, DNA damage response and repair, and tumorigenesis [1-3, 5-11]. The majority of mutant mice carrying targeted disruptions of SIRT1 die during embryonic development or at neonatal stages [8, 12, 13]; therefore tissue-specific approaches have been employed to overcome the lethality and to study SIRT1 function in adult animals. However, mice carrying a liver specific disruption of SIRT1 by two different groups exhibited opposite phenotypes. In both studies, the investigators deleted exon 4 of the Sirt1 gene by a Cre-loxP-mediated approach using an albumin-Cre transgene to generate liver-specific SIRT1 mutant mice (Sirt1flox4/flox4;Alb-Cre). The Sirt1flox4/flox4;Alb-Cre mice appeared normal under regular feeding conditions. However, upon feeding with a high fat diet, the mutant mice in the two different studies exhibited either accelerated [14] or attenuated [15] fatty liver formation when compared with controls. Liver steatosis is a complex disease and may be affected by a variety of intrinsic and environmental factors [16, 17]. While the causes for this discrepancy are currently unclear, it is noted that the Cre-mediated recombination generates a truncated protein in the liver of the Sirt1flox4/flox4;Alb-Cre mice [14, 15], which may further complicate the situation.
While investigating SIRT1 function, we generated a mutant strain carrying a Cre-mediated deletion of exons 5 and 6 of the Sirt1 gene in the liver. Unlike previously-reported phenotypes of the Sirt1flox4/flox4;Alb-Cre mice, our mutant mice (Sirt1flox5-6/flox5-6;Alb-Cre, or Sirt1LKO) gradually developed fatty liver disease even in the absence of a high-fat diet. Fatty liver developed when the animals were two months of age, and gradually increased in older animals. The steatosis is accompanied by increased expression of carbohydrate responsive element binding protein (ChREBP), which is a major regulator for lipid synthesis [16]. Thus, our data reinforced a critical role of SIRT1 in regulating lipid metabolism in the liver.
Results and Discussion
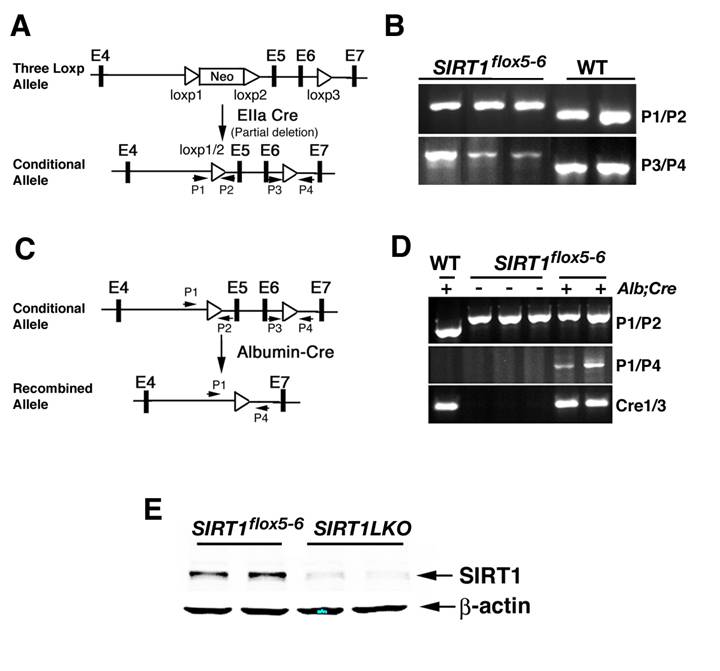
To generate a Sirt1 conditional mutant allele, we started with an existing mutant allele that carries a ploxPneo gene in the intron 4 and a third loxP in intron 6 of the Sirt1 locus [8]. Cre-mediated complete deletion of loxP floxed sequence created a null allele of Sirt1, leading to middle gestation lethality [8]. Using a strategy for screening Cre-mediated incomplete recombination in mice [18], we successfully deleted the ploxPneo specifically while leaving the third loxP intact, thus generating an allele that can be used for conditional knockout of SIRT1 (Sirt1flox5-6) (Fig. 1A, B). To study functions of SIRT1 in the liver, we generated liver- specific knockout of Sirt1 (Sirt1LKO) by crossing the Sirt1flox5-6 mice with mice that carry an album promoter driven Cre transgene (Alb-Cre) [19] (Fig. 1C, D). With an antibody against N-terminus of SIRT1, we did not detect any truncated or short isoforms of SIRT1 (Fig. 1E), indicating that our Sirt1LKO mouse is a liver SIRT1 null animal model.
Generation of liver specific SIRT1 knockout mice. (A) To generate a Sirt1 conditional mutant allele, an existing mutant allele that carries a ploxPneo gene in the intron 4 and a third loxP in the intron 6 of the Sirt1 locus was used (upper panel). After breeding mice carrying the 3-loxP allele of Sirt1 gene with EIIa-Cre transgenic mice, Cre-mediated recombination between loxP1 and loxP2 was identified using primers 1 and 2 (P1 and P2) to screen for offspring that has the neo gene deleted while still keeping the loxP3 (lower panel). (B) PCR analysis to identify offspring that carry Cre-mediated recombination between loxP1 and loxP2 (using primers 1 and 2: ~700 bp for Sirt1flox5-6 allele and ~640 bp for wild type allele) while keeping loxP3 (using primers 3 and 4: ~420 bp for mutant allele and ~360 bp for wild type allele). The first three lanes are liver DNA from Sirt1 flox5-6/flox5-6 mice and these primers only detect the 700 pb and 420 bp bands, while the last two lanes are samples from wild type mice and only detect the 640 bp and 360 bp bands. (C) Sirt1 flox5-6/flox5-6 mice were crossed with mice carrying an album promoter driving Cre transgene (Alb-Cre) to generate liver specific SIRT1 knockout mice (Sirt1flox5-6/flox5-6;Alb-Cre, or Sirt1LKO). (D) Sirt1LKO mice were validated by PCR analysis of genomic DNA isolated from liver tissue with primers P1/P4 to demonstrate the deletion of exon 5 and exon 6. (E) Western blot with an antibody against SIRT1 N-terminus to demonstrate that there is no truncated SIRT1 in our SIRT1 liver specific knockout mice. The samples were from 2 months old male mice. More than 20 pairs of animals were analyzed. Data from 2 pairs are shown here.
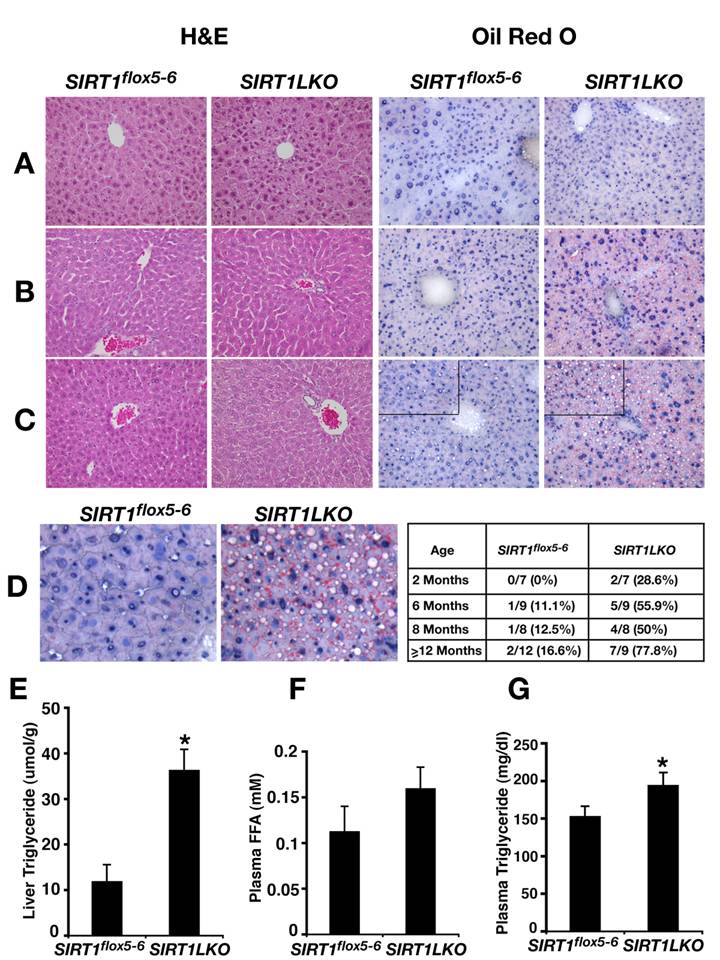
The livers from Sirt1LKO mice were examined at 4 time points, i.e. 2 months, 6 months, 8 months or over 12 months after birth. At 2 months, although the livers of Sirt1LKO mice were histologically normal (Fig. 2A), some (2/7) of them have started to accumulate lipid droplet as revealed by Oil-Red O staining (data not shown). At 6 months of age, 56% (5/9) of Sirt1LKO mice displayed fat liver as revealed by Oil-Red O staining (Fig. 2B). The frequency of fatty liver was also significantly increased as the animals aged, reaching 78% (7/9) when they were over 1 year of age (Fig. 2C,D). In contrast, only 17% (2/12) of control animals exhibited fatty liver during the same period of time (Fig. 2D). Consistent with the increased lipid deposition, we detected marked increase of triglyceride (TG) in the mutant liver (Fig. 2E). Although free fatty acid (FFA) in serum is only slightly increased (Fig. 2F), significant higher level of TG content was also observed in the serum of the mutant mice (Fig. 2G). Altogether, these data indicated that absence of SIRT1 in the liver causes fatty liver even without feeding these mice with high-fat diet.
SIRT1 liver specific knockout causes liver steatosis. (A-C) H&E staining and Oil Red O staining of 2 months (A), 6 months (B) and 14 months (C) old male mice liver respectively. (D) Oil Red O staining of 14 months old male liver with higher magnification. The inserted table is the summary of fatty liver cases at different age among control (Sirt1flox5-6) and liver specific SIRT1 knockout (Sirt1LKO) males. (E-G) Liver triglyceride (TG) content (E), plasma free fatty acid (FFA) amount (F) and plasma triglyceride (TG) content (G) of 9 months old male mice (n=11). *p<0.05.
Altered signaling in a number of biological pathways, including glycolysis, β oxidation, fatty acid synthesis, TG synthesis and fat uptake, could cause the liver steatosis [16, 20]. To understand the underlying mechanism(s) for the disease, we studied gene expression in Sirt1LKO mice. We reasoned that whatever the signaling pathway changed, if it is intrinsic to SIRT1 deletion in the liver, it should take place when the mice were young. Thus, liver RNA was extracted from 2-month mice and real time RT-PCR was performed on sets of genes that are involved in the pathways mentioned above. Our examination of two major glycolytic genes, glucokinase (Gk) and liver-specific pyruvate kinase (Lpk), did not detect an obvious alteration in their expression (Fig. 3A). The investigation of genes that are involved in β-oxidation, esterification and fat uptake did not detect any evident change either (Fig. 3A). Next, we examined the expression of genes that are involved in de novo lipid synthesis. We found the mutant mice had elevated expression of a number of genes, including fatty acid synthase (FAS), acetyl-CoA carboxylase (ACC), and elongase of long chain fatty acids family 6 (ELOVL6) (Fig. 3B). The liver samples from older Sirt1LKO mice also displayed increased expression levels of these genes (Fig. 3C), indicating that the absence of SIRT1 elevated de novo lipid synthesis.
SIRT1 liver specific knockout leads to increased expression of lipid metabolism genes. (A) The expression of genes involved in glycolysis, β-oxidation, esterificaiton and fat uptake is not changed in the livers of Sirt1LKO male mice at 2 months of age (n≥6). (B) Sirt1LKO mice display elevated expression of FAS, ACC1 and ELOVL6 at 2 months (n≥6). *p<0.05. (C) At 6 months of age, the expression of FAS, ACC1, ELOVL6 and SCD1 remains increased in Sirt1LKO mice (n≥6). *p<0.05. (D-E) The expression of SREBP-1c is down regulated in Sirt1LKO mice at 2 months of age at both mRNA level (C) and protein level (D). (F) In vitro, deletion of SIRT1 (compare last two bars) or knocking down SIRT1 by siRNA (the first two bars) reduces mRNA level of SREBP-1c, while over expressing SIRT1 (middle two bars) increases SREBP-1c mRNA level. * p<0.01
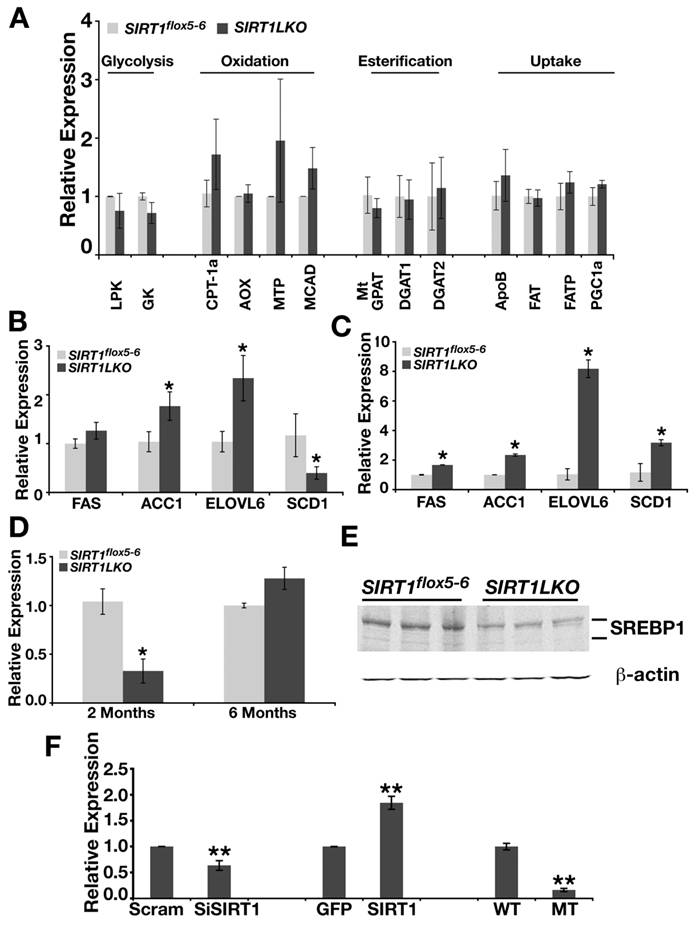
Hepatic de novo lipid synthesis in general is under the control of two major transcriptional factors: sterol regulatory element binding protein-1c (SREBP-1c) and carbohydrate response element binding protein (ChREBP) [16, 21]. Evidence is emerging that both SREBP-1c and ChREBP are positive regulators for FAS, ACC1 and ELOVL6. These two master regulators have a fundamental difference: SREBP-1c responds to insulin signaling while ChREBP is totally tied with the level of hepatic glucose. To our surprise, we found that SREBP-1c was reduced in the liver of Sirt1LKO mice at 2 months of age dramatically at both mRNA level and protein level (Fig. 3D, E). To further determine the potential relationship between SRIT1 and SREBP-1c, we quantified mRNA level of SREBP-1c in SIRT1 knockout, SIRT1 knockdown by siRNA and over-expressed conditions in mouse embryonic fibroblast cells (MEFs) (Fig. 3F). Both deletion and knockdown of SIRT1 decreased the expression level of SREBP-1c, while over expression of SIRT1 up-regulated the mRNA level of SREBP-1c (Fig. 3F). These data indicates that insulin dependent lipid synthesis pathway is impaired in Sirt1LKO mice due to the reduced expression of SREBP-1c. At 6 months of age, although levels of SREBP-1c mRNA increased to a level slightly higher that those of controls (Fig. 3D), this might be caused by a feedback mechanism from sensing overall signaling after long-term deficiency of SIRT1. This phenomenon has been manifested well by the expression of stearoyl-CoA desaturase-1 (SCD-1), which is a primary target of SREBP-1c [22]. In Sirt1LKO mice, at two month of age, when SREBP-1c is dramatically down regulated, SCD-1 mRNA level is significantly reduced compared with control (Fig. 3B). However, at 6 month of age, when SREBP-1c displayed a tendency to be induced, SCD-1 mRNA level becomes significantly increased (Fig. 3C).
Since CREB, C/EBP proteins and PPARγ can regulate genes in SREBP-1c pathway, we also examined the expression level of CREB, C/EBPα, C/EBPβ, C/EBPδ and PPARγ in 2-month and 6-month old Sirt1LKO mice livers. As shown in Fig. 4A, even though CREB, and C/EBPβ, C/EBPδ displayed elevated mRNA level at 2 months of age, but this trend did not stay when the mutant mice got older (6 months). Thus, we excluded them as primary effectors for increased lipid synthesis in Sirt1LKO mice.
SIRT1 liver-specific knockout leads to increased expression of ChREBP. (A) The expression of CREB, CEBPβ and CEBPδ is up-regulated in the liver of Sirt1LKO male mice at 2 months of age (n≥6). *p<0.05 (B) Sirt1LKO mice display consistent elevated mRNA level of ChREBP at both 2 months and 6 months of age (n≥6). *p<0.05 . (C) At 2months of age, Sirt1LKO mice contain higher ChREBP protein level. (D) In vitro, deletion of SIRT1 (last two bars) or knocking down SIRT1 by siRNA (first two bars) increased the mRNA level of ChREBP, while over expressing SIRT1 (middle two bars) reduces ChREBP mRNA level. * p<0.01. (E-G) Chromatin Immunoprecipitation (ChIP) assay demonstrates deletion of SIRT1 leads to increased acetylation of histone H3K9 and histone H4K16 on 3 fragments (365-539, 984-1173 and 1614-1752) upstream of starting codon ATG of ChREBP promoter.
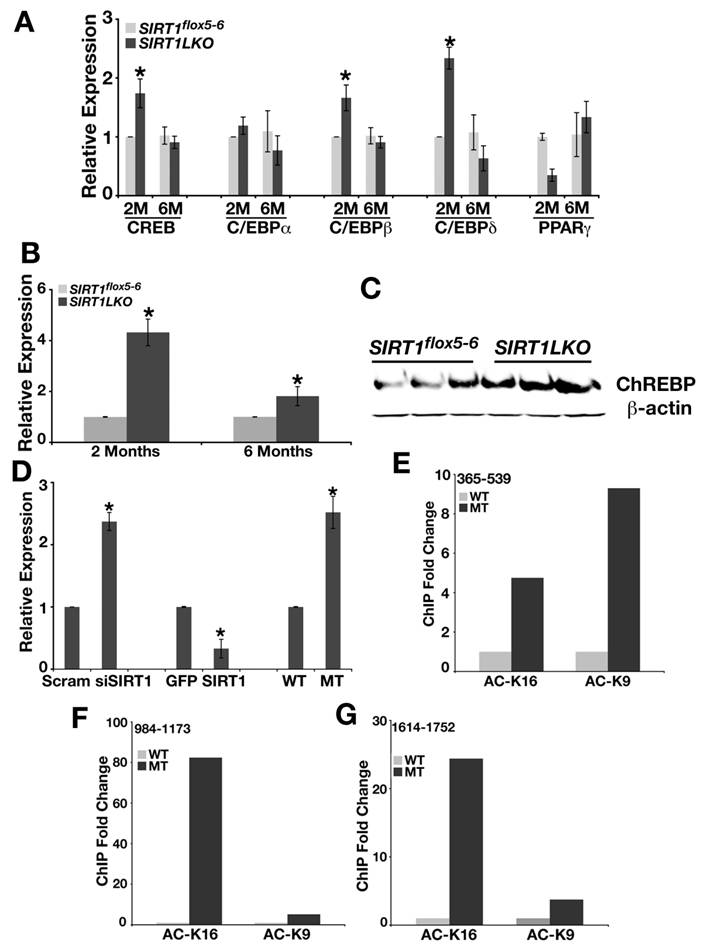
Next, we studied ChREBP, another major transcriptional factor that regulates expression of genes involved in fat metabolism and may be responsible for the increased expression of FAS, ACC1 and ELOVL6 [23, 24]. We detected up-regulated expression of ChREBP in the liver of both 2 months and 6 months old Sirt1LKO mice at mRNA level (Fig. 4B) and protein level (Fig. 4C). Studies in MEFs demonstrated that deletion of SIRT1 by either knockout or knockdown increased the expression of ChREBP by 2-3 folds, while over-expression of SIRT1 reduced its level by 2 folds (Fig. 4D). Chromatin immunoprecipitation (ChIP) with antibodies against acetylated histone H3 lysine 9 (Ac-H3K9) and histone H4 lysine 16 (Ac-H4K16) revealed elevated levels of acetylation on H3K9 and H4K16 (Fig. 4E-G). Thus, ChREBP promoter carries an open structure with increased transcription possibility, which is consistent with the absence of SIRT1 deacetylase.
In summary, we have constructed a liver specific knockout SIRT1 model by deleting exon5 and exon6. Unlike the previously described allele [14, 15], the mutant mice created here do not contain a smaller SIRT1 product. Although potential function of this product has not been tested, it remains possible that this protein, combined with different experimental conditions, may modify phenotypes. Nonetheless, the Sirt1LKO mice generated in our study develop into fatty liver that is accompanied by increased expression of ChREBP. The liver steatosis is developed under normal feeding condition when they are relatively young and the symptom is worsened while the animals are getting older. This mimics the human non-alcoholic fatty liver disease (NAFLD), which is the most common cause of liver dysfunction worldwide in human and it affects about 20 million people in USA [25, 26]. Thus, the Sirt1LKO mice may serve as an animal model for understanding mechanism of liver steatosis and may facilitate the development of effective therapeutic approaches for this disease.
Materials and Method
Generation of Sirt1 floxed allele and liver specific Sirt1 deletion animals
Mice that carry 3 loxP sites (a ploxPneo [27] in intron 4 and the third loxP in intron 6) were crossed with EIIa-Cre transgenic mice [28] to delete the ploxPneo using a protocol as described [18]. The resulting mice carrying the floxed exon 5 and exon 6 of the Sirt1 gene were bred with Albumin-Cre transgenic mice [19] to obtain Sirt1+/flox5-6;Alb-Cre strain. Afterwards, males and females of Sirt1+/flox5-6;Alb-Cre were intercrossed to generate Sirt1flox5-6/flox5-6;Alb-Cre mice that carry a liver specific deletion of SIRT1 (Sirt1LKO) and control mice with various genotypes. These mice are in a mixed genetic background of 129/FVB/Black Swiss and are genotyped with the following primers:
Floxed Sirt1 gene: p1- 5' CTT CCT TGC CAC AGT CAC TC 3'; p2- 5' CAT CTA AAC TTT GTT GGC TGC 3'; p3- 5' GTG GAG GTC AGA AGA TCA ACC 3'; p4- 5' CAG ACA TGC AGG CAA ACA CCC 3'. Alb-Cre: Forward- 5' CCT GTT TTG CAC GTT CAC CG 3'; Reverse- 5' ATG CTT CTG TCC GTT TGC CG 3'. Recombined allele was genotyped with p1 and p4. All experiments were approved by the Animal Care and Use Committee of the National Institute of Diabetes, Digestive and Kidney Diseases (ACUC, NIDDK).
Triglyceride (TG) and Free Fatty Acid (FFA) measurement
Liver was used for triglyceride content measurements with RA method. Serum TG and FFA were measured by Metabolic Core Facility of NIDDK.
Western blot analysis
Western blot was carried out with Licor (Lincoln, Nebraska) utilizing antibodies against SIRT1 (Upstate), SREBP-1c (Abcam) and ChREBP (Santa Cruz).
QRT-PCR
Liver tissue was dissected and immediately put into RNALater Solution (Ambion Inc.). Total RNA was isolated with STAT-60TM (TEL-TEST, Inc.) from these liver tissues and cleaned up with RNeasy Mini Kit (Qiagen). cDNA was synthesized with Cells-to-cDNA TMII (Ambion Inc.). Quantitative RT-PCR was performed using a SYBR green PCR Master Mix (Applied Biosystems) and 7500 Real Time PCR (Applied Biosystems). The primers are as following:
SREBP-1 F: 5' AAG CAA ATC ACT GAA GGA CCT GG 3', R 5' AAA GAC AAG GGG CTA CTC TGG GAG 3'; 18S(RAT, MUS HU) F: 5' AGT CCC TGC CCT TTG TAC ACA 3', R: 5' CGA TCC GAG GGC CTC ACT A 3'; ChREBP F: 5' GCATCCTCATCCGACCTTTA 3', R: 5' GATGCTTGTGGAAGTGCTGA 3'.
Histology and oil red O staining
Tissue was fixed in 10% neutral-buffered formalin (VWR) at 4oC overnight, dehydrated through a graded alcohol series, xylene and paraffin, and then embedded in paraffin. Sections of 5 μm were prepared for H&E. For Oil Red O staining, liver tissues, which were frozen in OCT compounds, were cut at 5 μm, mounted on slides and allowed to dry for 1-2 hrs. The sections were fixed with 10% formalin for 10 min and then the slides were rinsed with PBS (PH 7.4). After air dry, the slides were placed in 100% propylene glycol for 2 min, and stained in 0.5% Oil Red O solution in propylene glycol for 30 min. The slides were transferred to an 85% propylene glycol solution for 1 min., rinsed in distilled water for 2 changes, and processed for hematoxylin counter staining.
Transfection
Mammalian expression vector pUSE-SIRT1 was purchased from Upstate. Mouse SIRT1 siRNAs were used as previously (Mol Cell paper). All the transfections were performed with LipofectaminTM 2000 (invitrogen) on MEF cells.
Chromatin immunoprecipitation (ChIP) analysis
For ChIP analysis of cultured cells, cells were cross-linked with a final concentration of 1% formaldehyde for 15 min at RT, quenched with 125 mM of glycine for 5 min, and were suspended with 1 ml of lysis buffer (50 mM Tris-Cl at pH7.5, 150mM NaCl, 5mM EDTA, 1% Triton X-100, 0.5% NP-40) supplemented with protease inhibitors for 1 min at 4°C, and sonicated extensively. After centrifugation, the supernatant was used for ChIP with antibodies against Ac-H3K9 or AC-H4K16 together with rabbit IgG as a control. Final immunoprecipitated DNA was analyzed by Real Time PCR.
Acknowledgements
We thank members of Deng lab for their helpful discussion of this work and Dr. Cristine Chisholm for critical reading of this manuscript. This work was supported by the Intramural Research Program of the National Institute of Diabetes, Digestive and Kidney Diseases, National Institutes of Health, USA.
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. Saunders LR, Verdin E. Sirtuins: critical regulators at the crossroads between cancer and aging. Oncogene. 2007;26:5489-5504
2. Lavu S, Boss O, Elliott PJ. et al. Sirtuins--novel therapeutic targets to treat age-associated diseases. Nat Rev Drug Discov. 2008;7:841-853
3. Finkel T, Deng CX, Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature. 2009;460:587-591
4. Guarente L. Sirtuins in aging and disease. Cold Spring Harb Symp Quant Biol. 2007;72:483-488
5. Vaquero A, Scher M, Erdjument-Bromage H. et al. SIRT1 regulates the histone methyl-transferase SUV39H1 during heterochromatin formation. Nature. 2007;450:440-444
6. Deng CX. SIRT1, is it a tumor promoter or tumor suppressor? Int J Biol Sci. 2009;5:147-152
7. Wang RH, Zheng Y, Kim HS. et al. Interplay among BRCA1, SIRT1, and Survivin during BRCA1-associated tumorigenesis. Mol Cell. 2008;32:11-20
8. Wang RH, Sengupta K, Li C. et al. Impaired DNA damage response, genome instability, and tumorigenesis in SIRT1 mutant mice. Cancer Cell. 2008;14:312-323
9. Kim HS, Patel K, Muldoon-Jacobs K. et al. SIRT3 is a mitochondria-localized tumor suppressor required for maintenance of mitochondrial integrity and metabolism during stress. Cancer Cell. 2010;17:41-52
10. Jacobs KM, Pennington JD, Bisht KS. et al. SIRT3 interacts with the daf-16 homolog FOXO3a in the Mitochondria, as well as increases FOXO3a Dependent Gene expression. Int J Biol Sci. 2008;4:291-299
11. Byles V, Chmilewski LK, Wang J. et al. Aberrant cytoplasm localization and protein stability of SIRT1 is regulated by PI3K/IGF-1R signaling in human cancer cells. Int J Biol Sci. 2010;6:599-612
12. McBurney MW, Yang X, Jardine K. et al. The mammalian SIR2alpha protein has a role in embryogenesis and gametogenesis. Mol Cell Biol. 2003;23:38-54
13. Cheng HL, Mostoslavsky R, Saito S. et al. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc Natl Acad Sci U S A. 2003;100:10794-10799
14. Chen D, Bruno J, Easlon E. et al. Tissue-specific regulation of SIRT1 by calorie restriction. Genes Dev. 2008;22:1753-1757
15. Purushotham A, Schug TT, Xu Q. et al. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab. 2009;9:327-338
16. Postic C, Girard J. Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: lessons from genetically engineered mice. J Clin Invest. 2008;118:829-838
17. Savage DB, Semple RK. Recent insights into fatty liver, metabolic dyslipidaemia and their links to insulin resistance. Curr Opin Lipidol. 2010;21:329-336
18. Xu X, Li C, Garrett-Beal L. et al. Direct removal in the mouse of a floxed neo gene from a three-loxP conditional knockout allele by two novel approaches. Genesis. 2001;30:1-6
19. Yakar S, Liu JL, Stannard B. et al. Normal growth and development in the absence of hepatic insulin-like growth factor I. Proc Natl Acad Sci U S A. 1999;96:7324-7329
20. Kim H-S, Xiao C, Wang R-H. et al. Hepatic-specific disruption of SIRT6 in mice results in fatty liver formation due to enhanced glycolysis and triglyceride synthesis. Cell metabolism. 2010;12:224-236
21. Strable MS, Ntambi JM. Genetic control of de novo lipogenesis: role in diet-induced obesity. Crit Rev Biochem Mol Biol. 2010;45:199-214
22. Le Lay S, Lefrere I, Trautwein C. et al. Insulin and sterol-regulatory element-binding protein-1c (SREBP-1C) regulation of gene expression in 3T3-L1 adipocytes. Identification of CCAAT/enhancer-binding protein beta as an SREBP-1C target. J Biol Chem. 2002;277:35625-35634
23. Denechaud PD, Bossard P, Lobaccaro JM. et al. ChREBP, but not LXRs, is required for the induction of glucose-regulated genes in mouse liver. J Clin Invest. 2008;118:956-964
24. Yamashita H, Takenoshita M, Sakurai M. et al. A glucose-responsive transcription factor that regulates carbohydrate metabolism in the liver. Proc Natl Acad Sci U S A. 2001;98:9116-9121
25. Rogers CQ, Ajmo JM, You M. Adiponectin and alcoholic fatty liver disease. IUBMB Life. 2008;60:790-797
26. Ahmed MH, Byrne CD. Current treatment of non-alcoholic fatty liver disease. Diabetes Obes Metab. 2009;11:188-195
27. Deng CX, Xu X. Generation and analysis of Brca1 conditional knockout mice. Methods Mol Biol. 2004;280:185-200
28. Lakso M, Pichel JG, Gorman JR. et al. Efficient in vivo manipulation of mouse genomic sequences at the zygote stage. Proc Natl Acad Sci U S A. 1996;93:5860-5865
Author contact
![]() Corresponding author: Phone: 301-402-7225; Fax: 301-480-1135; Email: chuxiadniddk.nih.gov
Corresponding author: Phone: 301-402-7225; Fax: 301-480-1135; Email: chuxiadniddk.nih.gov